

Abstract
Colistin plays a key role in treatment of serious infections by Pseudomonas aeruginosa. The aims of this study were to (i) identify the pharmacokinetic/pharmacodynamic (PK/PD) index (i.e., the area under the unbound concentration-time curve to MIC ratio [ƒAUC/MIC], the unbound maximal concentration to MIC ratio [ƒCmax/MIC], or the cumulative percentage of a 24-h period that unbound concentrations exceed the MIC [ƒT>MIC]) that best predicts colistin efficacy and (ii) determine the values for the predictive PK/PD index required to achieve various magnitudes of killing effect. Studies were conducted in a one-compartment in vitro PK/PD model for 24 h using P. aeruginosa ATCC 27853, PAO1, and the multidrug-resistant mucoid clinical isolate 19056 muc. Six intermittent dosing intervals, with a range of ƒCmax colistin concentrations, and two continuous infusion regimens were examined. PK/PD indices varied from 0.06 to 18 for targeted ƒCmax/MIC, 0.36 to 312 for ƒAUC/MIC, and 0 to 100% for ƒT>MIC. A Hill-type model was fit to killing effect data, which were expressed as the log10 ratio of the area under the CFU/ml curve for treated regimens versus control. With ƒCmax values equal to or above the MIC, rapid killing was observed following the first dose; substantial regrowth occurred by 24 h with most regimens. The overall killing effect was best correlated with ƒAUC/MIC (R2 = 0.931) compared to ƒCmax/MIC (R2 = 0.868) and ƒT>MIC (R2 = 0.785). The magnitudes of ƒAUC/MIC required for 1- and 2-log10 reductions in the area under the CFU/ml curve relative to growth control were 22.6 and 30.4, 27.1 and 35.7, and 5.04 and 6.81 for ATCC 27853, PAO1, and 19056 muc, respectively. The PK/PD targets identified will assist in designing optimal dosing strategies for colistin.
Globally there is a growing threat from the emergence of multidrug-resistant (MDR) microorganisms (38), especially among a number of important Gram-negative bacterial pathogens (16, 29, 38). Colistin (polymyxin E) still retains significant activity against many of these MDR Gram-negative pathogens, including Pseudomonas aeruginosa, Acinetobacter baumannii, and Klebsiella pneumoniae, which often leaves it as the only therapeutic option available (19, 26). With very few new chemical entities against Gram-negative infections in the drug development pipeline (29, 30, 38), particularly against P. aeruginosa (38), the use of colistin, a once-neglected antibiotic, has increased dramatically over the last 5 years (11, 26).
Colistin is available commercially as colistin sulfate (hereafter referred to as colistin) and sodium colistin methanesulfonate (CMS), which is administered parenterally. CMS is an inactive prodrug of colistin (3) and, after parenteral administration, colistin is formed in vivo (21, 27, 33). Despite its newfound importance in therapy, there is a dearth of information on the pharmacokinetic (PK) and pharmacodynamic (PD) properties of colistin, a situation of significant concern given that resistance to colistin is beginning to emerge (1, 15, 18, 26, 28). Thus, the aims of the present study were to utilize an in vitro PK/PD model to (i) identify the PK/PD index (i.e., the area under the unbound concentration-time curve to MIC ratio [ƒAUC/MIC], the unbound maximal concentration to MIC ratio [ƒCmax/MIC], or the cumulative percentage of a 24-h period that unbound concentrations exceed the MIC [ƒT>MIC]) that best predicts colistin efficacy and (ii) determine the magnitude of the predictive PK/PD index required to achieve various magnitudes of killing effect.
(Parts of the present study were presented at the 48th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy [ICAAC], Washington, DC, 25 to 28 October 2008, and at the Second American Conference on Pharmacometrics, Mashantucket, CT, 4 to 7 October 2009.)
MATERIALS AND METHODS
Bacterial strains and media.
Three strains of P. aeruginosa were used in the present study: two reference strains, ATCC 27853 and PAO1 (American Type Culture Collection, Rockville, MD), and an MDR mucoid clinical isolate, 19056 muc. The MICs of colistin, as determined by broth microdilution (6), were 1 μg/ml for ATCC 27853 and PAO1 and 0.5 μg/ml for 19056 muc. All MIC determinations were performed in three replicates on separate days. Storage was in tryptone soy broth (Oxoid, Basingstoke, Hampshire, England) with 20% glycerol (Ajax Finechem, Seven Hills, New South Wales, Australia) at −80°C in cryovials (Simport Plastics, Boloeil, Quebec, Canada).
Chemicals and reagents.
Colistin sulfate was purchased from Sigma-Aldrich (lot 095K1048, 20,195 U/mg; St. Louis, MO). Immediately prior to each experiment, colistin stock solutions were prepared by using Milli-Q water (Millipore Australia, North Ryde, New South Wales, Australia), sterilized by filtration with a 0.22-μm-pore-size Millex-GP filter (Millipore, Bedford, MA), and then stored at 4°C before use; colistin is stable under these conditions (22). All other chemicals were from suppliers previously described (23).
Binding of colistin in growth medium.
