

Abstract
The orderly formation of the nervous system requires a multitude of complex, integrated and simultaneously occurring processes. Neural progenitor cells expand through proliferation, commit to different cell fates, exit the cell cycle, generate different neuronal and glial cell types, and new neurons migrate to specified areas and establish synaptic connections. Gestational and perinatal exposure to environmental toxicants, pharmacological agents and drugs of abuse produce immediate, persistent or late-onset alterations in behavioral, cognitive, sensory and/or motor functions. These alterations reflect the disruption of the underlying processes of CNS formation and development. To determine the neurotoxic mechanisms that underlie these deficits it is necessary to analyze and dissect the complex molecular processes that occur during the proliferation, neurogenesis and differentiation of cells. This symposium will provide a framework for understanding the orchestrated events of neurogenesis, the coordination of proliferation and cell fate specification by selected genes, and the effects of well-known neurotoxicants on neurogenesis in the retina, hippocampus and cerebellum. These three tissues share common developmental profiles, mediate diverse neuronal activities and function, and thus provide important substrates for analysis. This paper summarizes four invited talks that were presented at the 12th International Neurotoxicology Association meeting held in Jerusalem, Israel during the summer of 2009. Donald A. Fox described the structural and functional alterations following low-level gestational lead exposure in children and rodents that produced a supernormal electroretinogram and selective increases in neurogenesis and cell proliferation of late-born retinal neurons (rod photoreceptors and bipolar cells), but not Müller glia cells, in mice. Lisa Opanashuk discussed how dioxin [TCDD] binding to the arylhydrocarbon receptor [AhR], a transcription factor that regulates xenobiotic metabolizing enzymes and growth factors, increased granule cell formation and apoptosis in the developing mouse cerebellum. Alex Zharkovsky described how postnatal early postnatal lead exposure decreased cell proliferation, neurogenesis and gene expression in the dentate gyrus of the adult hippocampus and its resultant behavioral effects. Bernard Weiss illustrated how environmental endocrine disruptors produced age- and gender-dependent alterations in synaptogenesis and cognitive behavior.
Keywords: retina, cerebellum, hippocampus, lead, dioxin, endocrine disrupters
Low-Level Human Equivalent Gestational Lead Exposure Produces Supernormal Scotopic Electroretinograms, Increased Retinal Neurogenesis (Donald A. Fox)
The adverse effects of low-level developmental lead exposure ([BPb] ≤10 μg/dL) on cognitive, auditory and visual-motor function are well-documented [1–4]. In contrast, few studies examined the effect of low-level developmental lead exposure on retinal and visual function in children [5,6], despite findings that retinal and visual cortical abnormalities occur in animals following low (≤10 μg/dL) [7], moderate- ([BPb] 11–39 μg/dL) and high-level ([BPb] ≥40 μg/dL) developmental lead exposure [8–10].
Persistent rod photoreceptor-mediated (scotopic) electroretinographic (ERG) and behavioral deficits occur in monkeys, rats and mice following moderate- and high-level postnatal lead exposure [8–10]. The scotopic ERG alterations were characterized by decreases in a-wave and b-wave ERG amplitude (subnormality), sensitivity and temporal resolution. Similar ERG changes occurred in lead-exposed workers and isolated retinas exposed to Pb2+ [9,10]. In contrast, adult monkeys and rats with gestational lead exposure exhibited increased ERG b-wave amplitudes (supernormality) [7,11]. Moreover, prospective epidemiological study of children with lifetime low-level and moderate-level lead exposure revealed that only gestational lead exposure (GLE) showed a significant dose-dependent relationship with supernormal ERG a-waves and b-waves and increased b-wave sensitivity with no change in implicit times [6].
To determine the sites and mechanisms of the GLE-induced supernormal scotopic ERGs, we used a new dose-response model of GLE [12] and conducted scotopic ERG studies and retinal histological, and morphometric experiments in adult offspring [7]. Control dams received water and GLE dams received one of two lead drinking solutions: 27 ppm lead (low) or 55 ppm lead: (moderate) Lead drinking solutions were provided to dams two weeks prior to mating, throughout gestation and until PN10. The prenatal through PN10 period was selected because rodent brain and retinal development during this period is equivalent to that during human gestation [13–15]. Control, low- and moderate-level GLE produced concentration-dependent increases in [BPb] at PN0-10 with peak [BPb] of 1, 10–12 and 21–24 μg/dL, respectively. By PN30, [BPb] in GLE rats was not significantly different from controls.
As shown in Table 1, single-flash scotopic ERGs at PN90 revealed significant dose-dependent increases in a-wave amplitude measured at 10 msec (direct measure of rod function: [16], peak a-wave amplitude and peak b-wave amplitude (measure of depolarizing rod bipolar function: [17]. There were no changes in a-wave or b-wave latencies.
Table 1.
Gestational lead exposure produces scotopic ERG supernormality and increased proliferation of rod photoreceptors and bipolar cells in adult rats.
| DEPENDENT MEASURES AT POSTNATAL DAY 90 | HUMAN EQUIVALENT GESTATIONAL LEAD EXPOSURE | |
|---|---|---|
| LOW-DOSE (27 ppm) | MODERATE- DOSE (55 ppm) | |
| ERG A-wave amplitude at 10 msec (μV) | 30% | 30% |
| ERG peak a-wave amplitude (μV) | 12–16% | 22–26% |
| ERG peak b-wave amplitude (μV) | 14–19% | 28–31% |
| ERG a-wave latency (msec) | NSD | NSD |
| ERG b-wave latency (msec) | NSD | NSD |
| Outer nuclear layer thickness (μm) | 17% | 24% |
| Inner nuclear layer thickness (μm) | 40% | 50% |
| Total retinal thickness (μm) | 10% | 16% |
| Number of rods/100 μm length | 16–18% | 35–38% |
| Number of cones/100 μm length | NSD | NSD |
| Rod bipolar cells (PKCαβ content) | 21% | 37% |
| Retinal GFAP content (ng/mg protein) | NSD | NSD |
All percent data, analyzed with ANOVA and post-hoc from original ratio scale data, were significantly different from controls at p <0.05. NSD = not significantly different.
Representative images of DAPI (4′,6-diamidino-2-phenylindole) stained adult GLE retinas reveal that low-level and moderate-level GLE increased ONL and INL thickness (Figure 1). These increases reflect an increase of 1–3 nuclei per retinal layer. In controls, the ONL, INL and total central retinal thickness was 38.7 ± 3.0, 17.5 ± 1.6 and 201.2 ± 2.1 μm, respectively. Low-level and moderate-level GLE significantly increased ONL thickness, INL thickness and total retinal thickness (Table 1). In control central and peripheral retina, the number of rod nuclei was 137.8 ± 5.6 and 87.3 ± 4.3 per 100 μm, respectively, and of cone nuclei was 2.9 ± 0.3 and 1.7 ± 0.3 per 100 μm, respectively: values consistent with our previous results [18]. Low-level and moderate-level GLE exhibited significant dose-dependent increases in the number of rods in central and peripheral retina (Table 1). The number of cones was not changed significantly by GLE treatment.
Figure 1. Histological staining of adult control and gestationally lead-exposed rat with the nuclear stain DAPI.

Relative to controls, the thickness of the outer (ONL) and inner (INL) nuclear layer increased in the low-level and moderate-level gestationally lead-exposed rats. Scale bar equals 20 μm.
Morphometric analysis showed that low-level and moderate-level GLE increased INL thickness (Figure 1). Examination of the retinas suggested that the number of BCs was increased by GLE: consistent with our supernormal scotopic ERG b-wave results (Table 1). Control rat retinas immunostained for PKCαβ show that the entire rod BC selectively labeled with PKCαβ [7,19]. PKCαβ content was significantly increased in the low-level and moderate-level GLE groups (Table 1), consistent with the increase in INL thickness and supernormal scotopic ERB b-waves. Moreover, there were no significant effects of GLE on retinal glial fibrillary acid protein (GFAP) expression at any age [7], indicating that there was no change in the number of Müller glial cells and no retinal injury.
In summary, our results show that low-level and moderate-level GLE: produced supernormal scotopic ERGs in adult rats that are similar to our ERG findings in male and female children with GLE [6] and increased neurogenesis of rod photoreceptors and rod BCs without affecting Müller glial cells. The [BPb] in the low-level and moderate-level GLE groups were similar to that measured in pregnant mothers whose children had supernormal ERGs [6]. Additionally, the scotopic ERG is a sensitive non-invasive biomarker that identified and discriminated supernormal ERGs resulting from GLE from postnatal/adult lead exposure that produces subnormal ERGs [8–10]. The GLE-induced increase in rods and rod BCs likely explains the presence of supernormal scotopic ERGs, since this would significantly amplify the rod signal. The molecular mechanism responsible for this novel retinal phenotype is unknown. BrdU pulse-labeling immunohistochemical data indicates that it is due to increased proliferation of retinal progenitor cells and not due to decreased number of TUNEL- (terminal deoxynucleotidyl transferase dUTP nick end labeling) and propidium iodide-positive apoptotic cells during development (Fox and co-workers, unpublished data). Taken together, our results reveal dose-dependent and developmental stage-dependent effects of lead exposure as moderate- to high-level postnatal lead exposure produces rod-selective apoptosis [20–22] and decreased proliferation and/or neurogenesis in the rat hippocampus [23–25], whereas low- to moderate GLE increased retinal proliferation [7] and produced late-onset male-specific obesity [12].
