

Abstract
A new β-glucosidase from a novel strain of Terrabacter ginsenosidimutans (Gsoil 3082T) obtained from the soil of a ginseng farm was characterized, and the gene, bgpA (1,947 bp), was cloned in Escherichia coli. The enzyme catalyzed the conversion of ginsenoside Rb1 {3-O-[β-d-glucopyranosyl-(1-2)-β-d-glucopyranosyl]-20-O-[β-d-glucopyranosyl-(1-6)-β-d-glucopyranosyl]-20(S)-protopanaxadiol} to the more pharmacologically active rare ginsenosides gypenoside XVII {3-O-β-d-glucopyranosyl-20-O-[β-d-glucopyranosyl-(1-6)-β-d-glucopyranosyl]-20(S)-protopanaxadiol}, gypenoside LXXV {20-O-[β-d-glucopyranosyl-(1-6)-β-d-glucopyranosyl]-20(S)-protopanaxadiol}, and C-K [20-O-(β-d-glucopyranosyl)-20(S)-protopanaxadiol]. A BLAST search of the bgpA sequence revealed significant homology to family 3 glycoside hydrolases. Expressed in E. coli, β-glucosidase had apparent Km values of 4.2 ± 0.8 and 0.14 ± 0.05 mM and Vmax values of 100.6 ± 17.1 and 329 ± 31 μmol·min−1·mg of protein−1 against p-nitrophenyl-β-d-glucopyranoside and Rb1, respectively. The enzyme catalyzed the hydrolysis of the two glucose moieties attached to the C-3 position of ginsenoside Rb1, and the outer glucose attached to the C-20 position at pH 7.0 and 37°C. These cleavages occurred in a defined order, with the outer glucose of C-3 cleaved first, followed by the inner glucose of C-3, and finally the outer glucose of C-20. These results indicated that BgpA selectively and sequentially converts ginsenoside Rb1 to the rare ginsenosides gypenoside XVII, gypenoside LXXV, and then C-K. Herein is the first report of the cloning and characterization of a novel ginsenoside-transforming β-glucosidase of the glycoside hydrolase family 3.
Ginseng refers to the roots of members of the plant genus Panax, which have been used as a traditional medicine in Asian countries for over 2,000 years due to their observed beneficial effects on human health. Ginseng saponins, also referred to as ginsenosides, are the major active components of ginseng (27). Various biological activities have been ascribed to ginseng saponins, including anti-inflammatory activity (43), antitumor effects (23, 39), and neuroprotective and immunoprotective (15, 31) effects.
Ginsenosides can be categorized as protopanaxadiol (PPD), protopanaxatriol, and oleanane saponins, based on the structure of the aglycon, with a dammarane skeleton (29). The PPD-type ginsenosides are further classified into subgroups based on the position and number of sugar moieties attached to the aglycon at positions C-3 and C-20. For example, one of the largest PPD-type ginsenosides, Rb1 {3-O-[β-d-glucopyranosyl-(1-2)-β-d-glucopyranosyl]-20-O-[β-d-glucopyranosyl-(1-6)-β-d-glucopyranosyl]-20(S)-protopanaxadiol}, contains 4 glucose moieties, two each attached via glycosidic linkages to the C-3 and C-20 positions of the aglycon (Fig. 1).
FIG. 1.

Chemical structures of protopanaxadiol and protopanaxatriol ginsenosides (5). The ginsenosides represented here are all (S)-type ginsenosides. glc, β-d-glucopyranosyl; arap, α-l-arabinopyranosyl; araf, α-l-arabinofuranosyl; rha, α-l-rhamnopyranosyl; Gyp, gypenoside; C, compound.
Because of their size, low solubility, and poor permeability across the cell membrane, it is difficult for human body to directly absorb large ginsenosides (44), although these components constitute the major portion of the total ginsenoside in raw ginseng (30). Moreover, the lack of the availability of the rare ginsensoides limits the research on their biological and medicinal properties. Therefore, transformation of these major ginsenosides into smaller deglycosylated ginsenosides, which are more effective in in vivo physiological action, is required (1, 37).
The production of large amounts of rare ginsenosides from the major ginsenosides can be accomplished through a number of physiochemical methods such as heating (17), acid treatment (2), and alkali treatment (48). However, these approaches produce nonspecific racemic mixtures of rare ginsenosides. As an alternative, enzymatic methods have been explored as a way to convert the major ginsenosides into more pharmacologically active rare ginsenosides in a more specific manner (14, 20).
To date, three types of glycoside hydrolases, β-d-glucosidase, α-l-arabinopyranosidase, and α-l-arabinofuranosidase, have been found to be involved in the biotransformation of PPD-type ginsenosides. For example, a β-glucosidase isolated from a fungus converts Rb1 to C-K [20-O-(β-d-glucopyranosyl)-20(S)-protopanaxadiol] (45), and an α-l-arabinopyranosidase and α-l-arabinofuranosidase have been isolated from an intestinal bacterium that hydrolyze, respectively, Rb2 {3-O-[β-d-glucopyranosyl-(1-2)-β-d-glucopyranosyl]-20-O-[α-l-arabinopyranosyl-(1-6)-β-d-glucopyranosyl]-20(S)-protopanaxadiol} to Rd {3-O-[β-d-glucopyranosyl-(1-2)-β-d-glucopyranosyl]-20-O-β-d-glucopyranosyl-20(S)-protopanaxadiol} and Rc {3-O-[β-d-glucopyranosyl-(1-2)-β-d-glucopyranosyl]-20-O- [α-l-arabinofuranosyl-(1-6)-β-d-glucopyranosyl]-20(S)-protopanaxadiol} to Rd (34). Two recombinant enzymes that convert major ginsenosides into rare ginsenosides have been cloned and expressed in Escherichia coli: Solfolobus solfataricus β-glycosidase, which transforms Rb1 or Rc to C-K (28), and β-glucosidase from a soil metagenome, which transforms Rb1 to Rd (16). Both of these glycoside hydrolases are family 1 glycoside hydrolases.
