

Abstract
Guanine deaminase (GDA; cypin) is an important metalloenzyme that processes the first step in purine catabolism, converting guanine to xanthine by hydrolytic deamination. In higher eukaryotes, GDA also plays an important role in the development of neuronal morphology by regulating dendritic arborization. In addition to its role in the maturing brain, GDA is thought to be involved in proper liver function since increased levels of GDA activity have been correlated with liver disease and transplant rejection. Although mammalian GDA is an attractive and potential drug target for treatment of both liver diseases and cognitive disorders, prospective novel inhibitors and/or activators of this enzyme have not been actively pursued. In this study, we employed the combination of protein structure analysis and experimental kinetic studies to seek novel potential ligands for human guanine deaminase. Using virtual screening and biochemical analysis, we identified common small molecule compounds that demonstrate a higher binding affinity to GDA than does guanine. In vitro analysis demonstrates that these compounds inhibit guanine deamination, and more surprisingly, affect GDA (cypin)-mediated microtubule assembly. The results in this study provide evidence that an in silico drug discovery strategy coupled with in vitro validation assays can be successfully implemented to discover compounds that may possess therapeutic value for the treatment of diseases and disorders where GDA activity is abnormal.
1. Introduction
Over the last four decades, guanine deaminase (GDA) has been studied as a critical enzyme in the purine salvage pathway in both prokaryotes and eukaryotes. GDA is a metalloenzyme that catalyzes the first step in purine catabolism by converting guanine to xanthine by hydrolytic deamination. GDA also regulates the total cellular purine-derived nucleotide pool by converting adenylic derivatives to guanine [1-2]. Since GDA activity is involved in guanine metabolism, this enzyme is essential for the regulation of intracellular levels of guanylic derivatives [2]. Furthermore, in higher eukaryotes, GDA (also known as cypin) plays an important role in the development of neuronal morphology [3-5]. Promotion of dendrite branch formation by GDA is dependent on breakdown of guanine as substrate [3-6].
In addition, abnormally high levels of GDA activity occur in serum from patients suffering from liver diseases when compared to levels in healthy adults [7]. A strong correlation is observed between high GDA activity and patients with chronic hepatitis, biliary obstruction, and liver cirrhosis. In fact, GDA activity measurements are still currently used as a sensitive index for the diagnosis of acute liver diseases and liver transplant rejection [8-10].
Although GDA serves as an attractive drug target for the prospective treatment of purine metabolism deficiency, liver diseases, and cognitive disorders, novel ligands, which may act as clinically significant inhibitors and/or activators of the enzyme, have not been intensely investigated. A small number of guanine analogues have been studied in the past, and these analogues were selected primarily due to their structural similarity to guanine [11-13]. In addition, azepinomycin, an antibiotic and antitumor agent derived from the culture filtrate of Streptomyces sp. MF718 [14], acts as a GDA inhibitor by inhibiting the binding of guanine to GDA in a competitive manner [15-17]. Although azepinomycin analogues are potential inhibitors of GDA, all experimentally tested analogues show lower binding affinity to GDA than does guanine [17]. Consequently, there is significant interest in discovering novel GDA ligands that may lead to potential therapeutics for the treatment of liver disease and cognitive disorders.
Here, we report the discovery of novel ligands for human GDA using iterative methods in rational (computer-aided) drug design and in vitro biochemical evaluation. The availability of a high-resolution X-ray crystal structure of human GDA enabled us to employ methods in structure-based drug design (SBDD) and target-based virtual screening of potential ligands. Structural refinement using energy minimization and molecular dynamics simulations was performed to assess the structural integrity and plasticity of the GDA-guanine binding site in an aqueous environment. To calibrate the computational predictions and experimental measurements, a series of known competitive ligands were docked and scored for comparison with their known biochemically measured inhibitory activity. In addition, using rabbit GDA as a model mammalian enzyme, we performed kinetic experiments using untested GDA ligands and obtained a series of compounds for which the binding affinity for GDA was higher than guanine. Surprisingly, we also found that these compounds can inhibit GDA (cypin)-mediated microtubule polymerization, suggesting that they might modulate cellular microtubule assembly and dendrite development. The results of this study provide evidence that through the use of computational drug discovery approaches coupled with biological evaluation, small molecule ligands of GDA can be identified that modify GDA enzymatic activity, and hence, may be used for treating disorders with abnormal dendrite morphology or aberrant purine metabolism.
2. Materials and Methods
All calculations were conducted on 3.2 GHz RedHat Linux workstations. Development of the computer-aided inhibitor analysis on the guanine deaminase (GDA; cypin) structure proceeded in three steps: 1) sequence analysis, 2) energy minimization (EM) and molecular dynamics (MD) simulations, and 3) inhibitor docking and scoring.
2.1 Sequence Analysis
All sequence data were obtained from the National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/), while structural data were obtained from the Research Collaboratory for Structural Bioinformatics-Protein Data Bank (RCSB-PDB). Evaluation of the primary sequence in the X-ray crystal structure of human guanine deaminase (hGDA; cypin; PDB ID =2UZ9) [20] was conducted using the hGDA protein sequence (accession number: NP_004284). Pairwise sequence alignment between NP_004284 and hGDA was conducted using the ClustalW1.83 routine with default parameters [31-32]. This alignment revealed a 22-residue (MHHHHHHSSGVDLGTENLYFQS) insertion in the N-terminus of the hGDA structure. In order to reproduce the Reference Sequence (RefSeq) from the NCBI human protein sequence database, the inserted peptide was manually extracted from the hGDA structure using Sybyl v7.2 (http://www.tripos.com) [33].