The binding of colistin in cation-adjusted Mueller-Hinton broth (CAMHB; Ca2+ at 23.0 μg/ml and Mg2+ at 12.2 μg/ml; Oxoid, Hampshire, England) was measured by equilibrium dialysis using a Perspex dialysis cell unit containing two chambers (1 ml in each chamber) separated by a semipermeable membrane (Spectra/Por-2, lot 29300; Spectrum Laboratories, Rancho Dominguez, CA). Colistin (sulfate) was spiked into CAMHB (donor chamber) to achieve concentrations of 10 and 30 μg/ml and dialyzed at 37°C against the same volume of isotonic phosphate buffer (0.067 M, pH 7.3) (acceptor chamber); samples were prepared in triplicate. Samples of CAMHB and buffer were removed from each reservoir after 24 h (shown in preliminary studies to be the time required for equilibration) and stored at −80°C until analyzed as described below. The fraction of colistin unbound in CAMHB (ƒu) was calculated as follows: (acceptor colistin concentration)/(donor colistin concentration).
In vitro PK/PD model and colistin dosing regimens.
Experiments to examine the PK/PD indices driving the microbiological response to colistin were conducted over 24 h using a one-compartment in vitro PK/PD model (2). Briefly, the system consisted of four sealed containers (compartments), each containing 100 ml of CAMHB at 37°C and a magnetic stir bar to ensure adequate mixing. One compartment acted as a control to define growth dynamics in the absence of colistin, whereas colistin was delivered into the remaining compartments to achieve the desired intermittent injection or continuous infusion regimens (see below).
Prior to each experiment, strains were subcultured onto horse blood agar (Media Preparation Unit, The University of Melbourne, Parkville, Australia) and incubated at 35°C overnight. One colony was then selected and grown overnight in 10 ml of CAMHB, from which early-log-phase growth was obtained. A 1.0-ml aliquot of this early-log-phase bacterial suspension was inoculated into each compartment at the commencement of each experiment to yield approximately 106 CFU/ml.
Both intermittent and continuous infusion dosage regimens of colistin were examined. For dosage regimens involving intermittent administration of colistin, sterile drug-free CAMHB from a central reservoir was pumped through the system at a predetermined rate, displacing CAMHB from each compartment, thus simulating colistin elimination (half-life [t1/2] = 4 h) in healthy volunteers (12) and people with cystic fibrosis (21, 34, 35). Flow rates were calibrated prior to experiments and monitored throughout to ensure the system was performing optimally. The appropriate loading dose of colistin (sulfate) was injected into each treatment compartment following bacterial inoculation to achieve the desired steady-state Cmax (≅ƒCmax; see Results); intermittent maintenance doses were given at appropriate intervals to achieve the same ƒCmax as after the respective loading dose. This simulated steady-state PK with intermittent dosing. For the continuous-infusion regimens, colistin was spiked into the CAMHB within the central reservoir prior to initiation of the experiment such that all media flowing through the system (with the exception of the growth control compartment) contained a constant concentration of colistin. For both intermittent and continuous regimens, serial samples were collected aseptically, as shown in Table 1, for viable counting and determination of colistin concentrations. Viable counting was performed by using a Whitley automatic spiral plater (WASP; Don Whitley Scientific, West Yorkshire, United Kingdom) and a ProtoCOL colony counter (Synbiosis, Cambridge, United Kingdom); the limits of counting and quantification of the procedure were 20 and 400 CFU/ml, respectively, as specified in the ProtoCOL manual.
TABLE 1.
Colistin dosage regimens and sampling times in the in vitro PK/PD modela
| Parameter | Dosage regimenb |
|||||
|---|---|---|---|---|---|---|
| 3 h | 4 h | 8 h | 12 h | 24 h | CI | |
| Target ƒCmax(μg/ml) | ||||||
| ATCC 27853 | 0.50, 1.5 | 1.0, 2.0, 3.5, 9.0, 18 | 3.0* | 2.0, 4.5*, 9.0 | 0.20, 0.25, 0.30, 0.50, 1.0, 1.5, 3.5, 9.0*, 18 | 1.0, 4.5 |
| PAO1 | 3.0 | 3.0, 9.0, 18 | 0.06, 0.13, 0.25, 0.50, 1.0, 2.0 | 1.0 | ||
| 19056 mucc | 3.0* | 0.06, 0.13, 0.15, 0.30, 1.0, 1.5, 4.5* | 0.03, 0.06, 0.13, 0.15, 0.25, 0.35, 0.50, 1.0, 1.5, 2.0, 9.0* | |||
| Sampling times (h) for microbiological measurements | 0, 1, 2, 3, 4, 6, 7, 9, 10, 12, 13, 15, 16, 18, 19, 21, 22, 24 | 0, 1, 2, 4, 5, 6, 8, 9, 12, 13, 16, 17, 20, 21, 24 | 0, 1, 2, 4, 6, 8, 9, 16, 17, 24 | 0, 1, 2, 4, 6, 8, 12, 13, 24 | 0, 1, 2, 4, 6, 8, 24 | 0, 1, 2, 4, 6, 8, 12, 24 |
Dosage regimens involved intermittent administration at the dosage intervals indicated (3 to 24 h) to achieve target ƒCmax or constant concentrations simulating continuous infusion (CI).
*, results taken from the first 24 h of a previous study (2).
For this strain, an additional 6-hourly regimen with an ƒCmaxof 1 μg/ml was performed.