Here, we report the cloning and expression in E. coli of a gene (bgpA) encoding a new ginsenoside-hydrolyzing β-glucosidase from a novel bacterial strain, Terrabacter ginsenosidimutans sp. nov. Gsoil 3082, isolated from a ginseng farm in Korea. BgpA is a family 3 glycoside hydrolase, and the recombinant enzyme employs a different enzymatic pathway from ginsenoside-hydrolyzing family 1 glycoside hydrolases. BgpA preferentially and sequentially hydrolyzed the terminal and inner glucoses at the C-3 position of ginsenoside Rb1 and then the outer glucose at the C-20 position. Thus, BgpA could be effective in the biotransformation of ginsenoside Rb1 to gypenoside (Gyp) XVII {3-O-β-d-glucopyranosyl-20-O-[β-d-glucopyranosyl-(1-6)-β-d-glucopyranosyl]-20(S)-protopanaxadiol}, Gyp LXXV {20-O-[β-d-glucopyranosyl-(1-6)-β-d-glucopyranosyl]-20(S)-protopanaxadiol}, and C-K.
MATERIALS AND METHODS
Chemicals.
Ginsenosides Rb1, Rd, F2, Rg3, C-K, and Rh2 were purchased from Dalian Green Bio, Ltd. (Dalian, China). 5-Bromo-4-chloro-3-indolyl β-d-glucopyranoside (X-Glc) and p-nitrophenyl-β-d-glucopyranoside (PNPGlc) were obtained from Sigma. The other chemicals used in this study were at least of analytical reagent grade, and the sources are noted individually in the methods sections below.
Isolation and preparation of strain Gsoil 3082 for ginsenoside bioconversion.
Strain Gsoil 3082 was isolated from the soil of a ginseng farm in Korea (37°58′22"N, 127°15′27"E) by a three-step screening process consisting of isolation of bacteria from soil, screening of bacteria for β-glucosidase activity based on esculin hydrolysis, and then selection of strains with biotransformation activity for ginsenosides by thin-layer chromatography (TLC).
Soil samples taken from around ginseng root hairs were thoroughly suspended in 50 mM phosphate buffer (pH 7.0), serially diluted with 50 mM phosphate buffer (pH 7.0), and then spread on one-fifth-strength modified R2A agar plates (full-strength modified R2A agar contained 0.25 g of tryptone, 0.25 g of peptone, 0.25 g of yeast extract, 0.125 g of malt extract, 0125 g of beef extract, 025 g of Casamino Acids, 0.25 g of soytone, 0.5 g of dextrose, 0.3 g of soluble starch, 0.2 g of xylan, 0.3 g of sodium pyruvate, 0.3 g of K2HPO4, 0.05 g of MgSO4, 0.05 g of CaCl2, and 15 g of agar per liter). Plates were incubated at 30°C for 1 month. Bacterial isolates were obtained by transferring single colonies to one-half-strength modified R2A agar plates. Isolates were routinely cultured on R2A agar (Difco) at 30°C and stored as glycerol suspensions (20% [wt/vol]) at −70°C.
Isolated colonies were transferred to esculin-R2A plates (R2A agar containing 1.0 g of esculin and 0.5 g of ferric citrate per liter) to screen for strains with high β-glucosidase activity. Esculetin released from esculin by β-glucosidase activity reacts with ferric ions to produce a reddish-to-dark brown zone in the transparent background. Black colonies on esculin-R2A agar that exhibited β-glucosidase activity were tested for their ability to biotransform ginsenoside Rb1. Liquid R2A broth cultures of β-glucosidase-positive bacterial strains in the logarithmic phase of growth were mixed with 200 μl of autoclaved R2A broth containing 1 mg/ml Rb1 or other ginsenosides in a 1.5-ml Eppendorf tube and then incubated at 30°C. At 24-h intervals, a 50-μl aliquot was removed, extracted with the same volume of water-saturated n-butanol, and then analyzed by TLC. Strain Gsoil 3082, which was positive for ginsenoside-hydrolyzing β-glucosidase activity, was selected for gene cloning experiments.
Construction and screening of a fosmid library.
To clone the ginsenoside β-glucosidase gene from Gsoil 3082, a fosmid library was constructed using a CopyControl fosmid library production kit (Epicentre, Madison, WI), according to the manufacturer's protocol. Gsoil 3082 genomic DNA was randomly sheared into fragments of approximately 40 kb by passage through a 200-μl small-bore pipette. The fragments were separated by pulsed-field gel electrophoresis (CHEF-DRII apparatus; Bio-Rad), and the sheared DNA was repaired to produce blunt 5′-phosphorylated ends. End-repaired DNA fragments above 40 kb were obtained using low-melting-point agarose gel electrophoresis. Blunt-end DNA was ligated into pCC1FOS (Epicentre). In vitro packaging was performed with the MaxPlax lambda packaging extract kit (Epicentre), and the products were transformed into E. coli EPI300-T1R grown in Luria-Bertani (LB) broth containing 10 mM MgSO4. Infected bacteria were transferred onto LB plates containing 12.5 μg/ml chloramphenicol and 27 μg/ml X-Glc, and then the plates were incubated at 37°C. After a 16- to 20-h incubation period, putative β-glucosidase-producing clones were selected based on their blue color. A single clone containing a putative ginsenoside β-glucosidase gene was selected by TLC assay for ginsenoside-hydrolyzing activity.