2.2 Energy minimization (EM) and molecular dynamics (MD) simulations
EM and MD calculations were performed to refine the hGDA structure using the AMBER9 force field (http://amber.scripps.edu/) [21]. To study the preservation and integrity of the three-dimensional structure of hGDA, MD simulations were first run on the modified hGDA crystal structure (2UZ9). The structure was checked for missing atoms using residue templates. The AMBER9 package added 3531 unresolved hydrogen atoms, and 6 extra atoms were manually extracted (LPD1 and LPD2 from Met1 and Met6; LPG1 and LPG2 from Cys2) from the raw structure using Sybyl v.7.2 to pass the AMBER9 parameters to neutralize the unit structure 16 Na+ ions in the AMBER9 package. The xanthine molecule, Zn2+ ion, and water molecules associated with the retrieved structure were extracted, and the unit was solvated by a 9 Å radius shell of TIP3 water molecules [34]. Subsequently, the solvated system was energy-minimized in two phases: first, the atoms in the protein structure were restrained using a 500 kcal/mol/rad force for 1000 iterations of constrained steepest descent (SD), and the water molecules and ions were free to move in order to eliminate bad contacts; second, the entire system was energy-minimized for 1000 iterations of SD followed by 2500 iterations of conjugate gradient (CG). Finally, MD simulations were run on the system for electrostatic and van der Waals (vdW) interactions, using the standard force-field parameter set parm99 in AMBER9 with dielectric constant ε = 1 and cutoff distance = 12.0 Å. During the MD simulations, the solvated system was coupled to a Berendsen bath at 300 K by using a coupling constant τT = 2 ps [35], and the temperature was gradually increased from 0 to 300 K over 10 fs of simulation time with the volume held constant (ensemble NVT). The hGDA structures derived from the final 300 ps MD simulation were averaged and energy-minimized using 1000 iterations of SD and 4000 iterations of CG. The average structure was used for further molecular docking experiments.
Molecular Docking of Inhibitors- Guanine and a list of 188 guanine analogues, including previously studied GDA inhibitors [15, 17-18] and novel compounds, were docked to the hGDA structure using GOLD (Genetic Optimization of Ligand Docking) [23]. The IUPAC names and chemical properties of all of the compounds tested are described in the Supplementary Material. The majority of the small molecules were retrieved from the NCBI PubChem Compound database, and others were built with Sybyl v7.2 (http://www.tripos.com) [33]. All molecules were optimized with the MMFF94 force field and Gasteiger-Hückel atomic charges. The number of poses for each compound was set to 10, and the default algorithm speed was selected during the docking procedure. The hGDA residues within a 10-Å radius of the centroid of the guanine/xanthine binding site were designated as the hGDA substrate/ligand pocket. During molecular docking, early termination was allowed if the RMSD of the top five bound conformations of the ligand were within 1.5 Å. If the compound was previously assigned an experimental inhibition constant, the top-ranked conformation of this compound was selected and the corresponding GOLD score was then correlated with the known inhibition constant. The binding conformations of inhibitors in the ATP-binding site were displayed using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081) [34].
2.3 Kinetic Assay for GDA inhibition
GDA kinetic assays were performed as previously described [15, 17, 37]. Briefly, purified guanine deaminase from rabbit liver (purchased from MP Biomedicals) was used for biochemical studies. Commercially available purine and guanine analogues were used in our assays. With the exception of N2-acetylguanine (Toronto Research Chemicals, Ontario, Canada), all tested compounds were purchased from Sigma-Aldrich. The colorimetric assays were performed at 25 °C and pH 7.4 in Tris buffered saline buffer (TBS; 25 mM Tris-HCl, 137 mM NaCl, 2.7 mM KCl; Fisher Scientific). Guanine hydrolysis rates were measured by optical density changes at 245 nm. An enzyme unit was defined as the amount of enzyme that deaminates 1.0 μmole of guanine to xanthine per minute at pH 7.4 and 25 °C. In the assay, a fixed concentration of 8 × 10−3 U guanine deaminase was incubated with seven guanine concentrations ([S] = 2, 5, 7, 10, 12, 15, 17 and 20 μM) and each inhibitor concentration (8 or 20 μM). Lineweaver–Burk plots (1/V vs 1/S) were used to calculate kinetic constants.
2.4 Guanine Deaminase Assay
To measure GDA enzyme activity, COS-7 cells were transfected with the indicated plasmids using Lipofectamine 2000 (Invitrogen) and 2.5 μg of DNA per 100 mm culture dish of pEGFP-C1 vector alone, containing the Rattus norvegicus wild-type full length, or containing mutant sequences of cypin. After 48 hours of expression, cells were washed twice with phosphate-buffered saline (PBS) and scraped into GDA lysis buffer (150 mM NaCl, 25 mM Tris-HCl, pH 7.4 and 1 mM PMSF). Lysates were homogenized by passing them through a 25-gauge needle five times and centrifuged at 9,500 × g at 4°C for 10 min. Concentration of cytosolic proteins in the supernatant was measured using the Bradford Assay. Protein samples were resolved by SDS-PAGE for equal cypin expression verification (data not shown). Fifty micrograms of lysate was then used for each guanine deaminase sample assay in GDA lysis buffer containing 0.025 U/ml xanthine oxidase (Sigma-Aldrich), 0.002 U/ml peroxidase (Sigma-Aldrich), Amplex Red reagent (Molecular Probes, Eugene, OR), and 125 mM guanine (Sigma-Aldrich, St. Louis, MO). Negative controls were performed using protein lysates in assay solution with no guanine added. Negative controls and samples were incubated at 37°C, and absorbance at 571 nm using a single beam Genesys 10 UV/Vis Spectrophotometer (Spectronic, Garforth, UK) was measured during the indicated time intervals after samples were centrifuged at 9,500 × g for 1 minute to remove insoluble guanine. Experiments were performed in triplicate, and results were normalized by subtracting nonspecific background absorbance from the negative controls.