Dosing regimens were selected to maximally differentiate among the PK/PD indices under investigation (ƒAUC/MIC, ƒCmax/MIC, and ƒT>MIC). Overall, six intermittent dosing intervals (every 3, 4, 6, 8, 12, and 24 h) were examined with ƒCmax varied across each schedule; two continuous infusion (CI) regimens were also examined (Table 1). In all, 85 treatments across 37 different combinations of dosage frequency and ƒCmax were examined for the three strains. PK/PD indices varied from 0.06 to 18 for targeted ƒCmax/MIC, from 0.36 to 312 for ƒAUC/MIC, and from 0 to 100% for ƒT>MIC. The range of ƒCmax used extended to greater than that seen in humans (21) to explore the complete dose-response relationship from essentially no effect to maximum effect.
Quantification of colistin in CAMHB and buffer.
Samples (250 μl) collected from the in vitro PK/PD model and equilibrium dialysis experiments were analyzed as previously described (2). Concentrations of colistin were measured by using HPLC with derivatization and fluorescence detection (24) with an assay range for colistin sulfate of 0.10 to 6.00 μg/ml; samples were diluted when the expected colistin concentrations were higher than the upper limit of quantification. Analysis of quality control (QC) samples with nominal concentrations of 0.40, 4.00, 9.00, and 18.00 μg/ml (the latter two QC samples required dilution) demonstrated that the accuracy and coefficients of variation were within 15%.
Determination of predictive PK/PD index.
The following PK/PD indices were determined for each dosage regimen: ƒAUC/MIC, ƒCmax/MIC, and ƒT>MIC. The area under the unbound colistin concentration-versus-time curves over 24 h (ƒAUC; μg·h/ml) was determined by equation 1 (see below), where n is the number of dosing intervals in the 24-h period, and k is the elimination rate constant (0.17 h−1, corresponding to a 4-h half-life). The percentage of time that unbound concentrations exceeded the MIC (ƒT>MIC) was determined by equation 2. Targeted ƒCmax, trough (ƒCmin), and k values were used for all calculations.
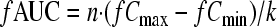 |
(1) |
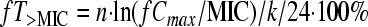 |
(2) |
The area under the curve (AUCCFU) of the time course profile of bacterial numbers (CFU/ml from 0 to 24 h) was calculated by using the linear trapezoidal rule. The killing effect (drug effect) chosen as the measure of efficacy (E) was quantified by the log ratio area method, which compensates for bacterial loss from our model (14):
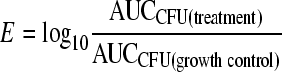 |
(3) |
The relationship between killing effect (E) and each of the three PK/PD indices was analyzed by using the Hill equation with a baseline and an inhibitory effect:
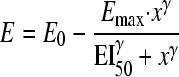 |
(4) |
where E is the observed effect, E0 is the baseline effect in the absence of colistin, Emax is the maximal effect, x is the PK/PD index under investigation, EI50 is the magnitude of the PK/PD index producing 50% of Emax, and γ is the sigmoidicity coefficient (Hill's constant).
The parameters of equation 4 were estimated by three different approaches: (i) uniformly weighted least-squares estimation in WinNonlin Professional (version 5.2.1; Pharsight Corp., Mountain View, CA), (ii) a pooled fitting approach based on maximum-likelihood estimation in NONMEM VI (level 1.2), and (iii) nonlinear mixed-effects modeling in NONMEM VI using the first-order conditional estimation method. An additive error model on a log scale was used for approaches ii and iii. The data for all regimens were fit separately for each of the nine combinations of strains and PK/PD indices for approaches i and ii. The data for all regimens and all three strains were comodeled for approach iii for each of the PK/PD indices. Median estimates and 90% nonparametric confidence intervals (5 to 95% percentile) were determined via nonparametric bootstrapping as described previously using 1,000 replicates for each analysis of approaches ii and iii (4). For each bootstrap data set, 44 regimens were randomly chosen for strain ATCC 27853, 26 regimens were randomly chosen for 19056 muc and 15 regimens were randomly chosen for PAO1. The P values for two-sided nonparametric comparisons between PK/PD indices were computed based on the pairwise differences in objective function values between two PK/PD indices for each of the 1,000 bootstrap replicates. Determination of the PK/PD index best characterizing killing effect was assessed by the coefficient of determination (R2), NONMEM's objective function (−2·log-likelihood), and visual inspection of the observed versus fitted effect plots.
The drug exposure (xnn log10 effect) required for 1- or 2-log10 reduction in the area under the CFU/ml curve relative to growth control (equation 5) and the drug exposure (EI90) causing 90% of maximal effect (equation 6) were calculated as follows:
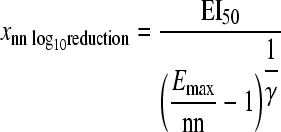 |
(5) |
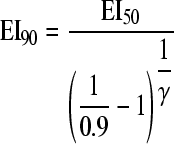 |
(6) |
The desired extent of the log10 reduction (nn) enters equation 5 as a positive number. Equation 5 yields exposure targets only if Emax is larger than nn.
RESULTS
Binding of colistin in CAMHB.
The fractions of colistin unbound in CAMHB (ƒu) at equilibrium, with initial concentrations for colistin sulfate of 10 and 30 μg/ml, were 0.96 and 0.95, respectively, indicating practical equivalence of total and unbound concentrations.
PK validation.