Fosmid sequencing.
The full sequence of the positive fosmid clone was determined by insertion of a transposon into the fosmid clone, followed by DNA sequencing using transposon-specific primers. The fosmid DNA was purified with a Fosmid MAX DNA purification kit (Epicentre). Transposon insertion was performed using a HyperMu KAN-1 insertion kit (Epicentre), according to the manufacturer's protocol. The complete sequence was obtained by bidirectional sequencing of the fosmid using two primers that were homologous to the ends of the inserted transposon. The DNA sequencing reactions were performed using ABI PRISM Bigdye terminator chemistry and ABI 3730XL capillary DNA sequencers (Applied Biosystems, Foster City, CA). Sequences were assembled using the SeqMan program in the DNASTAR package (DNASTAR, Madison, WI), yielding a 33.9-kb contig.
Molecular cloning, expression, and purification of recombinant BgpA.
The assembled DNA contig was analyzed using the ORF Finder program of the National Center for Biotechnology Information (NCBI) (www.ncbi.nlm.nih.gov). The sequence was subjected to a similarity search using BLASTP, and an open reading frame (ORF) was identified that encoded a putative β-glucosidase member of glycosyl hydrolase family 3. The gene, termed bgpA, was amplified using the following primers (with EcoRI and NotI restriction sites in boldface): bgpAF (5′-CGA ATT CAT GAC CAT GAT TAC GCC AAG CTA T-3′) and bgpAR (5′-TTT GCG GCC GCT CAC TCC GTG TCG AGA CGA AGC-3′). The amplified fragment was digested with EcoRI and NotI and then inserted pGEX-4T-1 (GE Healthcare) to generate a glutathione S-transferase (GST)-bgpA gene fusion.
Recombinant pGEX-bgpA was introduced into E. coli C41(DE3), and transformants were grown in LB-ampicillin medium at 37°C until the culture reached an optical density at 600 nm (OD600) of 0.6, at which point protein expression was induced by the addition of 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Bacteria were incubated for an additional 12 h at 20°C and then harvested by centrifugation at 5,000 × g for 20 min at 4°C. The cells were washed twice with a solution of 50 mM sodium phosphate, 5 mM EDTA, and 1% Triton X-100 (pH 7.0) and then resuspended in 50 mM sodium phosphate (pH 7.0). The cells were disrupted by ultrasonication (Vibra-cell; Sonics & Materials, Newtown, CT), and then intact cells and debris were removed by centrifugation at 24,000 × g for 40 min at 4°C to obtain a crude cell extract. The GST-tagged fusion protein was purified by affinity chromatography on a glutathione-Sepharose 4B column (GE Healthcare). The GST epitope was removed by incubation with thrombin, and then recombinant BgpA was purified by DEAE-cellulose DE-52 chromatography (Whatman) followed by Mono Q anion-exchange chromatography (GE Healthcare). The homogeneity of the protein was assessed by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by Coomassie blue staining. The purified protein was dialyzed against 50 mM sodium phosphate, pH 7.0, and then concentrated to 10 mg/ml using an Amicon Ultra-15 filter (Millipore, Temecula, CA). Enzymatic assays and kinetic analysis of the purified protein were performed in 50 mM sodium phosphate, pH 7.0. The molecular mass of the protein was determined by size exclusion chromatography using a Superose 6 10/300 GL column (GE Healthcare) followed by SDS-PAGE, with reference to protein standards obtained from Bio-Rad (catalog no. 151-1901).
Enzyme characterization.
The specific activity of purified BgpA was determined using PNPGlc as a surrogate substrate in 50 mM sodium phosphate buffer, pH 7.0 at 37°C. Reactions were stopped after 5 min by the addition of Na2CO3 at a final concentration of 0.5 M, and the release of p-nitrophenol was measured immediately using a microplate reader at 405 nm (model 680; Bio-Rad, Hercules, CA). One unit of activity was defined as the amount of protein required to generate 1 μmol of p-nitrophenol per min. Specific activity was expressed as units per milligram of protein. Protein concentrations were determined using the bicinchoninic acid (BCA) protein assay (Pierce, Rockford, IL), with bovine serum albumin (Sigma) as the standard. All assays were performed in triplicate.
The effect of pH on enzymatic activity was determined using 2.0 mM PNPGlc as a substrate in the following buffers (each at 50 mM): KCl-HCl (pH 2), glycine-HCl (pH 3), sodium acetate (pH 4 and 5), sodium phosphate (pH 6.0 and 7.0), Tris-HCl (pH 8.0 and 9.0), and glycine-sodium hydroxide (pH 10). In order to determine whether buffer composition had any influence on enzymatic activity, we also measured enzyme activity in McIlvaine buffer (100 mM citric acid, 200 mM disodium phosphate) at various pHs ranging from 6.0 to 8.0 (24). The pH stability of recombinant BgpA was determined by measuring enzymatic activity after incubation in each buffer (containing 2.0 mM PNPGlc in 50 mM potassium buffer as a substrate) for 24 h at 4°C. The results are expressed as a percentage of the activity obtained at the optimum pH. The effect of temperature on enzymatic activity was tested by incubating the enzyme at various temperatures ranging from 4 to 90°C at optimum pH for 5 min in 50 mM potassium phosphate buffer containing 2.0 mM PNPGlc. The thermostability of the enzyme was examined by incubating the enzyme in 50 mM potassium phosphate buffer for 30 min at different temperatures. After cooling the sample on ice for 10 min, activity was determined using PNPGlc as the substrate.