2.5 Tubulin Polymerization and Competition Assay
The tubulin polymerization assay kit was purchased from Cytoskeleton (Denver, CO). Purified tubulin (>97% pure) was mixed with general tubulin buffer (GTB, 80 mM PIPES pH 6.9, 2 mM MgCl2, 0.5 mM EGTA, and 1 mM GTP) in a 96-well plate at 37°C. Absorbances at 340 nm were measured every 1 min for 70 min by a VICTOR X3 Multilabel Plate Reader with Wallac 1420 v.3.0 software (PerkinElmer, Waltham, MA). GST or GST-cypin (0.1 nmoles) was incubated with or without guanine analogues (0.1mM). Tubulin (3-4 mg/mL) was added in GTB buffer at 37°C for 5 min. The analysis was performed with GraphPad Prism software (La Jolla, CA).
3. Results
3.1 Characterization of Structure-Binding of Guanine Deaminase/Cypin
The X-ray crystal structures of the Bacillus subtillis and human GDA proteins have been recently solved with a bound Zn2+ ion at 1.17-Å and 2.30-Å resolutions, respectively [18-20]. Although zinc binding is conserved from bacteria to higher eukaryotes, the specific ion coordination diverges between these homologs. An imidazole environment with three histidines and an aspartate residue dominates the Zn2+ ion coordination in human GDA [6]. In contrast, the Zn2+ ion coordination in the bacterial protein of B. subtilis is maintained by two cysteines, one histidine, and a water molecule for conservation of the tetrahedral coordination. Since the substrate binding site of the enzyme is directly adjacent to the Zn2+ ion, and the ion itself has been proposed to play a role during the mechanism of deamination [18-19], this coordination difference influences the stability of substrate binding and product release in the enzymatic reaction.
The hGDA X-ray crystal structure was solved with a xanthine molecule bound to the specific substrate-binding site [20]. To develop new substrates that show high affinity and specificity for hGDA, we performed a detailed characterization of the substrate-binding site. Analysis of the intramolecular interactions between the substrate or product molecules and the residues in hGDA within this site revealed that hydrophobic and van der Waals forces control the heterocyclic aromatic ring interactions in the guanine (Figure 1A) and xanthine molecule (Figure 1B). The carbonyl groups in the purine ring form hydrogen bonds with two nitrogen atoms in residues Arg213 and Gln87. Moreover, the amine group in the guanine molecule forms two hydrogen bonds with the imidazole ring in His279 and the carboxylic acid side chain in Glu243. These essential interactions between the substrate (guanine) and residues in hGDA must be considered during substrate modification so as to increase enzyme-compound specificity.
Figure 1.
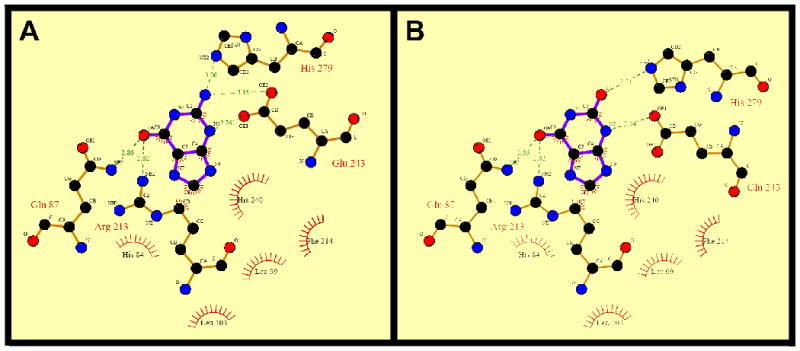
Protein-ligand interactions in the substrate-binding site of human GDA. (A) Guanine-hGDA interactions. (B) Xanthine-hGDA interactions. Two-dimensional representation of ligand–protein interactions were analyzed using Ligplot [54]. Hydrogen bonds formed between protein residues and ligands are indicated by dashed green lines and the corresponding distance (Å) and residues engaged in hydrophobic interactions are represented in red.
3.2 Energy Minimization and Molecular Dynamics Simulations
To refine the X-ray crystal structure coordinates and analyze the structure stability in a realistic water solvated system, we performed Energy Minimization (EM) to diminish possible interatomic clashes during molecular ligand docking experiments. We used the AMBER9 package and force field [21] to assess if the experimentally retrieved structure parameters preserve the three-dimensional structure of hGDA. As shown in Figure 2A, we compared EM results from a high-energy unstable structure to a low-energy structure. Molecular dynamics (MD) simulations on the hGDA structure raised confidence in structure stabilization, as evidenced by the rapid convergence of the root mean square deviation (RMSD) of the α-carbon backbone in the structure inside of the water solvated system (Figure 2B).
Figure 2.
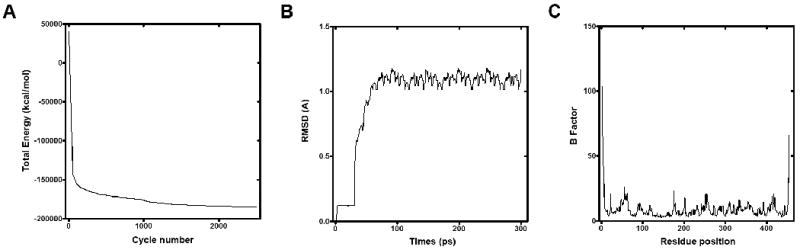
Energy Minimization and Molecular Dynamics studies of human GDA. (A) Total structure energy plot against the minimization cycle number. (B) The α-carbon backbone RMSD of hGDA determined from its starting conformation obtained from a 300ps simulation. (C) B factor plot against the primary sequence in hGDA.