Activation of the Ah Receptor by Dioxin Impacts Proliferation and Differentiation of Neural Precursor Cells (Lisa Opanashuk)
TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxin)-induced toxicity is mediated via binding to the aryl hydrocarbon receptor (AhR), a ligand-activated transcription factor that is a member of the basic helix-loop-helix/Per-Arnt-Sim (bHLH/PAS) superfamily [26]. These transcription factors are unique in that they serve as environmental sensors and transducers of physiological signals, particularly during development. Certain bHLH/PAS transcriptional regulators have been implicated in controlling cell fate determination, proliferation, and differentiation [27,28]. In the absence of ligand, the AhR is localized in the cytoplasm bound to several chaperone proteins [29,30]. Upon ligand binding, the AhR translocates into the nucleus and forms a heterodimer with the AhR nuclear translocator protein (Arnt), a related bHLH/PAS protein. The AhR/Arnt complex then binds to AhR-response elements (AhRE) located in DNA promoter regions and regulates the transcription of target genes [31,32]. AhR activation by TCDD is known to alter the expression of a wide array of drug metabolizing enzymes, cell cycle regulatory genes, and inflammatory mediators [33–35]. The list of genes that contain AhREs in their 5′ regulatory regions is expanding and many of these include growth modulatory proteins, such as Hes-1 and p27kip1 [36]. AhR participation in development is suggested by numerous defects observed in AhR null and nuclear localization mutant mice [37–45]. Despite much research, the normal functions of the AhR are unknown. Moreover, the search for an endogenous ligand that regulates transcriptional activity under physiological conditions has been largely unsuccessful. Nevertheless, most information regarding AhR function has been obtained from studies that involve the persistent environmental contaminant TCDD, which has the highest binding affinity among all of the reported ligands [46,47].
TCDD, via binding and activating AhR, alters normal gene expression and causes diverse biochemical and pathological abnormalities [46–52]. Various deficits have been observed in the nervous systems following perinatal exposure to TCDD and other dioxin-like compounds [53,54]. However, direct correlations between AhR-directed gene modulation and specific functional consequences have yet to be defined. Our laboratory determined that AhR is transcriptionally-active in the developing cerebellum [55]. We established that cerebellar granule neuron precursors (GNPs) expressed high levels of AhR during a critical period of neurogenesis and that TCDD interfered with maturation of these cells [40,55]. Our data also indicate that TCDD exposure altered the expression of several genes that mediate cell cycle regulation, transcriptional activity, mitogenic action, and apoptosis in GNPs (Table 2). Despite the existing information regarding molecular mechanisms by which TCDD regulates AhR target gene expression, specific roles for AhR during neurogenesis and in neurotoxicity are poorly understood.
Table 2.
TCCD exposure modifies gene expression in granule neuroblasts (GNP).
| GENES | FOLD LEVEL of EXPRESSION |
|---|---|
| Cyclin B2 | 1.9 |
| Cyclin D2 | 2.1 |
| n-myc | 2.6 |
| Akt1 | 1.7 |
| Integrin-like kinase | 2.4 |
| IGFB-6 | 2.4 |
| GLI2 | 2.8 |
| Zic1, Zic2 | 1.7 |
| Lamin receptor | 2.5 |
| Zipro 1 | 1.9 |
| EN1 | 1.9 |
| NF-kB p65 | 1.9 |
| Integrin a7 | 2.3 |
| Ephrin B1 | 2.3 |
| P55CDC | 2.9 |
| Bax | 2.3 |
Significant genes in boldface are associated with positive regulation of proliferation.
Recent evidence suggests that AhR participates in normal brain development. AhR and Arnt orthologues, are both expressed in C. elegans [56]. However, AhR lacks a ligand binding domain in invertebrates, so TCDD does not bind to AhR or induce a toxic response in these organisms. Evidence provided by loss of function experiments in C. elegans models indicate that AhR regulates neuronal fate decisions, differentiation, and migration during development [57,58]. More recent studies have demonstrated that Spineless, the mammalian AhR homologue in Drosophila, is important for controlling dendrite morphogenesis during neuronal development [59]. These observations suggest an ancestral role for the AhR family, which could even be ligand independent. Since AhR is present and coordinately regulated with Arnt in the embryonic mouse neuoroepithelium during prenatal neurogenesis [60–62] and in cerebellar GNPs during a critical window of postnatal maturation [55], it is conceivable that AhR mediates similar developmental events in mammalian systems.
Evidence at the behavioral, cellular and molecular levels indicates that the developing cerebellum is vulnerable to TCDD exposure. Epidemiological studies have suggested that accidental developmental exposures to PCB/dioxin mixtures is associated with delayed motor development, higher incidences of hypotonia, and increased activity levels [63–65]. However, these studies are confounded by co-exposures to other environmental contaminants. Therefore, studies in experimental animals are the only source of information regarding the specific effects related to TCDD exposure. For example, TCDD exposure resulted in delayed development of the righting reflex and impaired rotorod performance in rats [66]. behaviors that normally depend on proper cerebellar maturation and function [67,68]. A study from Japan recently reported that rats exposed to low doses of TCDD throughout gestation experienced delayed motor development during infancy [69]. Together these observations suggest that cerebellar development is adversely impacted following TCDD exposure. We have identified cerebellar GNPs as targets for AhR-mediated TCDD neurotoxicity [55]. Our in vitro and in vivo data also support the hypothesis that TCDD disrupts the balance between proliferation, differentiation, and apoptosis in GNPs during the early postnatal period [40,55]. Nevertheless, the primary cellular events associated with AhR activation by TCDD during cerebellar GNP maturation remain unclear.
Because AhR is robustly expressed in the cerebellum between postnatal days (PND) 6–10 [77], it is in an ideal position to mediate several important developmental events such as granule neuroblast expansion and differentiation. Our findings that TCDD exposure interferes with GNP proliferation and maturation suggest that AhR plays multiple roles during granule cell neurogenesis throughout this critical period [40,55]. The first three weeks of postnatal cerebellar development in the rodent represents a vulnerable phase for toxic insult because of the dynamic ongoing programs of neurogenesis, differentiation, apoptosis, and synaptogenesis [70]. This stage of rodent brain maturation corresponds to neuroanatomical development from the late fetal period to 1.5 years in humans The specific cellular signaling events associated with neurogenesis are orchestrated by a tightly regulated spatiotemporal program of gene expression [71–73]. Collectively our findings suggest that TCDD exposure is associated with abnormal proliferative activity, altered gene expression patterns and impaired GNP differentiation in the cerebellum [40,55] (Table 2; Figure 2). It is proposed that TCDD inappropriately activates AhR to disrupt the developmental genetic program that mediates cell cycle regulation and transcription factor activity during GNP maturation.
Figure 2. Genes associated with the regulation of GNP proliferation are increased following TCDD Exposure.
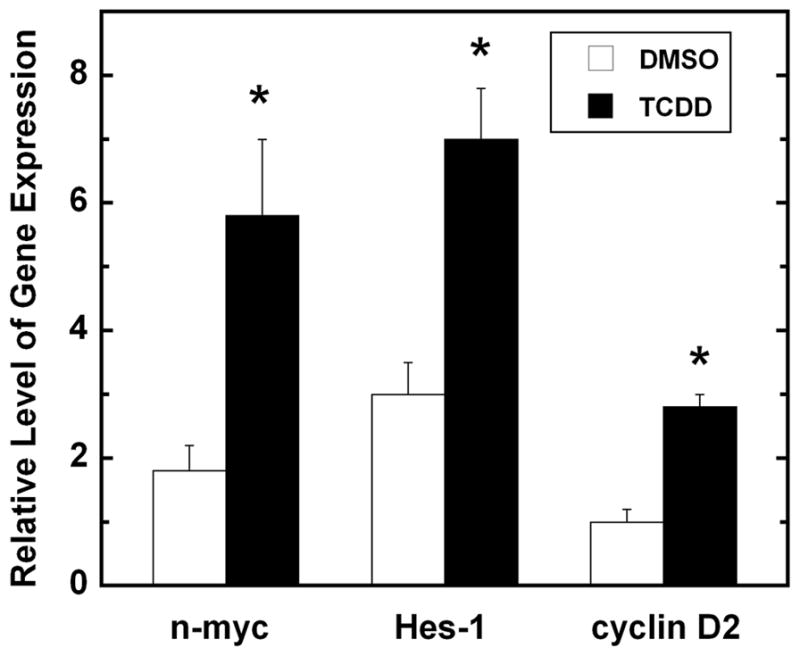
Mice were exposed to vehicle (DMSO) or TCDD (1.0 μg/kg) by oral gavage on postnatal day 6 and cerebella were removed 8 hours later. RNA was isolated and gene expression was analyzed by RT-PCR. Gene expression was normalized to GAPDH. Values represent mean ± SEM (n = 4) and were analyzed using a Student’s t-test. p< 0.05 relative to vehicle control.