The effects of metals and other chemicals on BgpA activity were also determined. BgpA activity was tested in the presence of 1 or 10 mM (final concentration) HgCl2, MnCl2, CaCl2, CoCl2, MgCl2, EDTA, NaCl, KCl, CuCl2, SDS, ZnSO4, dithiothreitol (DTT), and β-mercaptoethanol for 30 min at 25°C. The remaining activity was determined using PNPGlc as a substrate, and activities are expressed as a percentage of the activity obtained in the absence of compound.
Substrate preference was examined using 2.0 mM chromogenic o-nitrophenyl (ONP) and p-nitrophenyl (PNP) as substrates at 37°C for 5 min, with 1 activity unit being defined as the release of 1 μmol o-nitrophenol or p-nitrophenol per min. The following substrates were tested: PNP-β-d-glucopyranoside, PNP-β-d-galactopyranoside, PNP-β-d-fucopyranoside, PNP-N-acetyl-β-d-glucosaminide, PNP-β-l-arabinopyranoside, PNP-β-d-mannopyranoside, PNP-β-d-xylopyranoside, PNP-α-d-glucopyranoside, PNP-α-l-arabinofuranoside, PNP-α-l-arabinopyranoside, PNP-α-l-rhamnopyranoside, PNP-α-d-mannopyranoside, PNP-α-d-xylopyranoside, ONP-β-d-glucopyranoside, ONP-β-d-galactopyranoside, ONP-β-d-fucopyranoside, and ONP-α-d-galactopyranoside (Sigma).
Kinetic studies were performed with freshly purified enzyme using PNPGlc (0.05 mM to 5 mM) and ginsenoside Rb1 (0.05 mM to 1 mM) as substrates. For the former, the absorbance of p-nitrophenol at 405 nm was monitored for 20 min at 37°C; for the latter, the hydrolyzed products of ginsenoside Rb1 were determined by high-performance liquid chromatography (HPLC) analysis at early stages of hydrolysis. The data obtained were used to determine Km and Vmax using the enzyme kinetics program reported by Cleland (6).
Enzymatic hydrolysis of ginsenosides.
Initial biotransformation experiments using ginsenoside Rb1 as a substrate revealed that the presence of GST fused to BgpA did not affect the activity of the enzyme. Therefore, the GST fusion protein was used to determine the specificity and selectivity of hydrolysis of glucose moieties attached at the C-3 or C-20 positions of Rb1, Rd, and Rg3 by BgpA. Enzyme solution at a concentration of 0.1, 0.5 or 1.0 mg/ml in 50 mM sodium phosphate buffer (pH 7.0) was incubated with an equal volume of Rb1, Rd, and Rg3 solutions at a concentration of 0.1% (wt/vol) in 50 mM sodium phosphate buffer (pH 7.0) at 37°C. Samples were withdrawn at regular intervals. An equal volume of water-saturated n-butanol was added to stop the reaction, and the reactant(s) present in the n-butanol fraction was analyzed following pretreatment (see below).
Preparation of metabolites produced by BgpA.
The reaction mixture was extracted with an equal volume of water-saturated n-butanol, and then the n-butanol fraction was evaporated to dryness. Residual material was dissolved in CH3OH and then examined by TLC and high-performance liquid chromatography (HPLC). For the preparation of the reaction product of BgpA and Rb1, 500 ml of the reaction mixture (1 mg/ml of Rb1 and 0.1 mg/ml of the enzyme) was incubated at 37°C until all Rb1 was converted to metabolite 1, as confirmed by TLC. From this preparation, 0.37 g of metabolite 1 was obtained, 0.3 g of which was subjected to another round of catalysis (using 0.5 mg/ml enzyme) that produced 0.09 g of metabolite 2. This second reaction mixture was extracted twice with water-saturated n-butanol and evaporated in vacuo. To further purify the metabolites for nuclear magnetic resonance (NMR) analysis, 50 mg each of metabolites 1 and 2 was purified using a recycling preparative HPLC system (LC-9201; Japan Analytical Instrument) equipped with a UV/refractive index (RI) detector and a reverse-phase column (octyldecyl silane [ODS]) (500 by 20 mm; inside diameter [i.d.], 15 μm). An isocratic solvent system of CH3CN and deionized H2O (7:3) was used, and the detection wavelength was set at 203 nm. Using this procedure, 31 mg of metabolite 1 and 35 mg of metabolite 2 were obtained and subjected to NMR and mass spectrometry (MS) analysis after extraction.
TLC analysis.
TLC was performed using 60F254 silica gel plates (Merck, Darmstadt, Germany) with CHCl3-CH3OH-H2O (70:30:5 [vol/vol], lower phase) as the solvent. The spots on the TLC plates were detected by spraying with 10% (vol/vol) H2SO4, followed by heating at 110°C for 5 min.
HPLC analysis.
HPLC analysis of ginsenosides was performed using an HPLC system (Waters, Milford, MA) and a C18 (250 by 4.6 mm; i.d., 5 μm) column with acetonitrile (A) and distilled water (B) at A/B ratios of 15:85, 21:79, 58:42, 90:10, 90:10, and 15:85 and run times of 0 to 5, 5 to 25, 25 to 70, 70 to 72, 72 to 82, and 82 to 100 min, respectively. The flow rate was 1.6 ml/min, and detection was performed by monitoring absorbance at 203 nm.
NMR and MS analysis.
NMR spectral data were recorded on a Varian Unity 500 FT-NMR spectrometer at 500 MHz for 1H and 125 MHz for 13C using a Bruker Avance 800 spectrometer equipped with a TXi probe and xyz gradient shielding at 800 MHz for 1H and 200 MHz for 13C. Tetramethylsilane was used as an internal standard. Chemical shifts are presented in ppm. High-resolution electrospray ionization mass spectra (HR-ESIMS) were measured on a Waters Q-Tof Premier mass spectrometer.
Taxonomic characterization of strain Gsoil 3082.