Eukaryotic GDA has been implicated in diverse cellular signaling pathways, including the regulation of protein trafficking to the postsynaptic density (PSD) of excitatory neurons [3, 22]. GDA binds to the first and second PDZ domains of PSD-95, a scaffolding protein, via its PDZ-binding tail, consisting of the last four amino acids at the carboxyl terminus (-SSSV) [22]. To investigate the dynamics of these four carboxyl terminal residues, we determined the Debye-Waller factor (DWF) or B-factor of the hGDA crystal structure against the residue position in the primary sequence of the protein after the MD simulation. As shown in Fig. 3C, only the first and last amino acids demonstrated a fluctuation in atom-angle divergence about their average positions, suggesting that these residues are very dynamic compared to the other residue positions in the protein.
Figure 3.
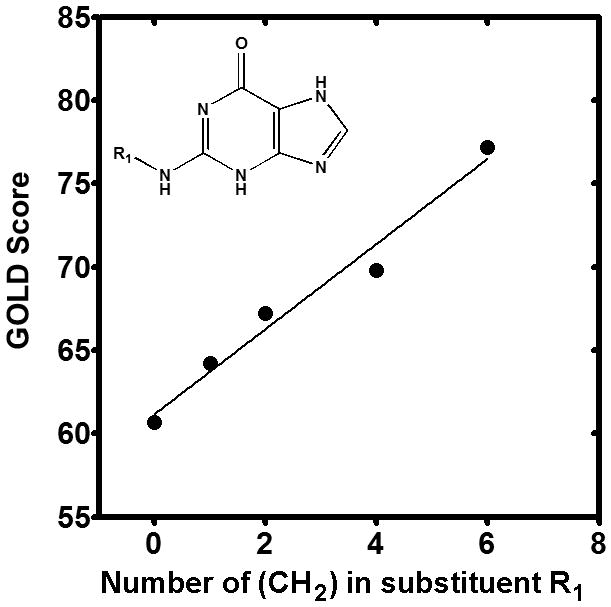
Alkane group substitutions in the guanine catalytic amine group increase binding affinity to hGDA. Molecular Docking results using the GOLD score are plotted against number of alkane groups added to the R1 position in the guanine molecule (inset). A linear correlation is noted (r2 = 0.78).
3.3 Molecular Docking of Guanine Analogues
To identify ligand candidates for the human GDA enzyme, an in silico molecular docking approach was conducted to assess the suitability of the hGDA structure for structure-based drug design and structure-activity relationship (SAR) studies. We used the docking and scoring algorithm implemented in the genetic algorithm for protein-ligand in GOLD [23]. Our system consisted of the water solvated and stable protein structure and a list of 188 guanine analogues, including previously studied GDA inhibitors [15, 17-18] and novel compounds. The chemical properties and docking scores of all of the compounds tested are described in the Supplementary Material. A total of 30 compounds (16%) show better fitness scores than the normal substrate (guanine). Among these compounds, we observed that five of them contain different alkane group numbers at the same position. When we compared the (CH3)n group number against their GOLD Score in hGDA, there was a strong linear correlation within the homologous series of alkane side chains (r2 = 0.78), suggesting that extension of the alkane group can increase the binding affinity of the ligand, possibly by increasing hydrophobic interactions with hGDA (Figure 3).
3.4 Kinetic Analysis of Guanine Analogues
To determine whether the ligands identified can modify the enzymatic activity of mammalian GDA, we performed competitive kinetic experiments using purified GDA enzyme with commercially available compounds, and determined their experimental binding affinity constants. Lineweaver–Burk plots were used to calculate inhibition constants (Ki) as shown in Figure 4, and results are tabulated in Table I. Our biochemical results demonstrate that guanine has a Km of 11.21 ± 0.11 μM. Interestingly, most of the tested compounds significantly and competitively inhibited guanine binding to GDA, with higher Ki values than the guanine Km, with the exception of caffeine, which has a lower Ki value than guanine. These results suggest that the compounds tested may be competitive inhibitors of the mammalian GDA and may bind in vivo with high affinity to alter normal GDA activities in guanine metabolism.
Figure 4.
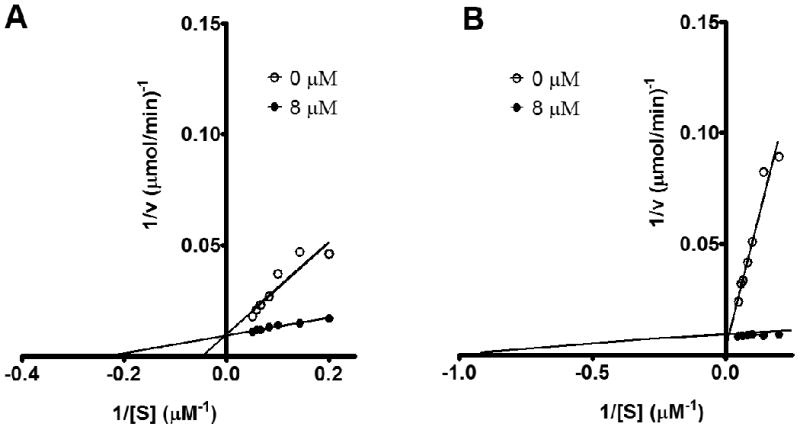
Kinetics of novel guanine deaminase susbtrates. Lineweaver-Burk plots were used to calculate kinetic constants and substrate type analysis using a competitive substrate assay with guanine. Seven guanine concentrations in the micromolar (μM) range were combined with 0 (no compound) or 8 μM of each inhibitor. Representative plots of (A) Xanthine and (B), 5-amino-4-imidazole carboxamide are shown.