The appropriate balance between proliferation and cell cycle withdrawal is critical for GNP differentiation. These developmental events are governed by diverse signaling systems that include soluble factors, membrane bound signals as well as downstream second messengers, transcritpion factors and genes. Although there have been several reports describing potential mitogenic factors for GNPs in vitro [74–77], the sonic hedgehog (Shh) pathway governs proliferation during the GNP expansion phase in vivo [78,79]. Shh is secreted by Purkinje neurons and binds to the patched (Ptc) receptors in GNPs to stimulate smoothened (Smo)-mediated transactivation of gene targets, most notably the the Gli transcription factor family [80]. Additional downstream effector molecules of the Shh pathway are reported to be the D- and B-type cyclins, as well as n-Myc. Our data suggest that AhR activation by TCDD interacts with the mitogenic signaling pathways to promote GNP proliferation in vitro (unpublished data). These observations are supported by the increased expression of Shh targets in our gene profiling studies, including n-myc, cyclin D1, and cyclin D2 (Table 2; Figure 2). Cell to cell contact in the EGL is also important for sustaining GNP proliferation [74]. For example, activation of Notch activity by its ligand Jagged1, a cell to cell interactive signaling pathway, was shown to maintain GNP proliferation in vitro [81]. The downstream effector of this signaling cascade is HES1, which was recently identified as an AhR target gene [57]. In our preliminary studies, TCDD was shown to increase HES-1 gene expression during the GNP expansion phase (Figure 2). If TCDD exposure intereferes with proliferation, there could be potential downstream consequences on granule neuron differentiation and apoptotic programs, which ultimately disrupts the final cell composition and function of the cerebellum.
In summary, perinatal exposure to TCDD and related chemicals causes neurological deficits that are consistent with improper cerebellar maturation [63–65]. Abnormal cerebellar maturation has also been connected to several neurodevelopmental disorders [83,84]. Therefore, the primary hypothesis of our future studies is that AhR intrinsically controls the balance between GNP proliferation, differentiation, and apoptosis during cerebellar development. It is proposed that AhR influences cell cycle regulation and transcription factor activity by modulating gene expression in GNPs. Consequently, TCDD exposure likely disrupts endogenous AhR-mediated spatiotemporal gene profiles associated with the proper development of cerebellar cytoarchitecture, which gives rise to functional abnormalities. Our ongoing studies will provide insight into the mechanisms by which AhR activation by TCDD disrupts cerebellar granule cell neurogenesis. The findings of this research will be particularly important to further our understanding of the effects of AhR activation on transcriptional and cell cycle regulation during GNP maturation, which may lead to identification of factors that contribute to the anatomical and functional deficits following exposure to TCDD.
Postnatal lead exposure reduces hippocampal neurogenesis and increases the number of PSA-NCAM expressing cells in the dentate gyrus of adult rat brain (Alex Zharkovsky)
Lead is still widely distributed in the environment, and the consequences of chronic exposure to low levels of lead in childhood has been extensively studied in recent years. All evidence indicates that there is no safe threshold for the adverse effects of lead on the nervous system [85–87]. The hippocampus plays an important role in learning and memory and several studies indicate that lead can induce alterations in the hippocampal plasticity [87–89]. In rodents, the hippocampal formation differs from other brain structures since approximately 85% of granule neurons of the dentate gyrus are produced during the post-natal period and adult neurogenesis persists through the whole life span. It has been hypothesized that adult hippocampal neurogenesis exists as a substrate for neuronal plasticity and is related to the memory formation, emotions and helps the brain to accommodate continued bouts of novelty. Several negative events that occur during early childhood, such as maternal deprivation, ethanol administration and impaired cognitive functions produce long-lasting alterations in hippocampal neurogenesis [90–92]. The persistent disruption of hippocampal neurogenesis might diminish the plasticity of the hippocampus and enhance the likelihood of mood and memory disorders [93,94].
Neural cell adhesion molecule (NCAM) plays an important role in the maintenance of neural plasticity, learning and memory consolidation [95]. Adhesive properties of NCAM are regulated through the addition of long linear homopolymers of 2,8-linked sialic acid residues (polysialic acid or PSA) attenuates NCAM mediated cell interaction and thereby creates plasticity in the positioning and movements of the cells and/or their processes [96,97]. PSA-NCAM is highly expressed in the hippocampal formation and hippocampal neurogenesis occurs in the environment provided by PSA-NCAM allowing newly born cells to migrate and make new synaptic contacts [93,94]. In the present study, we investigated whether low-level lead exposure, during the extended post-natal period, would induce deviations in hippocampus-dependent learning and affect the hippocampal neurogenesis and expression of PSA-NCAM in adulthood.
Wistar rat pups were exposed to 0.2% lead acetate from postnatal day (PND) 1 to PND30. Behavioral testing was performed on PND60 (anxiety testing) and PND80 (contextual fear conditioning). To assess neurogenesis, bromodeoxyuridine (BrdU) was administered in a dose of 300 mg/kg (ip) on PND80. Rats were killed 24 hours (proliferation study) or 3 weeks (survival/differentiation study) after administration of BrdU. The brain sections were processed for the immunohistochemical detection BrdU label (peroxidase method), or fluorescence double immunohistochemistry for BrdU and neuronal or glial markers. The number of PSA-NCAM expressing cells was estimated by counting all PSA-NCAM positive cells on every 20th section in a subgranula zone of dentate gyrus, starting randomly and multiplied by 20 to get the total number of PSA-NCAM expressing cells in the dentate gyrus of hippocampus. This exposure protocol resulted in pup blood lead levels that increased to 29.3 ± 5.0 μg/dL by PND15 and were 34.2 ± 5.8 μg/dL at weaning. Corresponding brain tissue lead levels were 456 ± 23 ng/g on PND15 and 781 ± 87 ng/g on PND30. At PND30, lead was removed from the drinking water and the animals were maintained on tap water until adulthood (PND80). At this time, the blood lead levels were 6.5 ± 1.2 μg/dL and brain tissue lead levels 6 ± 1 ng/g. Mean blood lead levels for the control group were 4.2 ± 1.7 μg/dL and brain tissue lead levels 6 ± 2 ng/g.
Lead-exposed animals demonstrated an increase in the level of anxiety as evidenced by a reduction of percent entries into and percent time spent in the open arms of the plus-maze. The increased anxiety was not attributed to the changes in the locomotor activity in the plus-maze since the total number of entries did not differ between control and lead-exposed rats. For the assessment of retention and extinction of fear, animals were placed in the same context without presentation of foot shock (unconditioned stimulus) at 24 hours after training session, and were scored for freezing during a 5 min observation period. Statistical analysis demonstrated a significantly lower freezing time in lead-exposed rats.
To examine the potential effect of post-natal lead exposure on the proliferation of the neuronal and/or glial precursors in control and lead-exposed rats during adulthood, the number of BrdU-positive cells in the dentate gyrus was estimated 24 hours after BrdU administration. The number of BrdU-labeled cells was significantly (p<0.01) lower in lead-exposed rats, compared to controls (Figure 3). The reduced proliferative activity in the dentate gyrus was not associated, however, with changes of the volume of dentate gyrus (Figure 3). To determine the survival rate of BrdU-positive cells after lead exposure, we counted the number of BrdU-positive cells in the dentate gyrus three weeks after BrdU administration. Experiments failed to demonstrate any differences in the survival rate of BrdU–positive cells between control and lead-exposed animals. The percentage of surviving cells in lead-exposed rats was 39.4%, which did not differ significantly from the 47.8% in control animals. To determine the phenotype of BrdU-positive cells, born in the dentate gyrus of control or lead-exposed rats, we performed double immunohistochemical labeling for BrdU and markers for neuronal or glial phenotypes 3 weeks after administration of BrdU. In control rats, 40.6 ± 3.4% of the BrdU-positive cells expressed calbindin (marker of adult granule neurons), whereas in lead-exposed animals only 28.7 ± 3.5% of cells were adult calbindin-positive neurons (p<0.05). In contrast, the proportion of young post-mitotic neurons expressing Tuj1 was significantly higher (p<0.01) in lead-exposed rats as compared with those in the controls. In addition, a significantly higher proportion of BrdU-positive cells in the dentate gyrus of lead-exposed rats differentiated into astroglia, compared to controls. Further analysis showed that lead exposed rats increased expression of polysialic acid linked neural call adhesion molecule (PSA-NCAM) in the dentate gyrus. Migration of the newly born cells within the dentate gyrus occurs in the environment provided by PSA-NCAM. Thus, an increased PSA-NCAM expression can facilitate migration of the new cells and their ectopic positioning within the hippocampal formation.