Strain Gsoil 3082 was cultivated on R2A agar at 30°C, unless noted otherwise. Cell morphology was observed under a Nikon light microscope at a ×1,000 magnification using cells grown for 3 days. The Gram reaction was performed using the nonstaining method of Buck (3). Physiological and biochemical characterization assays, including Gram staining, anaerobic growth, and catalase activity, were carried out as described by Smibert and Krieg (35). Enzymatic activities, the assimilation of substrates as the sole carbon source, acid production from the substrates, and other physiological and biochemical characteristics were determined using the API 20E, API 20NE, API 32GN, and API 50 CH galleries (bioMérieux). Growth at different temperatures (4, 15, 20, 25, 30, 37, 42, and 45°C) and various pH levels (pH 4.5 to 10.0 at intervals of 0.5 pH unit) was assessed after a 5-day incubation period. Salt tolerance was tested on R2A medium supplemented with 1 to 15% (wt/vol) NaCl after incubation for 5 days. Growth on nutrient agar and Trypticase soy agar (Difco, Detroit, MI) was also evaluated at 30°C. Isoprenoid quinones were extracted with chloroform-methanol (2:1 [vol/vol]), evaporated under vacuum and then reextracted in n-hexane-water (1:1 [vol/vol]). The crude n-hexane-quinone solution was purified using silica Sep-Pak Vac cartridges (Waters) and subsequently analyzed by HPLC, as previously described by Collins (7). Cellular fatty acid profiles were determined for strain Gsoil 3082T grown on Trypticase soy agar at 30°C for 3 days. Cellular fatty acids were saponified, methylated, and then extracted according to the Sherlock microbial identification system (MIDI) protocol. The fatty acids were then analyzed by gas chromatography (model 6890; Hewlett Packard) using the Microbial Identification software package (33). The presence of diaminopimelic acid (DAP) isomers in the cell wall peptidoglycan was determined using TLC after hydrolysis with 6 N HCl at 100°C for 18 h, as described by Komagata and Suzuki (21). Polar lipids were extracted and examined using two-dimensional TLC (26). The phylogenetic relatedness of strain Gsoil 3082 to other bacteria was determined by analyzing a portion of the 16S rRNA gene. The 16S rRNA gene was first amplified by PCR using the universal primers 27F (5′-AGAGTTTGATCMTGGCTCAG-3′) and 1492R (5′TACGGYTACCTTGTTACGACTT3′) and then sequenced (SolGent, Daejeon, Korea). A near-complete sequence of the 16S rRNA gene was compiled using the SeqMan program in the DNASTAR package. Sequences of the 16S rRNA genes of related taxa were obtained from the GenBank database. Multiple alignments were performed using the CLUSTAL_X program (38). Gaps were edited in the BioEdit program (11), and evolutionary distances were calculated using the Kimura two-parameter model (19). A phylogenetic tree was constructed using the neighbor-joining method (32) and maximum parsimony (10) using the MEGA4 program (36), with bootstrap values based on 1,000 replicates (9). To determine the GC content of Gsoil 3082T chromosomal DNA, genomic DNA was enzymatically degraded into nucleotides, as described by Mesbah et al. (25). The nucleotide mixture was separated by reverse-phase HPLC using a Waters Nova-pak C18 column and 0.2 M (NH4)H2PO4 containing 7.5% acetonitrile as a solvent. E. coli DNA (Sigma) was used as a calibration reference. DNA-DNA hybridization analysis was performed fluorometrically according to the method of Ezaki et al. (8) using photobiotin-labeled DNA probes (Sigma) and microdilution wells (Greiner). Five replicates were performed for each sample. The highest and lowest values obtained for each sample were excluded, and the means of the remaining three values were reported as the DNA-DNA-relatedness value.
Nucleotide sequence accession numbers.
The sequences for the 16S rRNA and bgpA genes from Terrabacter sp. strain Gsoil 3082 have been deposited into GenBank/EMBL/DDBJ under accession no. EU332827 and GU196785, respectively.
RESULTS AND DISCUSSION
Characterization of Gsoil 3082.
After three rounds of screening, 20 bacterial strains were isolated from the soil of a ginseng farm in Pocheon Province, Korea. The biotransformation activity of the isolates was tentatively determined by TLC analysis. One of the isolates, designated Gsoil 3082, was characterized by a polyphasic approach to clarify its taxonomic position and used to clone and characterize a putative ginsenoside-hydrolyzing β-glucosidase.
The Gsoil 3082 isolate was Gram positive, aerobic, nonmotile, non-spore forming, and short rod shaped. Phylogenetic analysis based on 16S rRNA gene sequences indicated that the isolate belongs to the genus Terrabacter in the phylum Actinobacteria, and is most closely related to Terrabacter tumescens DSM 20308T (98.4% similarity), followed by Terrabacter lapilli LR-26T (98.3%), Terrabacter aerolatus 5516J-36T (98.0%), Terrabacter terrigena ON10T (97.6%), and Terrabacter aeriphilus 5414T-18T (97.2%) (Fig. 2). The major polar lipids of strain Gsoil 3082T were diphosphatidylglycerol, phosphatidylglycerol, phosphatidylethanolamine, phosphatidylinositol, and other unidentified phospholipids. The predominant menaquinone was MK-8(H4), and the whole-cell sugars were galactose, ribose, fucose, and rhamnose. The fatty acids (>5% of total fatty acids) contained iso-C15:0 (37.6%), iso-C16:0 (9.3%), C16:0 (8.0%), anteiso-C15:0 (6.9%), and iso-C14:0 (6.7%) (see Table S1 in the supplemental material). The GC content of the genomic DNA was 69.2 mol%. DNA-DNA relatedness values between strains Gsoil 3082T and T. tumescens DSM 20308T, T. aerolatus 5516J-36T, T. lapilli LR-26T, T. aeriphilus 5414T-18T, T. terrigena ON10T, and T. terrae PPLBT were 62, 52, 36, 35, 28, and 26%, respectively.