Table 1. Properties of experimentally tested compounds for mammalian guanine deaminase activity.
| Structure | Name | Docking Score | Km or Ki (μM)a |
|---|---|---|---|
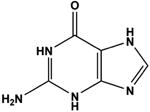 |
Guanine b | 61 | 11.21 ± 0.11 |
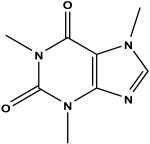 |
Caffeine c | 47 | 10.20 ± 0.16 |
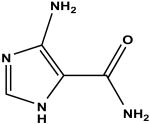 |
5-amino-4-imidazole carboxamide c | 51 | 4.44 ± 0.09 |
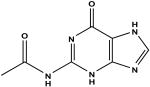 |
N-2-acetylguanine c | 54 | 3.44 ± 0.18 |
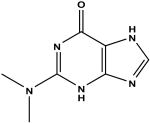 |
2-dimethylamino-6-hydroxypurine d | 71 | 1.09 ± 0.11 |
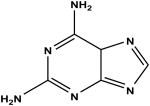 |
2,6-diaminopurine c | 50 | 1.88 ± 0.13 |
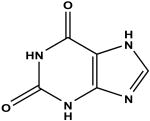 |
Xanthine c | 48 | 1.96 ± 0.07 |
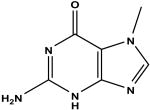 |
7-methylguanine c | 60 | 5.55 ± 0.14 |
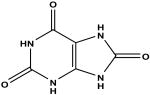 |
Uric acid c | 49 | 4.34 ± 0.21 |
Inhibition constants were derived from Lineweaver–Burk plots against guanine as substrate.
Guanine Deaminase Substrate
Guanine Deaminase Inhibitor
Enhances guanine deamination
3.5 Guanine Deaminase Activity is disrupted by Substrate Analogues
Competitive binding of ligands to the active site of GDA during deamination may affect enzymatic activity. Thus, we tested the identified small molecules in a GDA assay using GFP-tagged rat GDA-Cypin and purified rabbit guanine deaminase. COS-7 cells overexpressing GFP and GFP-tagged GDA (cypin) were lysed, and the lysates were subjected to a colorimetric GDA activity assay as previously described [3, 6]. Purified rabbit GDA was also used to test guanine analogues. Compounds that resemble the chemical structure of guanine and contain an amine group caused an attenuation of GDA activity measured over time (Figure 5). We were unable to test 6-thioguanine since the compound affected absorbance. Among the tested compounds, we found that 2,6-diaminopurine significantly attenuated GDA activity, suggesting that chemical substitutions in the 2- and 6-position in the guanine molecule can inhibit enzymatic activity. In contrast, 2-dimethylamino-6-hydroxypurine promoted GDA activity when guanine was added. The basis of this response may possibly due to a hydrolysis, resulting in the release of a dimethylamine molecule. These results indicate that only substrates that contain an amino group can compete with guanine for the GDA active site or that compounds with substitutions in the 2-position of the purine ring may enhance deamination.
Figure 5.

Guanine deaminase activity is disrupted by substrate analogues. (A) Lysates (50 μg) from COS-7 cells expressing GFP and GFP-cypin with guanine analogues (10μM) in DMSO were examined using the Amplex Red dye in a colorimetric GDA assay (n = 3). Lysates from cells expressing GFP do not contain GDA activity and were used as a negative control. (B) Purified rabbit GDA (8×10-4U) with guanine analogues (10μM) in DMSO was subjected to a GDA assay as in (A). Standard error is shown for each data point. Note that some standard error values are smaller than the data symbols.
3.6 GDA (Cypin)-Mediated Microtubule Assembly is regulated by Substrate Analogues
Conformational changes during substrate binding to GDA (cypin) may also affect microtubule assembly. Thus, we tested the possible regulation of cypin-promoted microtubule assembly by guanine analogues. We used a cell-free system, which permits the assessment of direct regulation by GDA substrates [3]. Purified tubulin was polymerized into microtubules in the presence of purified GST-tagged cypin with or without compounds dissolved in DMSO. The polymerization reaction path was monitored by changes in optical density over time (Figure 6). As established previously [3], we found that GST-cypin significantly increased the maximum velocity of microtubule polymerization compared to the GST control, suggesting that cypin promotes microtubule assembly in vitro. Among the tested compounds, we found that guanine and 7-methylguanine significantly inhibited cypin-promoted microtubule assembly. Xanthine and 2,6-diaminopurine had no effect, which is not inconsistent since microtubule assembly promoted by cypin is thought to be independent of guanine deamination. We were unable to test 6-thioguanine since the compound affected absorbance. Furthermore, none of the compounds shown in Figure 6 affected microtubule polymerization on their own (i.e. in the presence of GST). Together with the results shown in Figure 5, these results suggest that the substrate analogues can have differing effects on GDA activity and cypin-promoted microtubule assembly.
Figure 6.
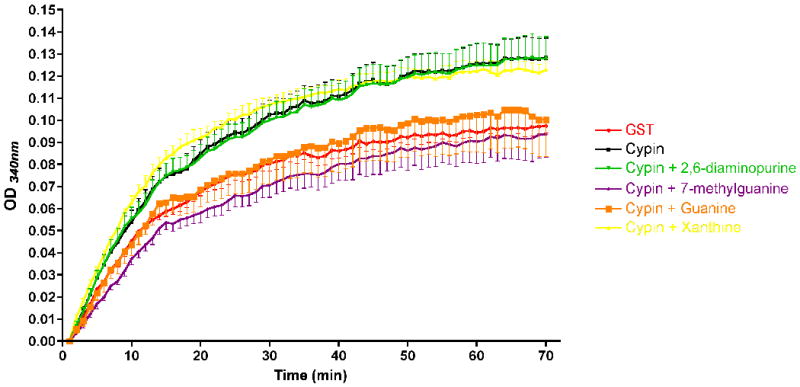
Cypin-mediated microtubule polymerization is affected by substrate analogues. A microtubule polymerization assay was performed using tubulin (>97% pure; 3 mg/mL) in a 96-well plate at 37°C. Absorbances at 340 nm were measured every 1 min for 70 min. GST or GST-cypin (0.1 nmoles) were incubated with or without guanine analogues (0.1 mM).