The mean ± the standard deviation (SD; n = 58) of the absolute percentage relative differences between targeted and achieved colistin ƒCmax concentrations as determined by high-pressure liquid chromatography (HPLC) was 9.76 ± 14.2, and the mean of the percentage relative differences was −4.64 ± 16.7. The observed mean t1/2 for the simulated intermittent dosage regimens was 4.06 ± 0.46 h (n = 47) for the targeted value of 4 h; since the ƒCmin for some dosage regimens was below the lower limit of quantification of the HPLC assay (0.10 μg/ml), t1/2 was not directly measured in all experiments.
Bacterial killing of P. aeruginosa in the in vitro PK/PD model.
Representative killing profiles for each strain are shown in Fig. 1. The initial inocula in control and treatment compartments (mean ± the SD) were 6.21 ± 0.09 (n = 15) and 6.18 ± 0.14 (n = 44) log10 CFU/ml for ATCC 27853, 6.39 (n = 2) and 6.29 ± 0.13 (n = 15) log10 CFU/ml for PAO1, and 5.88 ± 0.41 (n = 4) and 6.08 ± 0.32 (n = 26) log10 CFU/ml for 19056 muc. After 24 h, bacterial numbers in control compartments had increased to 8.06 ± 0.20 (n = 15) log10 CFU/ml for ATCC 27853, 8.21 (n = 2) log10 CFU/ml for PAO1, and 7.74 ± 0.07 (n = 4) log10 CFU/ml for 19056 muc. For all strains there was early dosage-dependent killing, followed by regrowth to various extents (Fig. 1).
FIG. 1.

Typical microbiological responses observed in the in vitro PK/PD model simulating the colistin pharmacokinetics of different dosage regimens using ATCC 27853 (MIC = 1 μg/ml) (A), PAO1 (MIC = 1 μg/ml) (B), and the MDR clinical isolate 19056 muc (MIC = 0.5 μg/ml) (C). The y axis starts from the limit of counting, and the limit of quantification is indicated by the horizontal broken line.
Relationships between killing effect and PK/PD indices.
Since the differences in the initial inocula were small (see above), we elected not to standardize AUCCFU by dividing by initial inoculum when calculating the killing effect. Parameter estimates for modeling approaches i, ii, and iii were consistent for all three PK/PD indices and robust for fAUC/MIC and fCmax/MIC; approach iii yielded the most robust estimates for fT>MIC. Modeling by approach iii (Table 2) indicated that between-strain variability was largest for EI50 and negligible for the other parameters. The confidence intervals for EI50 indicated significantly lower values for strain 19056 muc. The relationships between killing effect and ƒAUC/MIC, ƒCmax/MIC, or ƒT>MIC are shown in Fig. 2. Of the three indices, ƒAUC/MIC best described the killing effect (R2 = 0.931; Fig. 2A); the relationship between the killing effect and ƒCmax/MIC had a lower R2 of 0.868 (Fig. 2B). A poorer relationship existed between the killing effect and ƒT>MIC, where a high degree of scatter and systematic deviations from the curve fit was observed (R2 = 0.785; Fig. 2C). Median (nonparametric 90% confidence interval) objective function values from modeling approach iii were as follows: −79.6 (−111 to −54.4) for fAUC/MIC, −28.3 (−72.1 to 2.9) for fCmax/MIC, and 16.4 (−35.9 to 47.5) for fT>MIC. The objective function was significantly lower for fAUC/MIC compared to fCmax/MIC (P = 0.050, two-sided testing; from 1,000 nonparametric bootstrap replicates) and for fAUC/MIC compared to fT>MIC (P < 0.01). Differences between fCmax/MIC and fT>MIC were not significant (P = 0.2).
TABLE 2.
Median parameter estimates from 1,000 bootstrap replicates for each of the three PK/PD indicesa
| PK/PD index | Strain | Median parameter estimates (90% nonparametric confidence intervals) |
|||
|---|---|---|---|---|---|
| E0 | Emax | EI50 | γ | ||
| fAUC/MIC | ATCC 27853 | -0.232 (-0.349 to −0.139) | 3.05 (2.88-3.21) | 26.4 (23.8-28.9) | 4.77 (3.20-7.27) |
| PAO1 | 31.2 (28.6-35.4) | ||||
| 19056 muc | 5.91 (4.60-12.5) | ||||
| fCmax/MIC | ATCC 27853 | -0.110 (-0.319 to 0.025) | 3.12 (2.81-3.38) | 1.82 (1.54-2.25) | 3.13 (2.08-12.2) |
| PAO1 | 2.48 (1.74-3.27) | ||||
| 19056 muc | 0.834 (0.645-1.27) | ||||
| fT>MIC | ATCC 27853 | -0.365 (-0.558 to −0.212) | 3.07 (2.68-3.39) | 39.6 (31.9-47.1) | 2.13 (1.26-4.30) |
| PAO1 | 68.4 (41.0-135) | ||||
| 19056 muc | 13.6 (9.90-21.7) | ||||
Data for all three strains were comodeled for each PK/PD index (approach iii, see Materials and Methods). Initial models with between-strain variability for all four parameters showed that the variability in E0, Emax, and γ was negligible. Since exclusion of the variability for these three parameters did not affect the objective function significantly, the final model only included between-strain variability for EI50.
FIG. 2.

Relationship between killing effect (log area ratio) against P. aeruginosa ATCC 27853 (solid line and open circles), PAO1 (dashed line and solid triangles), and 19056 muc (dotted line and crosses) as a function of three PK/PD indices: ƒAUC/MIC (A), ƒCmax/MIC (B), and ƒT>MIC (C). Each data point represents the result from a single treatment run. Lines represent model-generated fits using modeling approach iii (see Materials and Methods and Table 2).