FIG. 2.

Rooted phylogenetic tree based on 16S rRNA sequences of strain Gsoil 3082T and related Actinobacteria. The phylogenetic tree was constructed using the neighbor-joining method (32) with a Kimura (19) two-parameter distance matrix and pairwise deletion. Bootstrap values (expressed as percentages of 1,000 replications) greater than 60% are shown at the branch points. The bar represents 10 nucleotide substitutions per 1,000 nucleotides.
Taxonomic position of strain Gsoil 3082T.
The physiological and chemotaxonomic characteristics of strain Gsoil 3082T suggested that it is a member of the genus Terrabacter. However, DNA-DNA relatedness tests showed that strain Gsoil 3082T should be classified as a novel species (40). Strain Gsoil 3082T was distinguishable from other recognized Terrabacter species by several phenotypic characteristics, shown in Table 1. The distinctive phylogenetic and genetic characteristics and differential phenotypic properties were highly suggestive of Gsoil 3082T as a novel species of the genus Terrabacter, for which the name Terrabacter ginsenosidimutans sp. nov. is proposed.
TABLE 1.
Biochemical characteristics of strain Gsoil 3082T and related taxa
| Characteristic | Result for straina: |
||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
| Growth at/on: | |||||||
| pH 4.1 | − | − | + | − | + | − | + |
| pH 12.1 | − | + | w | − | − | − | − |
| 10°C | + | + | + | − | + | + | + |
| 40°C | + | − | + | − (+) | − | − | − |
| 5% NaCl | + | + | − | − (+) | + | − | + |
| Nitrate reduction | + | + | + | − | + | − | − |
| Gelatin hydrolysis | w | + | + | − | + | + | + |
| Enzyme activities (API ZYM) | |||||||
| Esterase (C4) | + | − | − | − | + | − | + |
| Lipase (C14) | − | − | − | − (+) | + | − | − |
| Valine arylamidase | − | − | + | + | + | − | + |
| Cystine arylamidase | − | − | + | − | − | + | + |
| Naphthol-AS-BI-phosphohydrolase | + | − | − | − (+) | + | − | + |
| Acid phosphatase | + | + | + | − | + | + | + |
| Whole-cell sugarsb | Gal, Rib, Fuc, Rha | ND | Glc, Rha, Rib, Xyl, Ara | Fuc, Gal | Glc, Rib, Rha, Xyl, Gal | Glc, Man, Ara, Xyl | Glc, Man, Rib |
| Polar lipidsc | DPG, PG, PE, PI, PL | DPG, PE, PI, APGL | DPG, PE, PG, PI, PL | DPG, PE, PI, PGL | DPG, PE, PI, PL | DPG, PG, PE, PI, PL, L | DPG, PE, PI, PG, PGL |
| G+C content (mol%) | 69.2 | 69.2-72.4 | 72.6 | 71.0 | 71.7 | 71.6 | 73 |
Strains: 1, Terrabacter ginsenosidimutans Gsoil 3082T; 2, T. tumescens DSM 20308T; 3, T. lapilli LR-26T; 4, T. terrae PPLBT; 5, T. aerolatus 5516J-36T; 6, T. terrigena ON10T; and 7, T. aeriphilus 5414T-18T. Data in columns 2 to 5 are from Lee et al. (22) and Weon et al. (41), data in column 6 are from Yoon et al. (46), and data in column 7 are from Weon et al. (42). +, positive; −, negative; w, weak reaction; ND, no data available.
Ara, arabinose; Fuc, fucose; Gal, galactose; Glc, glucose; Rha, rhamnose; Rib, ribose; and Xyl, xylose.
DPG, diphosphatidylglycerol; PE, phosphatidylethanolamine; PG, phosphatidylglycerol; PI, phosphatidylinositol; PL, unknown phospholipid(s); PGL, unknown phosphoglycolipid; APGL, unknown amino-containing phosphoglycolipid(s); L, unknown polar lipid.
Description of Terrabacter ginsenosidimutans sp. nov.
Terrabacter ginsenosidimutans (gin.se.no.si.di.mu'tans. N.L. n. ginsenosidum, ginsenoside; L. part. adj. mutans, transforming, converting; N.L. part. adj. ginsenosidimutans, ginsenoside converting).