4. Discussion
Guanine deaminase plays an essential role in diverse cellular signaling pathways. Mammalian GDA is a potential drug target for treatment of purine metabolism deficiency and cognitive disorders by virtue of the fact that its enzymatic activity is necessary for normal purine salvage and brain development and function [3-5]. Previous kinetic studies using purified rabbit GDA were performed using guanine analogues selected simply for their structural similarity to guanine [11-13]. Although potential substrates can be revealed using this deductive method, more efficient and effective strategies are available to accelerate and streamline the process of finding potential inhibitors. The current availability of a high-resolution human GDA X-ray crystal structure affords the use of methods in rational (computer-aided) structure-based drug design for identifying effective inhibitors and/or activators of the mammalian GDA. In this study, we employed rational drug design approaches with the GDA X-ray crystal structure to study the ion and substrate coordination in human GDA. A major finding of our current work is that we identified residues involved in substrate binding to hGDA. Consequently, the identification of specific residues that play a major role in the deamination mechanism allowed us to implement rational drug design approaches using the described pharmacophore, rather than using substrate structural similarity selection, to obtain novel ligands candidates for the enzyme.
GDA orthologues are found in prokaryotes and lower eukaryotes, suggesting that purine metabolism is a critical cellular process. Although these orthologues share the same enzymatic activity, the mammalian GDA proteins acquired an additional task in neuronal development during evolution. A four-residue PDZ binding motif at the C-terminus is responsible for the interaction of and regulation of protein levels by GDA at the postsynaptic density [3-22]. These multi-functional activities of the mammalian GDA appear to be conferred by the specific three-dimensional amino acid arrangements of the internal ion and substrate binding sites.
Although a direct conclusive link between purine metabolism and cognitive function remains elusive, several studies revealed that neurobehavioral and cognitive impairment syndromes, including Lesch-Nyhan disease, are linked to changes in purine homeostasis resulting from purine salvage failure [24-25]. Abnormally high levels of guanine and derivatives have been shown to accumulate in media from Lesch-Nyhan disease cell line models [26]. A specific reduction in guanine to adenine nucleotides levels was also found in these cell lines [26]. In addition, other studies revealed that serum levels of uric acid, another guanine metabolite, are directly related to cerebral ischemia in adults [27]. These results suggest that enzymes involved in purine metabolism, like guanine deaminase, may play an essential role in regulating proper neuronal activity and communication.
Elevated GDA activity has been correlated with several liver diseases including hepatitis and liver transplantation rejection [9, 28-29]. As such, liver and serum GDA activities are currently used as molecular markers for liver disease diagnosis. Anomalous purine metabolism is reflected in clinical problems associated with nucleotide salvage during cellular and DNA repair, ranging from mild to severe and fatal disorders. Regulation of purine metabolism controls the cellular nucleotide pool, and the nucleotide levels available, directly affect DNA synthesis during cellular proliferation and differentiation. However, the major clinical manifestations of atypical purine catabolism arise from uric acid insolubility and its degradation, leading to inflammatory diseases like gout [30]. Small molecules, which can act as GDA inhibitors, could potentially attenuate uncontrolled purine metabolism. In addition, modulators of cypin (GDA)-promoted microtubule polymerization could impact neuronal development and morphology [3]. For example, attenuated cypin (GDA)-promoted microtubule polymerization may lead to too few dendrites and increased polymerization may lead to too many dendrites, thereby altering communication between neurons. Since anti-metabolic compounds have been used to treat purine metabolism-related diseases, our findings include a new array of molecules, which may inhibit overactivity of guanine catabolism seen in disease states. Similarly, our analysis has identified new GDA inhibitors with significantly lower binding constants (Ki) than those of previously described compounds, including azepinomycin, suggesting potential antimicrobial activity for these compounds.
Our current results show that the use of virtual screening is effective in narrowing down potential candidate molecules for drug targets. This screen is inexpensive and relatively quick. Since guanine deaminase and microtubule assembly assays are quite costly, choosing the top compounds (8 of 188 screened in silico) to use in these assays is efficient. Although the focus of this current study is to show the effectiveness of this screening, future experiments would focus on a more detailed analysis of Ki and IC50 values for compounds identified by the screen. Our work represents the first step in a general screening paradigm for analogues that alter guanine deaminase activity.
5. Conclusions
We have used a combination of computer-aided protein structure analysis and experimental kinetic studies to discover compounds as potential substrates or inhibitors of the human GDA for use in treating patients with purine metabolic disorders, liver disease, and/or cognitive dysfunction. Thus, our future studies will focus on identifying the specific activity of these newly identified compounds in models of neuronal and liver disease in order to screen candidates for therapies for these diseases.
Supplementary Material
Acknowledgments
We thank Vladyslav Kholodovych and Sandhya Kortagere for their technical support and the Brzustowicz laboratory for the use of the multilabel plate reader. We thank Dr. Erhard Bieberich for comments and critical reading of this manuscript. J.R.F. is a graduate trainee of the IGERT Program on Integratively Engineered Biointerfaces at Rutgers: NSF Grant DGE-0333196. E.S.S. is supported by NIH predoctoral training grant 5 T32 MH019957. This work was supported in part by a Busch Biomedical Grant, a New Jersey Governor's Council on Autism Pilot Grant, National Science Foundation Grant IBN-0548543, and March of Dimes Foundation Grants 1-FY04-107 and 1-FY08-464 (to B.L.F). This research was also supported in part by NIH Integrated Advanced Information Management Systems (IAIMS) Grant 2G08LM06230-03A1 from the National Library of Medicine (to W.J.W.).