Figure 3. Production of the new cells in the rat dentate gyrus, following postnatal lead exposure.
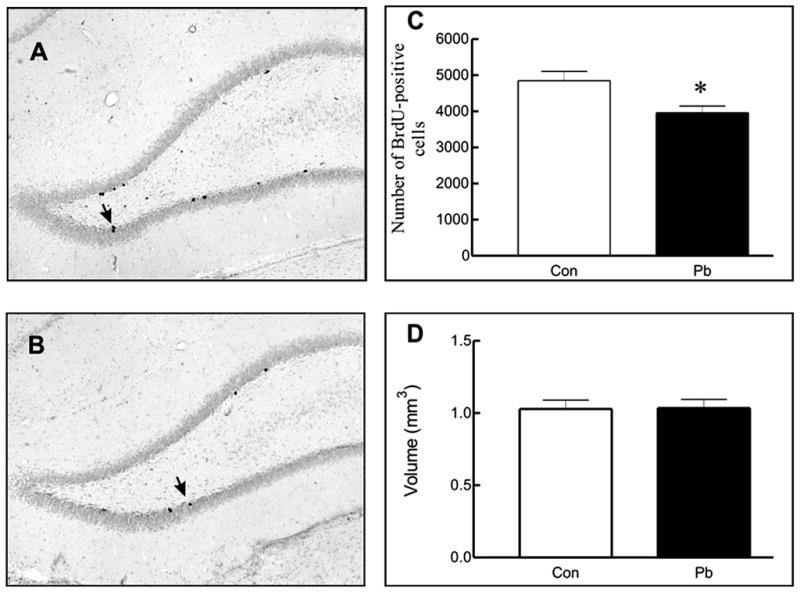
Light microscopic images of the BrdU-labeled cells in (A) control and (B) lead-exposed rats. The (C) number of BrdU-positive cells and (D) volume of dentate gyrus in control and lead-exposed rats. Values represent mean ± SEM (n = 6) and were analyzed using a Student’s t-test. p< 0.05 relative to vehicle control.
Our results demonstrate that post-natal lead exposure increased anxiety in the plus-maze test and disrupted contextual fear conditioning. The observed behavioral alterations persisted long after the termination of lead exposure. At that time, the blood lead levels were not different from controls. Therefore, we propose that lead exposure induced long-lasting or even permanent alterations in brain functions. The observed behavioral alterations following lead exposure were accompanied by the inhibition of the production of the new cells in the hippocampal dentate gyrus. Our novel studies also showed that not only proliferation of neuronal precursors was retarded, but that postnatal lead exposure also affected maturation of the newly born adult neuronal cells. Indeed, our study showed that three weeks after BrdU administration a lower proportion of BrdU-positive cells differentiated into adult calbindin-positive neurons, whereas the proportion of young post-mitotic neurons expressing Tuj1 was significantly higher in lead-exposed rats than in controls. We propose that a moderate level of postnatal lead exposure retarded the transition of young post-mitotic neurons into adult neurons. Further support for this proposal was obtained the experiments with the estimation of PSA-NCAM positive cells in the dentate gyrus of lead-exposed rats. Since newly generated cells start to express PSA-NCAM soon after they become post-mitotic, and the expression sharply reduces when they become adult neurons, the observed increase in the number of PSA-NCAM positive cells suggests that the pool of young post-mitotic cells was increased in lead-exposed animals. However, their transition into adult neurons was impaired following lead exposure. Our data also showed that observed alterations in neurogenesis were accompanied by an enhanced gliogenesis since a higher proportion of astroglial cells were generated from BrdU-positive cells. Subsequent work by others [25] confirmed our BrdU and behavioral findings. Thus, our data show that developmental lead exposure induced persistent inhibition of neurogenesis within the dentate gyrus, which produced behavioral deficits during adulthood.
The combination of age, gender and environmental endocrine disrupters presents a tangled puzzle for the process of neurogenesis. (Bernard Weiss)
Many neurobehavioral functions wane with aging. The process may be accelerated, or the loss of function exacerbated, by exposure to neurotoxicants. Metals such as lead, manganese and methylmercury, pesticides such as parathion and paraquat, and industrial chemicals such as PCBs can diminish functional capacities in aging populations. For example, levels of blood and bone lead accepted as risks for neurobehavioral functioning in children also diminish performance in elderly subjects [98]. Such declines in neurobehavioral function may arise from structural damage and functional disorders in the aging brain, as seen in extreme cases such as Parkinson’s disease and Alzheimer’s disease. Some of the more common, less dramatic, accompaniments of aging, such as impaired memory, may arise from a diminished capacity for neurogenesis and synaptogenesis, which grows less potent with time [99–100], but is still retained. Its retention is a crucial element in maintaining neurobehavioral function as we age.
Until about 15 years ago, the possibility of neurogenesis (which will also be used to describe synaptogenesis) in the adult brain was mostly dismissed except for regions such as the olfactory bulb. The prevailing dictum, following authorities such as Ramon y Cajal, asserted “Use it and lose it.” A number of developments, including new methods for tracing neurogenesis, came together to overthrow that dogma. Alvarez-Buylla and Lim [101] presented the new view in this way: “It is now becoming clear that pieces of the embryonic developmental puzzle are retained for adult neurogenesis.” Accompanying this revised view are data supporting the idea that continuing to challenge the brain by intellectual activities helps to maintain its vigor. The new view has been adopted by the public. Substantial sums are now being spent on methods and instruments, such as computer-based games, allegedly designed to foster brain activity.
One element that has received insufficient attention from neuroscientists is the role of endocrine function in neurogenesis. The underlying processes are known to depend, in part, on endocrine mechanisms, particularly the gonadal hormones estrogen and testosterone. Their roles in neurogenesis, in fact, were among the sources of data demonstrating that the process endured beyond early development [102–104]. The earlier papers demonstrated that the reduced density of apical dendrites in the brains of ovariectomized female rats could be restored by administering estradiol. More recently, Tanapat et al. [105] showed that estradiol administration to such rats also increased the number of BrdU-labeled cells in the dentate gyrus. In parallel, Leranth et al. [101] reported that the administration of testestosterone to gonadectomized male rats restored synaptic spine density in the CA1 hippocampal subfield.
Gonadal hormone levels, along with neurogenic capacity, also wane with aging, clearly seen in women after menopause, but in men as well [107] and seem to be a risk factor for depressed cognitive function [108–110]. We now know that supplemental doses can enhance both neurogenesis and synaptogenesis [111] and, under most, but not all circumstances, enhance neurobehavioral function in older men and women [112,113] and in adult and aging animals [106,114]. The Women’s Health Study seemed to indicate, in conflict with the studies cited above, that hormone replacement therapy in the form of equine estrogens exerted adverse consequences rather that elevating brain function. It is becoming increasingly apparent that the investigation was flawed. Its population consisted of women averaging twelve years since menopause. In women who adopted hormone replacement therapy at the advent of menopause, brain function, as reflected by neuropsychological tests, remained intact rather than declining. The phenomenon is termed a “window of opportunity” effect [113].
How do endocrine function, neurogenesis, and aging fit into the domain of neurotoxicology? Simply stated, by virtue of the toxic properties of those environmental chemicals now recognized as endocrine disruptors. These are chemicals that interfere with the biological actions of hormones by blocking, mimicking, displacing, or acting through a variety of other mechanisms to subvert their natural roles. Many of the chemicals labeled as environmental endocrine disruptors (EEDs) have the potential to interfere with the action of gonadal hormones on neurobehavioral function. Two of them, Bisphenol A and phthalate esters, have achieved recent prominence because they have been targets of regulation and legislation. Both are plasticizers, are produced by the millions of kilograms annually, and appear in a large number of products (Table 3). Their metabolites are detected in virtually all humans. Bisphenol A has been known as an estrogenic chemical for decades, and because of its now-documented effects on prostate and brain development, has been banned by Canada from products, such as polycarbonate baby bottles, that bring it into contact with children. Phthalates have been banned from products such as toys, where they are used to soften plastics, by California and the European Union. Phthalates are anti-androgenic and prenatal exposure has been documented to feminize boys.
Table 3.
Sources of Exposure to Bisphenol A and Phthalates
| Bisphenol A (Estrogenic) | Phthalate esters (Anti-androgenic) |
|---|---|
| Children’s tooth sealant | Perfumes |
| Baby bottles | Hair sprays |
| Can linings | Soaps, shampoos |
| Nail polish | Nail polish |
| Polycarbonate water bottles | Food packaging |
| Microwave ovenware | Plastic wrap |
| Flame retardants | Detergents |
| PVC stabilizers | Adhesives |
| Artificial teeth | Pesticides (“inert” ingredients) |
| Adhesives | Medical tubing |
| Enamels and varnishes | Skin moisturizers |
Given the pervasive contamination of the environment by chemicals with estrogenic, antiestrogenic, and antiandrogenic properties, threats to neurobehavioral function in aging populations by such agents warrant serious consideration by neuroscientists. For example, Bisphenol A, although classified as an estrogenic agent, and shown to masculinize the developing female brain, also has the unexpected capacity to eliminate the synaptogenic response elicited by estradiol, and does so at levels below those regarded as free of adverse effects by conventional assessment methods and standards. MacLusky et al [115] showed that, at the EPA Reference Dose of 50 μg/kg, it significantly reduced the increase in pyramidal cell dendritic spine synaptic density elicited by estradiol in rats. Leranth et al. [116] showed corresponding effects in the nonhuman primate. It can also inhibit synaptogenesis induced by testosterone in the adult male brain [117]. Lang et al. [118] also found that, in adult men, levels of Bisphenol A metabolites in urine were correlated with indices of cardiovascular disease.