The magnitudes of the ƒAUC/MIC index required for 1- and 2-log10 reduction in the area under the CFU/ml curve relative to growth control for each strain are shown in Table 3. Near-maximal killing was achieved with ƒAUC/MIC ratios of approximately 40, 50, and 9 for ATCC 27853, PAO1, and 19056 muc, respectively (Table 3 and Fig. 2A).
TABLE 3.
Median target values from 1,000 bootstrap replicates of colistin ƒAUC/MIC for 1- and 2-log10 reductions in the area under the CFU/ml curve relative to growth control and for 90% (EI90) of maximal effect
| Killing effect | Median target values (90% nonparametric confidence intervals) |
||
|---|---|---|---|
| ATCC 27853 | PAO1 | 19056 muc | |
| 1-log10 reduction | 22.6 (19.9-25.7) | 27.1 (23.6-29.9) | 5.04 (3.93-10.5) |
| 2-log10 reduction | 30.4 (27.2-33.0) | 35.7 (32.6-41.7) | 6.81 (5.21-14.3) |
| EI90 | 42.0 (35.3-52.1) | 49.3 (40.8-68.5) | 9.78 (6.71-20.3) |
DISCUSSION
Colistin, which first became clinically available more than 50 years ago, was never subjected to many of the drug development procedures required of new drugs today. As a consequence, current dosage regimens for CMS/colistin are chosen empirically, and much is still to be learned about the PK, PD, and the PK/PD index that best correlates with antibacterial activity of colistin (26). Such information is important for rational design of optimal dosing strategies.
Previous animal or in vitro pharmacodynamic studies have reported regrowth of P. aeruginosa with a range of colistin (2, 17) or polymyxin B (39) dosage regimens. Similarly, this was generally observed in the present study despite unbound colistin concentrations far in excess of clinically achievable concentrations in some experiments. Even for the regimens that achieved very extensive bacterial killing after the first dose, substantially less net killing occurred after subsequent doses (Fig. 1). We have shown previously the presence of resistant subpopulations after a 72-h exposure to colistin in the same in vitro PK/PD model (2). We are currently developing a mechanism-based mathematical model that can describe and predict the time course of bacterial growth and killing and which incorporates the emergence of multiple bacterial populations with various colistin susceptibilities.
Three previous studies have addressed issues around the exposure-response relationships for polymyxins. Using a limited dose fractionation design, Tam et al. (39) investigated the PD of polymyxin B against P. aeruginosa in an in vitro PK/PD hollow-fiber model and suggested that activity was most likely linked to AUC/MIC. The study by Tam et al. (39), however, was not specifically designed to examine the relationship between efficacy and each PK/PD index. In a study of colistin against P. aeruginosa in a neutropenic mouse infection model, Ketthireddy et al. (17) concluded that once-daily dosing was most effective and that the data were consistent with Cmax/MIC being the PK/PD index most predictive of efficacy; PK data, however, were not included in that study. In neutropenic mouse thigh and lung infection models, Dudhani et al. (10) found that ƒAUC/MIC was the index most predictive of efficacy. In the present study, we used a much larger dose fractionation design in an in vitro dynamic model to distinguish between PK/PD indices determining colistin efficacy. A Hill-type model was fit to the data using an area-based method whereby all CFU/ml versus time data for each regimen were taken into account. This approach allowed for a measure of the time-averaged drug effect and has been implemented in a previous investigation with vancomycin against Staphylococcus aureus (14). The analysis demonstrated that ƒAUC/MIC was most closely correlated with bacterial killing (Fig. 2). Estimates of PD parameters were precise and consistent between all three estimation approaches.
In order to design dosage regimens rationally, it is necessary to know not only which PK/PD index is most predictive of bacterial killing but also the magnitude of that index needed to achieve various extents of kill (7). In the present study, respective values of ƒAUC/MIC of ∼25 and 35 for the reference strains were required to achieve 1- and 2-log reductions in the area under the CFU/ml curve relative to growth control (Table 3). These results are in extremely good agreement with those obtained by Dudhani et al. (10) in the neutropenic mouse thigh infection model against the same two strains (ƒAUC/MIC values of ∼23 and 34 for 1- and 2-log reductions, respectively). Interestingly, the corresponding ƒAUC/MIC values for the MDR clinical isolate were somewhat lower in our in vitro model compared to the neutropenic mouse thigh infection model (∼6 and 7 in vitro compared to ∼16 and 28 in vivo for 1- and 2-log reductions, respectively). The explanation for this difference is not known but may relate to differences in growth dynamics of this mucoid strain between in vitro and in vivo systems. We acknowledge the presence of a washout effect on bacteria with the use of open one-compartment PK/PD systems such as in our study; however, the generally good level of agreement across all strains between the present in vitro study and infection models involving neutropenic mice (10) is very reassuring in relation to future clinical applications of the ƒAUC/MIC targets for colistin.