Abbreviations
- GDA
Guanine Deaminase
- hGDA
human Guanine Deaminase
- GOLD
Genetic Optimization of Ligand Docking
- MD
molecular dynamics
- EM
energy minimization
- RMSD
root mean square deviation
- SBDD
structure-based drug design
- PSD
postsynaptic density
- DWF
Debye-Waller factor
Footnotes
Supporting Information Available. The chemical properties and docking scores of all of the compounds tested are described.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nygaard P, Bested SM, Andersen KA, Saxild HH. Microbiology. 2000;146(Pt 12):3061–9. doi: 10.1099/00221287-146-12-3061. [DOI] [PubMed] [Google Scholar]
- 2.Saint-Marc C, Daignan-Fornier B. Yeast. 2004;21:1359–63. doi: 10.1002/yea.1186. [DOI] [PubMed] [Google Scholar]
- 3.Akum BF, Chen M, Gunderson SI, Riefler GM, Scerri-Hansen MM, Firestein BL. Nat Neurosci. 2004;7:145–52. doi: 10.1038/nn1179. [DOI] [PubMed] [Google Scholar]
- 4.Chen M, Lucas KG, Akum BF, Balasingam G, Stawicki TM, Provost JM, Riefler GM, Jornsten RJ, Firestein BL. Mol Biol Cell. 2005;16:5103–14. doi: 10.1091/mbc.E05-02-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen H, Firestein BL. J Neurosci. 2007;27:8378–86. doi: 10.1523/JNEUROSCI.0872-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fernandez JR, Welsh WJ, Firestein BL. Proteins. 2008;70:873–81. doi: 10.1002/prot.21683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li P, Xu S, Gao B, Cao F. Hua Xi Yi Ke Da Xue Xue Bao. 1996;27:189–91. [PubMed] [Google Scholar]
- 8.Kuzmits R, Seyfried H, Wolf A, Muller MM. Enzyme. 1980;25:148–52. doi: 10.1159/000459240. [DOI] [PubMed] [Google Scholar]
- 9.Shiota G, Fukada J, Ito T, Tsukizawa M, Yamada M, Sato M. Jpn J Med. 1989;28:22–4. doi: 10.2169/internalmedicine1962.28.22. [DOI] [PubMed] [Google Scholar]
- 10.Matsunaga H, Honda H, Kubo K, Sannomiya K, Cui X, Toyota Y, Mori T, Muguruma N, Okahisa T, Okamura S, Shimizu I, Ito S. J Med Invest. 2003;50:64–71. [PubMed] [Google Scholar]
- 11.Baker BR, Santi DV. J Med Chem. 1967;10:62–4. doi: 10.1021/jm00313a012. [DOI] [PubMed] [Google Scholar]
- 12.Baker BR, Wood WF. J Med Chem. 1967;10:1101–5. doi: 10.1021/jm00318a026. [DOI] [PubMed] [Google Scholar]
- 13.Baker BR. J Med Chem. 1967;10:59–61. doi: 10.1021/jm00313a011. [DOI] [PubMed] [Google Scholar]
- 14.Isshiki K, Takahashi Y, Iinuma H, Naganawa H, Umezawa Y, Takeuchi T, Umezawa H, Nishimura S, Okada N, Tatsuta K. J Antibiot (Tokyo) 1987;40:1461–3. doi: 10.7164/antibiotics.40.1461. [DOI] [PubMed] [Google Scholar]
- 15.Rajappan V, Hosmane RS. Bioorg Med Chem Lett. 1998;8:3649–52. doi: 10.1016/s0960-894x(98)00672-6. [DOI] [PubMed] [Google Scholar]
- 16.Wang L, Hosmane RS. Bioorg Med Chem Lett. 2001;11:2893–6. doi: 10.1016/s0960-894x(01)00591-1. [DOI] [PubMed] [Google Scholar]
- 17.Ujjinamatada RK, Bhan A, Hosmane RS. Bioorg Med Chem Lett. 2006;16:5551–4. doi: 10.1016/j.bmcl.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 18.Liaw SH, Chang YJ, Lai CT, Chang HC, Chang GG. J Biol Chem. 2004;279:35479–85. doi: 10.1074/jbc.M405304200. [DOI] [PubMed] [Google Scholar]
- 19.Chang YJ, Huang CH, Hu CY, Liaw SH. Acta Crystallogr D Biol Crystallogr. 2004;60:1152–4. doi: 10.1107/S0907444904008558. [DOI] [PubMed] [Google Scholar]
- 20.Moche M, Welin M, Arrowsmith C, Berglund H, Busam R, Collins R, Dahlgren LG, Edwards A, Flodin S, Flores A, Graslund S, Hammarstrom M, Hallberg BM, Holmberg-Schiavone L, Johansson I, Kallas A, Karlberg T, Kotenyova T, Lehtio L, Nyman T, Ogg D, Persson C. Structural Genomics Consortium (Sgc) 2007. [Google Scholar]
- 21.Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. J Comput Chem. 2005;26:1668–88. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Firestein BL, Brenman JE, Aoki C, Sanchez-Perez AM, El-Husseini AE, Bredt DS. Neuron. 1999;24:659–72. doi: 10.1016/s0896-6273(00)81120-4. [DOI] [PubMed] [Google Scholar]
- 23.Jones G, Willett P, Glen RC, Leach AR, Taylor R. J Mol Biol. 1997;267:727–48. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 24.Schretlen DJ, Harris JC, Park KS, Jinnah HA, del Pozo NO. J Int Neuropsychol Soc. 2001;7:805–12. doi: 10.1017/s135561770177703x. [DOI] [PubMed] [Google Scholar]