Phthalate esters, in addition to their effects on male genital development, also modify sexually dimorphic behaviors in rats (Weiss, unpublished data). In adult men, exposed at prevailing environmental levels, they also reduce testosterone levels and increase waist circumference, so it would not be inconsistent for them to impair neurobehavioral function as well [119,120]. Because of the role of diminished endocrine function in neurodegeneratve disease, at least partly due to how they modulate neurogenesis, EEDs must be regarded as threats to the capabilities of the aging brain.
Summary
The speakers in this symposia showed that a diverse array of environmental neurotoxicants can produce immediate as well as long-term central and sensory nervous system alterations and deficits in children, non-human primates and rodents. The adverse findings were dose-dependent, although nonmonotonic dose-dependent responses were evident with the lowest level often producing the most adverse effects. These data reinforce the idea that lifetime measures of dose-response toxicant exposure should be a component of the neurotoxic risk assessment process.
Acknowledgments
Supported in part by Grants RO1ES012482 (DAF), Core Grant EY07551 (DAF), RO1ES016357 (LO), R21ES013512 (LO), RO1ES015509 (BW), Center Grant ES01247 (BW; LO) and Training Grant ES07026 (BW; LO). The authors also acknowledge the financial support of the local organizers, the US-Israel Binational Science Foundation and the US Environmental Protection Agency.
Footnotes
CONFLICTS OF INTEREST
None of the authors has competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Donald A. Fox, Email: dafox@uh.edu.
Lisa Opanashuk, Email: lisa_opanashuk@urmc.rochester.edu.
Aleksander Zharkovsky, Email: aleksander.zarkovski@ut.ee.
Bernie Weiss, Email: bernard.weiss@urmc.rochester.edu.
References
- 1.Canfield RL, Henderson CR, Jr, Cory-Slechta DA, Cox C, Jusko TA, Lanphear BP. Intellectual impairment in children with blood lead concentrations below 10 microg per deciliter. N Engl J Med. 2003;348:1517–1526. doi: 10.1056/NEJMoa022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Osman K, Pawlas K, Schutz A, Gazdik M, Sokal JA, Vahter M. Lead exposure and hearing effects in children in Katowice, Poland. Environ Res. 1999;80:1–8. doi: 10.1006/enrs.1998.3886. [DOI] [PubMed] [Google Scholar]
- 3.Rothenberg SJ, Poblano A, Schnaas L. Brainstem auditory evoked response at five years and prenatal and postnatal blood lead. Neurotoxicol Teratol. 2000;22:503–510. doi: 10.1016/s0892-0362(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 4.Wasserman GA, Liu X, Popovac D, Factor-Litvak P, Kline J, Waternaux C, et al. The Yugoslavia Prospective Lead Study: contributions of prenatal and postnatal lead exposure to early intelligence. Neurotoxicol Teratol. 2000;22:811–818. doi: 10.1016/s0892-0362(00)00106-9. [DOI] [PubMed] [Google Scholar]
- 5.Altmann L, Sveinsson K, Kramer U, Weishoff-Houben M, Turfeld M, Winneke G, et al. Visual function in 6-year old children living in relation to lead and mercury levels. Neurotoxicol Teratol. 1998;20:9–17. doi: 10.1016/s0892-0362(97)00070-6. [DOI] [PubMed] [Google Scholar]
- 6.Rothenberg SJ, Schnaas L, Salgado-Valladares M, Casanueva E, Geller AM, Hudnell HK, et al. Increased ERG a- and b-wave amplitudes in 7- to 10-year-old children resulting from prenatal lead exposure. Invest Ophthalmol Vis Sci. 2002;43:2036–2044. [PubMed] [Google Scholar]
- 7.Fox DA, Kala SV, Hamilton WR, Johnson JE, O’Callaghan JP. Low-level human equivalent gestational lead exposure produces supernormal scotopic electroretinograms, increased retinal neurogenesis and decreased dopamine utilization in rats. Environ Hlth Perspect. 2008;116:618–625. doi: 10.1289/ehp.11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fox DA, Farber DB. Rods are selectively altered by lead: I. Electrophysiology and biochemistry. Exp Eye Res. 1988;46:597–611. doi: 10.1016/s0014-4835(88)80016-2. [DOI] [PubMed] [Google Scholar]
- 9.Otto DA, Fox DA. Auditory and visual dysfunction following lead exposure. Neurotoxicology. 1993;14:191–207. [PubMed] [Google Scholar]
- 10.Fox DA, Boyes WK. Toxic responses of the ocular and visual system. In: Klaassen CD, editor. Casarett & Doull’s Toxicology: The Science of Poisons. 7. McGraw-Hill Press; New York: 2007. pp. 665–697. [Google Scholar]
- 11.Lilienthal H, Kohler K, Turfeld M, Winneke G. Persistent increases in scotopic B-wave amplitudes after lead exposure in monkeys. Exp Eye Res. 1994;59:203–209. doi: 10.1006/exer.1994.1098. [DOI] [PubMed] [Google Scholar]
- 12.Leasure LJ, Giddabasappa A, Chaney S, Johnson JE, Pothakos K, Lau YS, et al. Low-level human equivalent gestational lead exposure produces gender-specific motor and coordination abnormalities and late-onset obesity in year-old mice. Environ Hlth Persp. 2007;116:355–361. doi: 10.1289/ehp.10862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3:79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- 14.Raedler A, Sievers J. The development of the visual system of the albino rat. Adv Anat Embryol Cell Biol. 1975;50:3–88. doi: 10.1007/978-3-642-45461-5. [DOI] [PubMed] [Google Scholar]
- 15.Rice D, Barone S. Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect. 2000;108(Suppl 3):511–533. doi: 10.1289/ehp.00108s3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bui BV, Fortune B. Ganglion cell contributions to the rat full-field electroretinogram. J Physiol. 2004;555:153–173. doi: 10.1113/jphysiol.2003.052738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robson JG, Frishman LJ. Dissecting the dark-adapted electroretinogram. Doc Ophthalmol. 1999;95:187–215. doi: 10.1023/a:1001891904176. [DOI] [PubMed] [Google Scholar]
- 18.Fox DA, Chu LW. Rods are selectively altered by lead: II. Ultrastructure and quantitative histology. Exp Eye Res. 1988;46:613–625. doi: 10.1016/s0014-4835(88)80017-4. [DOI] [PubMed] [Google Scholar]
- 19.Greferath U, Grünert U, Wässle H. Rod bipolar cells in the mammalian retina show protein kinase C-like immunoreactivity. J Comp Neurol. 1990;301:433–442. doi: 10.1002/cne.903010308. [DOI] [PubMed] [Google Scholar]
- 20.Fox DA, Campbell ML, Blocker YS. Functional alterations and apoptotic cell death in the retina following developmental or adult lead exposure. Neurotoxicology. 1997;18:645–665. [PubMed] [Google Scholar]
- 21.He L, Poblenz AT, Medrano CJ, Fox DA. Lead and calcium produce rod photoreceptor cell apoptosis by opening the mitochondrial permeability transition pore. J Biol Chem. 2000;275:12175–12184. doi: 10.1074/jbc.275.16.12175. [DOI] [PubMed] [Google Scholar]
- 22.He L, Perkins GA, Poblenz AT, Harris JB, Hung M, Ellisman MH, et al. Bcl-xL overexpression blocks bax-mediated mitochondrial contact site formation and apoptosis in rod photoreceptors of lead-exposed mice. Proc Natl Acad Sci USA. 2003;100:1022–1027. doi: 10.1073/pnas.0333594100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilbert ME, Kelly ME, Samsam TE, Goodman JH. Chronic developmental lead exposure reduces neurogenesis in adult rat hippocampus but does not impair spatial learning. Toxicol Sci. 2005;86:365–374. doi: 10.1093/toxsci/kfi156. [DOI] [PubMed] [Google Scholar]