Unfortunately, it is currently not possible to compare the ƒAUC/MIC targets from the present and other (10) preclinical studies with the ƒAUC/MIC values achieved in infected patients receiving currently recommended CMS dosage regimens. The inability to undertake this comparison arises because recent studies have shown that colistin binding in plasma involves the acute-phase reactant α1-acid glycoprotein (AAG), and the unbound fraction of colistin is influenced by the concentrations of both colistin and AAG (9). Since plasma AAG concentrations are influenced by pathophysiological stresses including infection (31, 40), the ƒu of colistin in patients is likely to vary depending on the severity and stage of infection and magnitude of plasma colistin concentration. Although the knowledge of total plasma colistin concentrations achieved in patients is increasing (21, 25, 27, 33), there is no information on unbound plasma concentrations. As such information is forthcoming it will be possible to not only assess the ability of current CMS dosage regimens to meet the above-mentioned ƒAUC/MIC targets but also to design optimized dosage regimens.
The use of once-daily doses of CMS has recently been reported (13, 36), presumably based upon the concentration-dependent killing of colistin observed in vitro (28). However, we suggest caution with this approach. First, in vitro data suggest that the toxicity of colistin is concentration and time dependent (20). Moreover, greater nephrotoxicity was observed in rats with a dosage regimen mimicking once-daily dosing of CMS in humans compared to a twice-daily regimen that delivered the same daily dose (41). Second, for both colistin and polymyxin B larger, infrequent doses in in vitro models led to greater emergence of resistance in P. aeruginosa compared to lower-dose/higher-frequency regimens (2, 39). Third, colistin lacks a significant postantibiotic effect in vitro (28, 32), although in a brief report such a phenomenon has been suggested to occur in vivo (17); additional studies are needed. Given the recent emergence of resistance to colistin (1, 15, 18, 26, 28), the ability to choose regimens which not only maximize killing but also suppress or minimize the development of resistance may prove crucial in preventing this trend.
In the present study, we simulated a 4-h colistin half-life as observed in healthy volunteers (12) and people with cystic fibrosis (21, 34, 35); a longer half-life has been reported in critically ill patients (27, 33, 37). The PK/PD indices described here are specific for P. aeruginosa and may differ for other Gram-negative pathogens; species specific differences in the magnitude of a particular index required to achieve certain levels of killing have been demonstrated for other anti-infectives (8). In addition, in vitro PK/PD models lack the defense mechanisms present in patients with intact immune systems; however, they may more adequately reflect drug-related antimicrobial activity in an immunocompromised host. Finally, higher PK/PD target values (e.g., for fAUC/MIC) may be required for infections with a high initial inoculum (5).
To our knowledge, this is the first in vitro investigation specifically designed to elucidate the relationship between bacterial killing and PK/PD indices for colistin against any organism. We have demonstrated that for colistin fAUC/MIC is the PK/PD index most closely correlated with the killing of P. aeruginosa. Our findings are in good agreement with those from recent studies in neutropenic mouse infection models. As information on the pharmacokinetics of unbound colistin in patients is obtained, the PK/PD targets reported here will assist in designing optimal dosing strategies for this increasingly important therapeutic option.
Acknowledgments
The assistance of Chun-Hong Tan and Hui He of the Facility for Anti-infective Drug Development and Innovation, Monash University, Melbourne, Australia, and of Charlemagne Lacza of the School of Pharmacy and Pharmaceutical Sciences, State University of New York at Buffalo, Buffalo, is gratefully acknowledged.
The project described was supported by awards R01AI079330 and R01AI070896 from the National Institute of Allergy and Infectious Diseases. J.L. is an Australian National Health and Medical Research Council R. Douglas Wright Research Fellow.
The content here is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
We have no conflicts of interest to declare.
Footnotes
Published ahead of print on 28 June 2010.
REFERENCES
- 1.Antoniadou, A., F. Kontopidou, G. Poulakou, E. Koratzanis, I. Galani, E. Papadomichelakis, P. Kopterides, M. Souli, A. Armaganidis, and H. Giamarellou. 2007. Colistin-resistant isolates of Klebsiella pneumoniae emerging in intensive care unit patients: first report of a multiclonal cluster. J. Antimicrob. Chemother. 59:786-790. [DOI] [PubMed] [Google Scholar]
- 2.Bergen, P. J., J. Li, R. L. Nation, J. D. Turnidge, K. Coulthard, and R. W. Milne. 2008. Comparison of once-, twice-, and thrice-daily dosing of colistin on antibacterial effect and emergence of resistance: studies with Pseudomonas aeruginosa in an in vitro pharmacodynamic model. J. Antimicrob. Chemother. 61:636-642. [DOI] [PubMed] [Google Scholar]
- 3.Bergen, P. J., J. Li, C. R. Rayner, and R. L. Nation. 2006. Colistin methanesulfonate is an inactive prodrug of colistin against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 50:1953-1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bulitta, J. B., S. B. Duffull, M. Kinzig-Schippers, U. Holzgrabe, U. Stephan, G. L. Drusano, and F. Sorgel. 2007. Systematic comparison of the population pharmacokinetics and pharmacodynamics of piperacillin in cystic fibrosis patients and healthy volunteers. Antimicrob. Agents Chemother. 51:2497-2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bulitta, J. B., J. C. Yang, L. Yohonn, N. S. Ly, S. V. Brown, R. E. D'Hondt, W. J. Jusko, A. Forrest, and B. T. Tsuji. 2010. Attenuation of colistin bactericidal activity by high inoculum of Pseudomonas aeruginosa characterized by a new mechanism-based population pharmacodynamic model. Antimicrob. Agents Chemother. 54:2051-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clinical and Laboratory Standards Institute. 2005. Performance standards for antimicrobial susceptibility testing; 15th informational supplement (M100-S15). Clinical and Laboratory Standards Institute, Wayne, PA.