Bacterial cell dimensions are 0.6 to 1.0 by 1.0 to 1.6 μm, and colonies on R2A agar plates at 30°C were round and white. On R2A agar medium, the strain is able to grow at 10 to 40°C, but not at 4 or 45°C. Growth occurs at pH 5.0 to 10.0 and up to 5% NaCl, but is optimal at pH 7.0 in the absence of NaCl. Acetate, N-acetyl-d-glucosamine, l-alanine, glycogen, gluconate, d-glucose, histidine, 3-hydroxybutyrate, inositol, malate, maltose, mannitol, mannose, d-melibiose, l-proline, propionate, rhamnose, salicin, l-serine, d-sorbitol, d-sucrose, and valerate could all be assimilated by the bacterial strain, and the cells are positive for hydrolysis of esculin and gelatin, as well as for nitrate reduction and β-galactosidase activity. Activity tests for indole production, arginine dihydrolase and urease activities, and glucose fermentation are negative, and the cells could not assimilate adipate, l-arabinose, caprate, citrate, l-fucose, 4-hydroxybenzoate, itaconate, 2-ketogluconate, 3-hydroxybenzoate, and 5-ketogluconate, lactate, malonate, phenylacetate, d-ribose, or suberate. According to the API ZYM gallery, the strain is positive for acid phosphatase, alkaline phosphatase, esterase (C4), esterase lipase (C8), α-galactosidase, β-galactosidase, β-glucuronidase, leucine arylamidase, naphthol-AS-BI-phosphohydrolase, and β-glucosidase and negative for N-acetyl-β-glucosaminidase, α-chymotrypsin, cystine arylamidase, α-fucosidase, α-glucosidase, lipase (C14), α-mannosidase, trypsin, and valine arylamidase. The whole-cell sugars that were detected in strain Gsoil 3082T are galactose, ribose, fucose, and rhamnose, and the major polar lipids are diphosphatidylglycerol, phosphatidylglycerol, phosphatidylethanolamine, phosphatidylinositol, and other unidentified phospholipids. The predominant menaquinone is MK-8(H4). The major fatty acids were iso-C15:0 (37.6%), iso-C16:0 (9.3%), C16:0 (8.0%), anteiso-C15:0 (6.9%), iso-C14:0 (6.7%), and others (less than 5%). The DNA G+C content is 69.2 mol%. The type strain is Gsoil 3082T, which was isolated from the soil of a ginseng farm in Pocheon Province, South Korea. Strain Gsoil 3082T has been deposited with the Korean Collection for Type Cultures (KCTC) under accession no. KCTC 19421T.
Cloning of bgpA.
Genomic DNA from Terrabacter sp. strain Gsoil 3082 was isolated by phenol-chloroform extraction and used to generate a fosmid library. The average insert size of the fosmid library was approximately 40 kb, and the percentage of clones that contained inserts was approximately 93%. Thirty out of 1,500 clones in the fosmid library conferred a blue color to E. coli transformants growing on LB agar plates containing 12.5 μg/ml of chloramphenicol and 27 μg/ml of X-Glc. One of these fosmid clones was positive for ginsenoside Rb1 transformation to unknown smaller ginsenosides, and this clone was fully sequenced.
The insert of the positive clone was a 33.9-kb genomic fragment, and the nucleotide sequence revealed a total of 14 putative ORFs (data not shown). Two of the ORFs were homologous to glycoside hydrolase genes, with identities ranging from 26 to 68%. The first ORF contained a gene that was homologous (66% similarity) to the Kribbella flavida α-arabinofuranosidase of glycohydrolase family 51 (13); the second ORF encoded a putative β-glucosidase gene, termed bgpA, that was highly similar to bacterial β-glucosidases of glycoside hydrolase family 3. Glycosyl hydrolases are classified according to amino acid sequence similarity, as this has been shown to reflect the structural features of the enzymes in addition to their substrate specificities (13) (http://www.cazy.org/fam/acc_GH.html). Family 3 glycoside hydrolases have been subdivided into six subfamilies by Harvey et al. (12). In order to determine the evolutionary position of BgpA within family 3 glycoside hydrolases, phylogenetic analysis was carried out using the neighbor-joining method (32) and maximum parsimony (10) using the MEGA4 program (36) with bootstrap values based on 1,000 replications (9). The resulting consensus tree is presented in Fig. S1 in the supplemental material. The consensus tree topology is largely congruent with the results obtained by Harvey et al. (12). BgpA clustered within subfamily 6 and formed a separate, well-supported clade (bootstrap value of 100) with Erwinia chrysanthemi β-glucosidase/xylosidase, Azospirillum irakense β-glucosidase SalB, unidentified microorganism β-glucosidase, and Bacillus sp. strain GL1 β-glucosidase. To date, only two ginsenoside-hydrolyzing glycoside hydrolases have been successfully cloned: a β-glucosidase from a soil metagenome (16) and a β-galactosidase from archaeon Solfolobus solfataricus (18). However, both of these genes code for family 1 glycoside hydrolases. BgpA is the first family 3 ginsenoside-converting β-glucosidase to be successfully cloned.
Expression and purification of recombinant BgpA.
The putative β-glucosidase gene, bgpA, was amplified by PCR and then subcloned into pGEX4T-1 to generate a GST-bgpA gene fusion that could be expressed in E. coli C41(DE3) under the control of the IPTG (isopropyl-β-d-thiogalactopyranoside)-inducible promoter Ptac.
To maximize the yield of the fusion protein, we tested different induction conditions. Induction with 0.5 mM IPTG at 20°C for 12 h produced the maximum level of soluble active fusion enzyme (data not shown). GST-BgpA fusion protein purified by glutathione-Sepharose 4B chromatography was digested by thrombin to remove the GST moiety, and the resulting recombinant BgpA was purified by successive chromatography on DEAE-cellulose and Mono Q anion-exchange columns. This procedure resulted in 20.8-fold purification and a recovery of 13% from crude extract (Table 2). The molecular masses of the native β-glucosidase were 193,000 Da, as determined by size exclusion chromatography, and 70,000 Da, as determined by SDS-PAGE (Fig. 3). The results of SDS-PAGE agreed with the size estimate based on the translated polypeptide sequence (71,140 Da), which suggested that the β-glucosidase was physiologically active as a trimeric protein.
TABLE 2.
Purification scheme
| Purification step | Vol (ml) | Total protein (mg) | Sp act (U/mg) | Total activity (U)a | Yield (%) | Purification (fold) |
|---|---|---|---|---|---|---|
| Crude extract | 5 | 191.1 | 3.6 | 696 | 100 | 1.0 |
| Glutathione-Sepharose | 12 | 5.5 | 63.5 | 351 | 50 | 17.4 |
| DEAE-cellulose | 27 | 2.7 | 67.3 | 182 | 26 | 18.5 |
| Mono Q | 5 | 1.2 | 75.9 | 87 | 13 | 20.8 |
One unit of BgpA was defined as the amount of enzyme liberating 1 μmol/min of p-nitrophenol.