- 24.Jaako-Movits K, Zharkovsky T, Romantchik O, Jurgenson M, Merisalu E, Heidmets LT, et al. Developmental lead exposure impairs contextual fear conditioning and reduces adult hippocampal neurogenesis in the rat brain. Int J Dev Neurosci. 2005;23:627–635. doi: 10.1016/j.ijdevneu.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 25.Verina T, Rohde CA, Guilarte TR. Environmental lead exposure during early life alters granule cell neurogenesis and morphology in the hippocampus of young adult rats. Neuroscience. 2007;145:1037–1047. doi: 10.1016/j.neuroscience.2006.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu YZ, Hogenesch JB, Bradfield CA. The PAS superfamily: sensors of environmental and developmental signals. Annu Rev Pharmacol Toxicol. 2000;40:519–561. doi: 10.1146/annurev.pharmtox.40.1.519. [DOI] [PubMed] [Google Scholar]
- 27.Crews ST. Control of cell lineage-specific development and transcription by bHLH-PAS proteins. Genes Dev. 1998;12:607–620. doi: 10.1101/gad.12.5.607. [DOI] [PubMed] [Google Scholar]
- 28.Crews ST, Fan C-M. Remembrance of things PAS: regulation of development by bHLH-PAS proteins. Current Opinion in Genetics & Development. 1999;9:580–587. doi: 10.1016/s0959-437x(99)00003-9. [DOI] [PubMed] [Google Scholar]
- 29.Hankiinson O. The aryl hydrocarbon receptor complex. Ann Rev Pharmacol Toxicol. 1995;35:307–340. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- 30.Mimura J, Fujii-Kuriyama Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim Biophys Acta. 2003;1619:263–268. doi: 10.1016/s0304-4165(02)00485-3. [DOI] [PubMed] [Google Scholar]
- 31.Fukunaga BN, Probst MR, Reisz-Porszasz S, Hankinson O. Identification of functional domains of the aryl hydrocarbon receptor. J Biol Chem. 1995;270:29270–29278. doi: 10.1074/jbc.270.49.29270. [DOI] [PubMed] [Google Scholar]
- 32.Schmidt JV, Bradfield CA. Ah receptor signaling pathways. Annu Rev Cell Dev Biol. 1996;12:55–89. doi: 10.1146/annurev.cellbio.12.1.55. [DOI] [PubMed] [Google Scholar]
- 33.Lai ZW, Pineau T, Esser C. Identification of dioxin-responsive elements (DREs) in the 5′ regions of putative dioxin-inducible genes. Chem Biol Interact. 1996;100:97–112. doi: 10.1016/0009-2797(96)03691-5. [DOI] [PubMed] [Google Scholar]
- 34.Nebert DW, Roe AL, Dieter MZ, Solis WA, Yang Y, Dalton TP. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem Pharmacol. 2000;59:65–85. doi: 10.1016/s0006-2952(99)00310-x. [DOI] [PubMed] [Google Scholar]
- 35.Tan Z, Chang X, Puga A, Xia Y. Activation of mitogen-activated protein kinases (MAPKs) by aromatic hydrocarbons, role in the regulation of aryl hydrocarbon receptor (AHR) function. Biochem Pharmacol. 2002;64:771–780. doi: 10.1016/s0006-2952(02)01138-3. [DOI] [PubMed] [Google Scholar]
- 36.Bock KW, Kohle C. Ah receptor, dioxin-mediated toxic responses as hints to deregulated physiologic functions. Biochem Pharmacol. 2006;72:393–404. doi: 10.1016/j.bcp.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 37.Abbott BD, Schmid JE, Pitt JA, Buckalew AR, Wood CR, Held GA, Diliberto JJ. Adverse reproductive outcomes in the transgenic Ah receptor-deficient mouse. Toxicol Appl Pharmacol. 1999;155:62–70. doi: 10.1006/taap.1998.8601. [DOI] [PubMed] [Google Scholar]
- 38.Benedict JC, Lin TM, Loeffler IK, Peterson RE, Flaws JA. Physiological role of the aryl hydrocarbon receptor in mouse ovary development. Toxicol Sci. 2000;56:382–388. doi: 10.1093/toxsci/56.2.382. [DOI] [PubMed] [Google Scholar]
- 39.Bunger MK, Moran SM, Glover E, Thomae TL, Lahvis GP, Lin BC, Bradfield CA. Resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity and abnormal liver development in mice carrying a mutation in the nuclear localization sequence of the aryl hydrocarbon receptor. J Biol Chem. 2003;278:17767–17774. doi: 10.1074/jbc.M209594200. [DOI] [PubMed] [Google Scholar]
- 40.Collins LL, Williamson MA, Thompson BD, Dever DP, Gasiewicz TA, Opanashuk LA. 2,3,7,8-Tetracholorodibenzo-p-dioxin exposure disrupts granule neuron precursor maturation in the developing mouse cerebellum. Toxicol Sci. 2008;103:125–136. doi: 10.1093/toxsci/kfn017. [DOI] [PubMed] [Google Scholar]
- 41.Fernandez-Salguero P, Pineau T, Hilbert DM, McPhail T, Lee SS, Kimura S, Nebert DW, Rudikoff S, Ward JM, Gonzalez FJ. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science. 1995;268:722–726. doi: 10.1126/science.7732381. [DOI] [PubMed] [Google Scholar]
- 42.Lin TM, Ko K, Moore RW, Buchanan DL, Cooke PS, Peterson RE. Role of the aryl hydrocarbon receptor in the development of control and 2,3,7,8-tetrachlorodibenzo-p-dioxin-exposed male mice. J Toxicol Environ Health A. 2001;64:327–342. doi: 10.1080/152873901316981312. [DOI] [PubMed] [Google Scholar]
- 43.Lin TM, Ko K, Moore RW, Simanainen U, Oberley TD, Peterson RE. Effects of aryl hydrocarbon receptor null mutation and in utero and lactational 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure on prostate and seminal vesicle development in C57BL/6 mice. Toxicol Sci. 2002;68:479–487. doi: 10.1093/toxsci/68.2.479. [DOI] [PubMed] [Google Scholar]
- 44.Lund AK, Goens MB, Kanagy NL, Walker MK. Cardiac hypertrophy in aryl hydrocarbon receptor null mice is correlated with elevated angiotensin II, endothelin-1, and mean arterial blood pressure. Toxicol Appl Pharmacol. 2003;193:177–187. doi: 10.1016/j.taap.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 45.Schmidt JV, Su GH, Reddy JK, Simon MC, Bradfield CA. Characterization of a murine Ahr null allele, involvement of the Ah receptor in hepatic growth and development. Proc Natl Acad Sci USA. 1996;93:6731–6736. doi: 10.1073/pnas.93.13.6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Denison MS, Pandini A, Nagy SR, Baldwin EP, Bonati L. Ligand binding and activation of the Ah receptor. Chem Biol Interact. 2002;141:3–24. doi: 10.1016/s0009-2797(02)00063-7. [DOI] [PubMed] [Google Scholar]
- 47.Whitlock JP., Jr Induction of cytochrome P4501A1. Annu Rev Pharmacol Toxicol. 1999;39:103–125. doi: 10.1146/annurev.pharmtox.39.1.103. [DOI] [PubMed] [Google Scholar]
- 48.Bock KW, Kohle C. Ah receptor- and TCDD-mediated liver tumor promotion, clonal selection and expansion of cells evading growth arrest and apoptosis. Biochem Pharmacol. 2005;69:1403–1408. doi: 10.1016/j.bcp.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 49.Mandal PK. Dioxin, a review of its environmental effects and its aryl hydrocarbon receptor biology. J Comp Physiol [B] 2005;175:221–230. doi: 10.1007/s00360-005-0483-3. [DOI] [PubMed] [Google Scholar]
- 50.Matsumura F. On the significance of the role of cellular stress response reactions in the toxic actions of dioxin. Biochem Pharmacol. 2003;66:527–540. doi: 10.1016/s0006-2952(03)00157-6. [DOI] [PubMed] [Google Scholar]
- 51.Rifkind AB. CYP1A in TCDD toxicity and in physiology-with particular reference to CYP dependent arachidonic acid metabolism and other endogenous substrates. Drug Metab Rev. 2006;38:291–335. doi: 10.1080/03602530600570107. [DOI] [PubMed] [Google Scholar]
- 52.Safe S. Molecular biology of the Ah receptor and its role in carcinogenesis. Toxicol Lett. 2001;120:1–7. doi: 10.1016/s0378-4274(01)00301-0. [DOI] [PubMed] [Google Scholar]
- 53.Theobald H, KImmel GL, Peterson RE. In: Developmental and reproductive toxicity of dioxins and related chemicals. Gasiewicz T, Schecter A, editors. John Wiley & Sons, Inc; New Jersey: 2003. pp. 329–432. [Google Scholar]
- 54.Birnbaum LS, Tuomisto J. Non-carcinogenic effects of TCDD in animals. Food Addit Contam. 2000;17:275–288. doi: 10.1080/026520300283351. [DOI] [PubMed] [Google Scholar]
- 55.Williamson MA, Gasiewicz TA, Opanashuk LA. Aryl hydrocarbon receptor expression and activity in cerebellar granule neuroblasts: implications for development and dioxin neurotoxicity. Toxicol Sci. 2005;83:340–348. doi: 10.1093/toxsci/kfi031. [DOI] [PubMed] [Google Scholar]