- 7.Craig, W. A. 2003. Basic pharmacodynamics of antibacterials with clinical applications to the use of beta-lactams, glycopeptides, and linezolid. Infect. Dis. Clin. N. Am. 17:479-501. [DOI] [PubMed] [Google Scholar]
- 8.Craig, W. A. 1995. Interrelationship between pharmacokinetics and pharmacodynamics in determining dosage regimens for broad-spectrum cephalosporins. Diagn. Microbiol. Infect. Dis. 22:89-96. [DOI] [PubMed] [Google Scholar]
- 9.Dudhani, R. V., J. Li, and R. Nation, L. 2009. Plasma binding (PB) of colistin (C) involves multiple proteins and is concentration dependent: potential clinical implications, abstr. A1-576. Abstr. 49th Annu. Intersci. Conf. Antimicrob. Agents Chemother.
- 10.Dudhani, R. V., J. D. Turnidge, K. Coulthard, R. W. Milne, C. R. Rayner, J. Li, and R. L. Nation. 2010. Elucidation of the pharmacokinetic/pharmacodynamic determinant of colistin activity against Pseudomonas aeruginosa in murine thigh and lung infection models. Antimicrob. Agents Chemother. 54:1117-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falagas, M. E., and S. K. Kasiakou. 2005. Colistin: the revival of polymyxins for the management of multidrug-resistant gram-negative bacterial infections. Clin. Infect. Dis. 40:1333-1341. [DOI] [PubMed] [Google Scholar]
- 12.Gregoire, N., P. Gobin, C. Grignon, D. Frasca, P. Saulnier, W. Couet, and O. Mimoz. 2008. Pharmacokinetic modeling of colistin methanesulfonate (CMS) and colistin in healthy volunteers after intravenous infusion of CMS, abstr. A-1668, p. 27. Abstr. 48th Intersci. Conf. Antimicrob. Agents Chemother.
- 13.Gunderson, B. W., K. H. Ibrahim, L. B. Hovde, T. L. Fromm, M. D. Reed, and J. C. Rotschafer. 2003. Synergistic activity of colistin and ceftazidime against multiantibiotic-resistant Pseudomonas aeruginosa in an in vitro pharmacodynamic model. Antimicrob. Agents Chemother. 47:905-909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harigaya, Y., J. B. Bulitta, A. Forrest, G. Sakoulas, A. J. Lesse, J. M. Mylotte, and B. T. Tsuji. 2009. Pharmacodynamics of vancomycin at simulated epithelial lining fluid concentrations against methicillin-resistant Staphylococcus aureus (MRSA): implications for dosing in MRSA pneumonia. Antimicrob. Agents Chemother. 53:3894-3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johansen, H. K., S. M. Moskowitz, O. Ciofu, T. Pressler, and N. Hoiby. 2008. Spread of colistin resistant non-mucoid Pseudomonas aeruginosa among chronically infected Danish cystic fibrosis patients. J. Cyst. Fibros. 7:391-397. [DOI] [PubMed] [Google Scholar]
- 16.Jones, R. N. 2001. Resistance patterns among nosocomial pathogens: trends over the past few years. Chest 119:397S-404S. [DOI] [PubMed] [Google Scholar]
- 17.Ketthireddy, S., D. G. Lee, Y. Murakami, T. Stamstad, D. R. Andes, and W. A. Craig. 2007. In vivo pharmacodynamics of colistin against Pseudomonas aeruginosa in thighs of neutropenic mice, abstr. A-4, p. 1. Abstr. 47th Intersci. Conf. Antimicrob. Agents Chemother.
- 18.Ko, K. S., J. Y. Suh, K. T. Kwon, S. I. Jung, K. H. Park, C. I. Kang, D. R. Chung, K. R. Peck, and J. H. Song. 2007. High rates of resistance to colistin and polymyxin B in subgroups of Acinetobacter baumannii isolates from Korea. J. Antimicrob. Chemother. 60:1163-1167. [DOI] [PubMed] [Google Scholar]
- 19.Landman, D., C. Georgescu, D. A. Martin, and J. Quale. 2008. Polymyxins revisited. Clin. Microbiol. Rev. 21:449-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis, J. R., and S. A. Lewis. 2004. Colistin interactions with the mammalian urothelium. Am. J. Physiol. Cell Physiol. 286:C913-C922. [DOI] [PubMed] [Google Scholar]
- 21.Li, J., K. Coulthard, R. Milne, R. L. Nation, S. Conway, D. Peckham, C. Etherington, and J. Turnidge. 2003. Steady-state pharmacokinetics of intravenous colistin methanesulphonate in patients with cystic fibrosis. J. Antimicrob. Chemother. 52:987-992. [DOI] [PubMed] [Google Scholar]
- 22.Li, J., R. W. Milne, R. L. Nation, J. D. Turnidge, and K. Coulthard. 2003. Stability of colistin and colistin methanesulfonate in aqueous media and plasma as determined by high-performance liquid chromatography. Antimicrob. Agents Chemother. 47:1364-1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li, J., R. W. Milne, R. L. Nation, J. D. Turnidge, K. Coulthard, and D. W. Johnson. 2001. A simple method for the assay of colistin in human plasma, using pre-column derivatization with 9-fluorenylmethyl chloroformate in solid-phase extraction cartridges and reversed-phase high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 761:167-175. [DOI] [PubMed] [Google Scholar]