- 25.Jinnah H, Friedmann T. In: The Metabolic and Molecular Basis of Inherited Disease. Scriver C, B A, Sly W, Valle D, editors. McGraw-Hill; New York: 2000. pp. 2537–2570. [Google Scholar]
- 26.Shirley TL, Lewers JC, Egami K, Majumdar A, Kelly M, Ceballos-Picot I, Seidman MM, Jinnah HA. J Neurochem. 2007;101:841–53. doi: 10.1111/j.1471-4159.2007.04472.x. [DOI] [PubMed] [Google Scholar]
- 27.Schretlen DJ, Inscore AB, Vannorsdall TD, Kraut M, Pearlson GD, Gordon B, Jinnah HA. Neurology. 2007;69:1418–23. doi: 10.1212/01.wnl.0000277468.10236.f1. [DOI] [PubMed] [Google Scholar]
- 28.Nishikawa Y, Fukumoto K, Watanabe F, Yoshihara H. Rinsho Byori. 1989;37:1392–4. [PubMed] [Google Scholar]
- 29.Crary GS, Yasmineh WG, Snover DC, Vine W. Transplant Proc. 1989;21:2315–6. [PubMed] [Google Scholar]
- 30.Kutzing MK, Firestein BL. J Pharmacol Exp Ther. 2008;324:1–7. doi: 10.1124/jpet.107.129031. [DOI] [PubMed] [Google Scholar]
- 31.Thompson JD, Higgins DG, Gibson TJ. Nucleic Acids Res. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Nucleic Acids Res. 2003;31:3497–500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sybyl. 7.2. Tripos, Inc.; St. Louis, MO: [Google Scholar]
- 34.Ryckaert JP, Ciccotti G, Berendsen HJC. Journal of Computational Physics. 1977;23:327–341. [Google Scholar]
- 35.Berendsen HJC, Postm JPM, Van Gunsteren WF, Di Nola A, Haak JR. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 36.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 37.Lewis AS, Glantz MD. J Biol Chem. 1974;249:3862–6. [PubMed] [Google Scholar]
- 38.Ali S, Sitaramayya A, Kumar KS, Krishnan PS. Biochem J. 1974;137:85–92. doi: 10.1042/bj1370085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gupta NK, Glantz MD. Arch Biochem Biophys. 1985;236:266–76. doi: 10.1016/0003-9861(85)90626-5. [DOI] [PubMed] [Google Scholar]
- 40.Kanzawa F, Hoshi A, Kuretani K. Chem Pharm Bull (Tokyo) 1971;19:1737–8. doi: 10.1248/cpb.19.1737. [DOI] [PubMed] [Google Scholar]
- 41.Kanzawa F, Hoshi A, Nishimoto T, Kuretani K. Chem Pharm Bull (Tokyo) 1970;18:392–4. doi: 10.1248/cpb.18.392. [DOI] [PubMed] [Google Scholar]
- 42.Kidder GW, Nolan LL. Mol Biochem Parasitol. 1981;3:265–9. doi: 10.1016/0166-6851(81)90001-3. [DOI] [PubMed] [Google Scholar]
- 43.Kumar KS, Krishnan PS. Biochem Biophys Res Commun. 1970;39:600–8. doi: 10.1016/0006-291x(70)90246-9. [DOI] [PubMed] [Google Scholar]
- 44.Kumar S. Arch Biochem Biophys. 1969;130:692–4. doi: 10.1016/0003-9861(69)90093-9. [DOI] [PubMed] [Google Scholar]
- 45.Kumar S. Experientia. 1970;26:217. doi: 10.1007/BF01895595. [DOI] [PubMed] [Google Scholar]
- 46.Kumar S, Josan V, Sanger KC, Tewari KK, Krishnan PS. Biochem J. 1967;102:691–704. doi: 10.1042/bj1020691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar S, Rathi M. Neurochem Res. 1980;5:547–50. doi: 10.1007/BF00964991. [DOI] [PubMed] [Google Scholar]
- 48.Lucacchini A, Montali U, Segnini D. Ital J Biochem. 1977;26:27–36. [PubMed] [Google Scholar]
- 49.Nolan LL, Kidder GW. Antimicrob Agents Chemother. 1980;17:567–71. doi: 10.1128/aac.17.4.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Solaini G, Rossi CA. Ital J Biochem. 1982;31:253–60. [PubMed] [Google Scholar]
- 51.Takano Y, Hase-Aoki K, Horiuchi H, Zhao L, Kasahara Y, Kondo S, Becker MA. Life Sci. 2005;76:1835–47. doi: 10.1016/j.lfs.2004.10.031. [DOI] [PubMed] [Google Scholar]
- 52.Thuillier L, Perignon JL, Houllier AM, Munier A, Cartier P. Biochim Biophys Acta. 1984;798:343–9. doi: 10.1016/0304-4165(84)90108-9. [DOI] [PubMed] [Google Scholar]
- 53.Wallace AC, Laskowski RA, Thornton JM. Protein Eng. 1995;8:127–34. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 54.Wang W, Hedstrom L. Biochemistry. 1998;37:11949–52. doi: 10.1021/bi981132w. [DOI] [PubMed] [Google Scholar]
- 55.Yao L, Cukier RI, Yan H. J Phys Chem B. 2007;111:4200–10. doi: 10.1021/jp0673056. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.