- 56.Powell-Coffman JA, Bradfield CA, Wood WB. Caenorhabditis elegans orthologs of the aryl hydrocarbon receptor and its heterodimerization partner the aryl hydrocarbon receptor nuclear translocator. Proc Natl Acad Sci USA. 1998;95:2844–2849. doi: 10.1073/pnas.95.6.2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qin H, Zhai Z, Powell-Coffman JA. The Caenorhabditis elegans AHR-1 transcription complex controls expression of soluble guanylate cyclase genes in the URX neurons and regulates aggregation behavior. Dev Biol. 2006;298:606–615. doi: 10.1016/j.ydbio.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 58.Huang X, Powell-Coffman JA, Jin Y. The AHR-1 aryl hydrocarbon receptor and its co-factor the AHA-1 aryl hydrocarbon receptor nuclear translocator specify GABAergic neuron cell fate in C. elegans. Development. 2004;131:819–828. doi: 10.1242/dev.00959. [DOI] [PubMed] [Google Scholar]
- 59.Kim MD, Jan LY, Jan YN. The bHLH-PAS protein Spineless is necessary for the diversification of dendrite morphology of Drosophila dendritic arborization neurons. Genes Dev. 2006;20:2806–2819. doi: 10.1101/gad.1459706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abbott BD, Birnbaum LS, Perdew GH. Developmental expression of two members of a new class of transcription factors, I. Expression of aryl hydrocarbon receptor in the C57BL/6N mouse embryo. Dev Dyn. 1995;204:133–143. doi: 10.1002/aja.1002040204. [DOI] [PubMed] [Google Scholar]
- 61.Abbott BD, Perdew GH, Birnbaum LS. Ah receptor in embryonic mouse palate and effects of TCDD on receptor expression. Toxicol Appl Pharmacol. 1994;126:16–25. doi: 10.1006/taap.1994.1085. [DOI] [PubMed] [Google Scholar]
- 62.Abbott BD, Probst MR. Developmental expression of two members of a new class of transcription factors, II. Expression of aryl hydrocarbon receptor nuclear translocator in the C57BL/6N mouse embryo. Dev Dyn. 1995;204:144–155. doi: 10.1002/aja.1002040205. [DOI] [PubMed] [Google Scholar]
- 63.Needham LL, Gerthoux PM, Patterson DG, Jr, Brambilla P, Smith SJ, Sampson EJ, Mocarelli P. Exposure assessment, serum levels of TCDD in Seveso, Italy. Environ Res. 80(1999):S200–S206. doi: 10.1006/enrs.1998.3928. [DOI] [PubMed] [Google Scholar]
- 64.Neuberger M, Rappe C, Bergek S, Cai H, Hansson M, Jager R, Kundi M, Lim CK, Wingfors H, Smith AG. Persistent health effects of dioxin contamination in herbicide production. Environ Res. 1999;81:206–214. doi: 10.1006/enrs.1999.3983. [DOI] [PubMed] [Google Scholar]
- 65.Rogan WJ, Gladen BC. Neurotoxicology of PCBs and related compounds. Neurotoxicology. 1992;13:27–35. [PubMed] [Google Scholar]
- 66.Thiel R, Koch E, Ulbrich B, Chahoud I. Peri- and postnatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin, effects on physiological development, reflexes, locomotor activity and learning behaviour in Wistar rats. Arch Toxicol. 1994;69:79–86. doi: 10.1007/s002040050141. [DOI] [PubMed] [Google Scholar]
- 67.Ferguson SA. Neuroanatomical and functional alterations resulting from early postnatal cerebellar insults in rodents. Pharmacol Biochem Behav. 1996;55:663–671. doi: 10.1016/s0091-3057(96)00253-5. [DOI] [PubMed] [Google Scholar]
- 68.Holson RR, Gazzara RA, Ferguson SA, Ali SF, Laborde JB, Adams J. Gestational retinoic acid exposure, a sensitive period for effects on neonatal mortality and cerebellar development. Neurotoxicol Teratol. 1997;19:335–346. doi: 10.1016/s0892-0362(97)00039-1. [DOI] [PubMed] [Google Scholar]
- 69.Nishijo M, Kuriwaki J, Hori E, Tawara K, Nakagawa H, Nishijo H. Effects of maternal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin on fetal brain growth and motor and behavioral development in offspring rats. Toxicol Lett. 2007;173:41–47. doi: 10.1016/j.toxlet.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 70.Altman J, Bayer SA. Development of the Cerebellar System in Relation to its Evolution, Structure, and Functions. CRC Press; Boca Raton: 1997. [Google Scholar]
- 71.Goldowitz D, Hamre K. The cells and molecules that make a cerebellum. Trends Neurosci. 1998;21:375–382. doi: 10.1016/s0166-2236(98)01313-7. [DOI] [PubMed] [Google Scholar]
- 72.Millen KJ, Millonig JH, Wingate RJ, Alder J, Hatten ME. Neurogenetics of the cerebellar system. J Child Neurol. 1999;14:574–581. doi: 10.1177/088307389901400905. [DOI] [PubMed] [Google Scholar]
- 73.Wang VY, Zoghbi HY. Genetic regulation of cerebellar development. Nat Rev Neurosci. 2001;2:484–491. doi: 10.1038/35081558. [DOI] [PubMed] [Google Scholar]
- 74.Gao WO, Heintz N, Hatten ME. Cerebellar granule cell neurogenesis is regulated by cell-cell interactions in vitro. Neuron. 1991;6:705–715. doi: 10.1016/0896-6273(91)90168-y. [DOI] [PubMed] [Google Scholar]
- 75.Hatten ME. Neuronal regulation of astroglial morphology and proliferation in vitro. J Cell Biol. 1985;100:384–396. doi: 10.1083/jcb.100.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Opanashuk LA, Hauser KF. Opposing actions of the EGF family and opioids, heparin binding-epidermal growth factor (HB-EGF) protects mouse cerebellar neuroblasts against the antiproliferative effect of morphine. Brain Res. 1998;804:87–94. doi: 10.1016/s0006-8993(98)00647-7. [DOI] [PubMed] [Google Scholar]
- 77.Wu J. Granule cells of the internal granule layer have increased expression of GABA(A) receptor beta 2/beta 3 subunits. J Neurosci Res. 1998;51:697–711. doi: 10.1002/(SICI)1097-4547(19980315)51:6<697::AID-JNR4>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 78.Wallace VA. Purkinje-cell-derived sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol. 1999;9:445–448. doi: 10.1016/s0960-9822(99)80195-x. [DOI] [PubMed] [Google Scholar]
- 79.Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by sonic hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- 80.Ruiz I, Altaba A, Palma V, Dahmane N. Hedgehog-Gli signalling and the growth of the brain. Nat Rev Neurosci. 2002;3:24–33. doi: 10.1038/nrn704. [DOI] [PubMed] [Google Scholar]
- 81.Solecki DJ, Liu XL, Tomoda T, Fang Y, Hatten ME. Activated Notch2 signaling inhibits differentiation of cerebellar granule neuron precursors by maintaining proliferation. Neuron. 2001;31:557–568. doi: 10.1016/s0896-6273(01)00395-6. [DOI] [PubMed] [Google Scholar]
- 82.Thomsen JS, Kietz S, Strom A, Gustafsson JA. HES-1, a Novel Target Gene for the Aryl Hydrocarbon Receptor. Mol Pharmacol. 2004;65:165–171. doi: 10.1124/mol.65.1.165. [DOI] [PubMed] [Google Scholar]
- 83.Courchesne E, Redcay E, Morgan JT, Kennedy DP. Autism at the beginning, microstructural and growth abnormalities underlying the cognitive and behavioral phenotype of autism. Dev Psychopathol. 2005;17:577–597. doi: 10.1017/S0954579405050285. [DOI] [PubMed] [Google Scholar]
- 84.Kern JK, Jones AM. Evidence of toxicity, oxidative stress, and neuronal insult in autism. J Toxicol Environ Health B Crit Rev. 2006;9:485–499. doi: 10.1080/10937400600882079. [DOI] [PubMed] [Google Scholar]
- 85.Canfield RL, Henderson CR, Cory-Sletcha DA, Cox C, Jusko TA, Lanphear BP. Intellectual impairment in children with blood lead concentrations below 10 μg per deciliter. New Engl J Med. 2003;348:1517–1526. doi: 10.1056/NEJMoa022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Finkelstein Y, Markowitz ME, Rosen JF. Low-level lead-induced neurotoxicity in children: an update on central nervous system effects. Brain Res Rev. 1998;27:168–176. doi: 10.1016/s0165-0173(98)00011-3. [DOI] [PubMed] [Google Scholar]
- 87.Murphy KJ, Regan CM. Low level lead exposure in the early postnatal period results in persisting neuroplastic deficits associated with memory consolidation. J Neurochem. 1999;72:2099–2104. doi: 10.1046/j.1471-4159.1999.0722099.x. [DOI] [PubMed] [Google Scholar]