- 24.Li, J., R. W. Milne, R. L. Nation, J. D. Turnidge, T. C. Smeaton, and K. Coulthard. 2003. Use of high-performance liquid chromatography to study the pharmacokinetics of colistin sulfate in rats following intravenous administration. Antimicrob. Agents Chemother. 47:1766-1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li, J., and R. L. Nation. 2006. Comment on: pharmacokinetics of inhaled colistin in patients with cystic fibrosis. J. Antimicrob. Chemother. 58:222-223. [DOI] [PubMed] [Google Scholar]
- 26.Li, J., R. L. Nation, J. D. Turnidge, R. W. Milne, K. Coulthard, C. R. Rayner, and D. L. Paterson. 2006. Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect. Dis. 6:589-601. [DOI] [PubMed] [Google Scholar]
- 27.Li, J., C. R. Rayner, R. L. Nation, R. Deans, R. Boots, N. Widdecombe, A. Douglas, and J. Lipman. 2005. Pharmacokinetics of colistin methanesulfonate and colistin in a critically ill patient receiving continuous venovenous hemodiafiltration. Antimicrob. Agents Chemother. 49:4814-4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li, J., J. Turnidge, R. Milne, R. L. Nation, and K. Coulthard. 2001. In vitro pharmacodynamic properties of colistin and colistin methanesulfonate against Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Antimicrob. Agents Chemother. 45:781-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Livermore, D. M. 2004. The need for new antibiotics. Clin. Microbiol. Infect. 10(Suppl. 4):1-9. [DOI] [PubMed] [Google Scholar]
- 30.Livermore, D. M. 2003. The threat from the pink corner. Ann. Med. 35:226-234. [DOI] [PubMed] [Google Scholar]
- 31.Morita, K., and A. Yamaji. 1995. Changes in the serum protein binding of vancomycin in patients with methicillin-resistant Staphylococcus aureus infection: the role of serum α1-acid glycoprotein levels. Ther. Drug Monit. 17:107-112. [DOI] [PubMed] [Google Scholar]
- 32.Owen, R. J., J. Li, R. L. Nation, and D. Spelman. 2007. In vitro pharmacodynamics of colistin against Acinetobacter baumannii clinical isolates. J. Antimicrob. Chemother. 59:473-477. [DOI] [PubMed] [Google Scholar]
- 33.Plachouras, D., M. Karvanen, L. E. Friberg, E. Papadomichelakis, A. Antoniadou, I. Tsangaris, I. Karaiskos, G. Poulakou, F. Kontopidou, A. Armaganidis, O. Cars, and H. Giamarellou. 2009. Population pharmacokinetic analysis of colistin methanesulfonate and colistin after intravenous administration in critically ill patients with gram-negative bacterial infections. Antimicrob. Agents Chemother. 53:3430-3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ratjen, F., E. Rietschel, D. Kasel, R. Schwiertz, K. Starke, H. Beier, S. van Koningsbruggen, and H. Grasemann. 2006. Pharmacokinetics of inhaled colistin in patients with cystic fibrosis. J. Antimicrob. Chemother. 57:306-311. [DOI] [PubMed] [Google Scholar]
- 35.Reed, M. D., R. C. Stern, M. A. O'Riordan, and J. L. Blumer. 2001. The pharmacokinetics of colistin in patients with cystic fibrosis. J. Clin. Pharmacol. 41:645-654. [DOI] [PubMed] [Google Scholar]
- 36.Rosenvinge, A., T. Pressler, and N. Hoiby. 2005. Colistin intravenously is a safe and effective treatment of multidrug-resistant microorganisms in cystic fibrosis. J. Cyst. Fibros. 4:S32. [Google Scholar]
- 37.Skiada, A., J. Pavleas, C. Vafiadi, K. Salatas, A. Markoyannakis, P. Tofas, K. Tzanetou, G. Thomopoulos, A. Rigas, K. Rigas, E. Vafiadi, G. L. Petrikkos, and G. L. Daikos. 2008. Pharmacokinetics of three different dosing regimens of colistin with meaning for optimum use, abstr. A-1670, p. 27. 48th Annu. Intersci. Conf. Antimicrob. Agents Chemother.
- 38.Talbot, G. H., J. Bradley, J. E. Edwards, Jr., D. Gilbert, M. Scheld, and J. G. Bartlett. 2006. Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin. Infect. Dis. 42:657-668. [DOI] [PubMed] [Google Scholar]
- 39.Tam, V. H., A. N. Schilling, G. Vo, S. Kabbara, A. L. Kwa, N. P. Wiederhold, and R. E. Lewis. 2005. Pharmacodynamics of polymyxin B against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 49:3624-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Voulgari, F., P. Cummins, T. I. Gardecki, N. J. Beeching, P. C. Stone, and J. Stuart. 1982. Serum levels of acute phase and cardiac proteins after myocardial infarction, surgery, and infection. Br. Heart J. 48:352-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wallace, S. J., J. Li, R. L. Nation, C. R. Rayner, D. Taylor, D. Middleton, R. W. Milne, K. Coulthard, and J. D. Turnidge. 2008. Subacute toxicity of colistin methanesulfonate in rats: comparison of various intravenous dosage regimens. Antimicrob. Agents Chemother. 52:1159-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]