- 88.Bourljeily N, Suszkiw JB. Developmental cholinotoxicity of lead: loss of septal cholinergic neurons and long-term changes in cholinergic innervation of the hippocampus in perinatally lead-exposed rats. Brain Res. 1997;771:319–328. doi: 10.1016/s0006-8993(97)00828-7. [DOI] [PubMed] [Google Scholar]
- 89.Moreira EG, Vassilieff I, Vassilieff VS. Developmental lead exposure: behavioral alterations in the short and long term. Neurotoxicol Teratol. 2001;23:489–495. doi: 10.1016/s0892-0362(01)00159-3. [DOI] [PubMed] [Google Scholar]
- 90.Ikonomidou C, Bittigau P, Ishimaru M, Woznjak DF, Koch C, Genz K, Price MT, Stefovska V, Hörster F, Tenkova T, Dikranian K, Olney J. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- 91.Jaako K, Zharkovsky T, Kaasik A, Zharkovsky A. Ethanol intoxication reduces whereas ethanol withdrawal transiently enhances, production of the neural progenitor cells in the adult mouse dentate gyrus. Neurosci Res Comm. 2003;33:158–167. [Google Scholar]
- 92.Roceri M, Hendriks W, Racagni G, Ellenbrock BA, Riva MA. Early maternal deprivation reduces the expression of BDNF and NMDA receptor subunits in rat hippocampus. Mol Psychiat. 2002;7:609–616. doi: 10.1038/sj.mp.4001036. [DOI] [PubMed] [Google Scholar]
- 93.Aonurm-Helm A, Jurgenson M, Zharkovsky T, Sonn K, Berezin V, Bock E, Zharkovsky A. Depression-like behaviour in neural cell adhesion molecule (NCAM)-deficient mice and its reversal by an NCAM-derived peptide. FGL Eur J Neurosci. 2008;28:1618–1628. doi: 10.1111/j.1460-9568.2008.06471.x. [DOI] [PubMed] [Google Scholar]
- 94.Jacobs BL, van Praag H, Gage FH. Adult hippocampal neurogenesis and psychiatry: novel theory of depression. Mol Psychiat. 2000;5:262–269. doi: 10.1038/sj.mp.4000712. [DOI] [PubMed] [Google Scholar]
- 95.Crossin KL, Krushel LA. Cellular signaling by neural cell adhesion molecules of the immunoglobulin superfamily. Dev Dyn. 2000;218:260–279. doi: 10.1002/(SICI)1097-0177(200006)218:2<260::AID-DVDY3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 96.Bruses JL, Rutishauser U. Roles, regulation, and mechanism of polysialic acid function during neural development. Biochimie. 2001;83:635–643. doi: 10.1016/s0300-9084(01)01293-7. [DOI] [PubMed] [Google Scholar]
- 97.Rutishauser U. Polysialic acid and the regulation of cell interactions. Curr Opin Cell Biol. 1996;8:679–84. doi: 10.1016/s0955-0674(96)80109-8. [DOI] [PubMed] [Google Scholar]
- 98.Weisskopf MG, Proctor SP, Wright RO, Schwartz J, Spiro A, Sparrow D, et al. Cumulative lead exposure and cognitive performance among elderly men. Epidemiology. 2007;18:59–66. doi: 10.1097/01.ede.0000248237.35363.29. [DOI] [PubMed] [Google Scholar]
- 99.Duan H, Wearne SL, Rocher AB, Macedo A, Morrison JH, Hof PR. Age-related dendritic and spine changes in corticocortically projecting neurons in macaque monkeys. Cereb Cortex. 2003;13:950–961. doi: 10.1093/cercor/13.9.950. [DOI] [PubMed] [Google Scholar]
- 100.Taupin P. Adult neurogenesis in mammals. Curr Opin Mol Ther. 2006;8:345–351. [PubMed] [Google Scholar]
- 101.Alvarez-Buylla A, Lim DA. For the long run: Maintaining germinal niches in the adult brain. Neuron. 2004;41:683–686. doi: 10.1016/s0896-6273(04)00111-4. [DOI] [PubMed] [Google Scholar]
- 102.Woolley CS, Gould E, Frankfurt M, McEwen BS. Naturally occurring fluctuation in dendritic spine density on adult hippocampal pyramidal neurons. J Neurosci. 1990;10:4035–4039. doi: 10.1523/JNEUROSCI.10-12-04035.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Woolley CS, McEwen BS. Estradiol mediates fluctuation in hippocampal synapse density during the estrous cycle in the adult rat. J Neurosci. 1992;12:2549–2554. doi: 10.1523/JNEUROSCI.12-07-02549.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cooke BM, Woolley CS. Gonadal hormone modulation of dendrites in the mammalian CNS. J Neurobiol. 2005;64 doi: 10.1002/neu.20143. [DOI] [PubMed] [Google Scholar]
- 105.Tanapat P, Hastings NB, Gould E. Ovarian steroids influence cell proliferation in the dentate gyrus of the adult female rat in a dose- and time-dependent manner. J Comp Neurol. 2005;481:252–265. doi: 10.1002/cne.20385. [DOI] [PubMed] [Google Scholar]
- 106.Leranth C, Petnehazy O, MacLusky NJ. Gonadal hormones affect spine synaptic density in the CA1 hippocampal subfield of male rats. J Neurosci. 2003;23:1588–1592. doi: 10.1523/JNEUROSCI.23-05-01588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hijazi RA, Cunningham GR. Andropause: Is androgen replacement therapy indicated for the aging male? Annu Rev Med. 2005;56:117–137. doi: 10.1146/annurev.med.56.082103.104518. [DOI] [PubMed] [Google Scholar]
- 108.Yaffe K, Lui LY, Zmuda J, Cauley J. Sex hormones and cognitive function in older men. J Am Geriatr Soc. 2002;50:707–712. doi: 10.1046/j.1532-5415.2002.50166.x. [DOI] [PubMed] [Google Scholar]
- 109.Yaffe K. Testosterone and the brain: Uncharted territory. Lancet Neurol. 2004;3:270. doi: 10.1016/S1474-4422(04)00732-X. [DOI] [PubMed] [Google Scholar]
- 110.Moffat SD. Effects of testosterone on cognitive and brain aging in elderly men. Ann NY Acad Sci. 2005;1055:80–92. doi: 10.1196/annals.1323.014. [DOI] [PubMed] [Google Scholar]
- 111.MacLusky NJ, Hajszan T, Prange-Kiel J, Leranth C. Androgen modulation of hippocampal synaptic plasticity. Neuroscience. 2006;138:957–965. doi: 10.1016/j.neuroscience.2005.12.054. [DOI] [PubMed] [Google Scholar]
- 112.Zandi PP, Carlson MC, Plassman BL, Welsh-Bohmer KA, Mayer LS, Steffens DC, et al. Hormone replacement therapy and incidence of Alzheimer disease in older women: The cache county study. JAMA. 2002;288:2123–2129. doi: 10.1001/jama.288.17.2123. [DOI] [PubMed] [Google Scholar]
- 113.Weiss B. Can endocrine disruptors influence neuroplasticity in the aging brain? Neurotoxicology. 2007;28:938–950. doi: 10.1016/j.neuro.2007.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hof PR. Morrison JH. The aging brain: Morphomolecular senescence of cortical circuits. Trends Neurosci. 2004;27:607–613. doi: 10.1016/j.tins.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 115.MacLusky NJ, Hajszan T, Leranth C. The environmental estrogen bisphenol a inhibits estradiol-induced hippocampal synaptogenesis. Environ Health Perspect. 2005;113:675–679. doi: 10.1289/ehp.7633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Leranth C, Hajszan T, Szigeti-Buck K, Bober J, MacLusky NJ. Bisphenol A prevents the synaptogenic response to estradiol in hippocampus and prefrontal cortex of ovariectomized nonhuman primates. Proc Natl Acad Sci USA. 2008a;105:14187–14191. doi: 10.1073/pnas.0806139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Leranth C, Szigeti-Buck K, Maclusky NJ, Hajszan T. Bisphenol A prevents the synaptogenic response to testosterone in the brain of adult male rats. Endocrinology. 2008b;149:988–994. doi: 10.1210/en.2007-1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lang IA, Galloway TS, Scarlett A, Henley WE, Depledge M, Wallace RB, et al. Association of urinary bisphenol A concentration with medical disorders and laboratory abnormalities in adults. JAMA. 2008;300:1303–1310. doi: 10.1001/jama.300.11.1303. [DOI] [PubMed] [Google Scholar]
- 119.Stahlhut RW, van Wijngaarden E, Dye TD, Cook S, Swan SH. Concentrations of urinary phthalate metabolites are associated with increased waist circumference and insulin resistance in adult U.S. males. Environ Health Perspect. 2007;115:876–882. doi: 10.1289/ehp.9882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Meeker JD, Calafat AM, Hauser R. Urinary metabolites of di(2-ethylhexyl) phthalate are associated with decreased steroid hormone levels in adult men. J Androl. 2009;30:287–297. doi: 10.2164/jandrol.108.006403. [DOI] [PMC free article] [PubMed] [Google Scholar]