FIG. 3.

SDS-PAGE analysis of recombinant BgpA. Lanes: M, molecular mass standard; 1, crude extract of C41(DE3) carrying pGEX-bgpA; 2, GST-BgpA enzyme fraction after glutathione-Sepharose chromatography; 3, enzyme fraction after DEAE anion-exchange chromatography and removal of the GST epitope by thrombin; 4, recombinant BgpA after Mono Q anion-exchange chromatography.
Enzyme characterization.
BgpA was active over a broad pH range (pH 5.0 to 10.0). The optimum pH was pH 7.0 in both sodium phosphate and McIlvaine buffers (Fig. 4 A). The enzyme retained over 82% of its optimal activity between pH values 6.0 and 8.0; it exhibited residual activity at pHs 5.0, 9.0, and 10.0, and it showed no activity at pH 4.0. The optimal temperature for BgpA activity was 45°C. The enzyme was stable at temperatures lower than 37°C, and about 32% of the activity was lost after incubation at 45°C for 30 min (Fig. 4).
FIG. 4.

Effect of pH (A) and temperature (B) on the stability and activity of recombinant BgpA. For pH, enzyme solutions containing 2.0 mM PNPGlc were incubated in buffers of various pHs ranging from pH 2 to 10 for 24 h at 4°C, after which residual activity was determined. The following buffers (50 mM each) were tested: KCl-HCl (pH 2), glycine-HCl (pH 3), sodium acetate (pHs 4 and 5), sodium phosphate (pHs 6 and 7), Tris-HCl (pHs 8 and 9), and glycine-sodium hydroxide (pH 10). The influence of buffer composition on the pH effect was also determined using McIlvaine buffer (▴) at pH 6, 7, or 8. The thermo-dependence of BgpA activity was assayed in 50 mM potassium phosphate buffer (pH 7.0) at various temperatures ranging from 4 to 90°C. Thermostability was tested by incubating aliquots of the enzyme in 50 mM potassium phosphate buffer (pH 7.0) for 30 min at different temperatures. After cooling the sample on ice for 10 min, residual activity was determined.
The effects of metal ions, EDTA, β-mercaptoethanol, and SDS on BgpA activity were also investigated (Table 3). The enzyme did not require Mg2+, Mn2+, Co2+, Zn2+, Ca2+, or Cu2+ for activity and was significantly inhibited by Hg2+. BgpA activity was not affected by DTT or β-mercaptoethanol, which are well-known thiol group inhibitors, suggesting that sulfhydryl groups may not be involved in the catalytic center of the enzyme, but rather may be essential for maintenance of the three-dimensional structure of the active protein. The chelating agent EDTA did not inhibit BgpA activity, indicating that divalent cations are not required for enzymatic activity. Enzymatic activity was greatly inhibited in the presence of 10 mM SDS.
TABLE 3.
Effects of metal ions and chemical agents on the activity of purified recombinant BgpA
| Metal ion or reagent | Relative activity ± SD (%) ata: |
|
|---|---|---|
| 1 mM | 10 mM | |
| NaCl | 102.3 ± 5.15 | 97.7 ± 4.51 |
| KCl | 102.7 ± 4.65 | 96.2 ± 4.64 |
| MgCl2 | 94.8 ± 5.10 | 95.2 ± 5.14 |
| MnCl2 | 101.1 ± 5.45 | 96.9 ± 4.79 |
| CoCl2 | 100.9 ± 8.43 | 97.0 ± 5.04 |
| ZnCl2 | 92.8 ± 4.72 | 72.8 ± 4.27 |
| CaCl2 | 91.8 ± 4.59 | 101.5 ± 5.23 |
| CuCl2 | 84.7 ± 4.48 | 71.7 ± 5.07 |
| HgCl2 | 0 | 0 |
| SDS | 94.8 ± 6.98 | 2.7 ± 3.22 |
| EDTA | 98.6 ± 6.49 | 96.6 ± 4.52 |
| β-Mercaptoethanol | 98.4 ± 4.89 | 102.5 ± 4.57 |
| DTT | 96.7 ± 5.59 | 96.6 ± 5.14 |
| Control | 100.0 ± 5.17 | 100.0 ± 5.11 |
The specific activity at 100% was 44.3 U/mg protein.
The substrate specificity of BgpA was tested using 2.0 mM PNP- and ONP-glycosides with α and β configurations. The results, summarized in Table 4, showed that BgpA was maximally active against ONPGlc, followed by PNPGlc, and was almost inert toward various PNP- and ONP-glycosides, with the exception of PNP- and ONP-β-d-fucopyranoside. The Km and Vmax for the hydrolysis of PNPGlc by β-glucosidase were 4.24 mM and 0.10 mmol·min−1·mg of protein−1, respectively, and for ginsenoside Rb1, they were 0.14 ± 0.05 mM and 329 ± 31 μmol·min−1·mg of protein−1, respectively.
TABLE 4.
Relative activity of purified recombinant BgpA toward various chromogenic substrates as measured by ONP or PNP release at 37°C
| Substrate | Relative activity ± SD (%)a |
|---|---|
| PNP-α-d-glucopyranoside | 1.12 ± 0.21 |
| PNP-α-d-mannopyranoside | 0 |
| PNP-α-d-xylopyranoside | 0 |
| PNP-α-l-arabinofuranoside | 0 |
| PNP-α-l-arabinopyranoside | 0 |
| PNP-α-l-rhamnopyranoside | 0 |
| PNP-β-d-fucopyranoside | 6.98 ± 0.06 |
| PNP-β-d-galactopyranoside | 0 |
| PNP-β-d-glucopyranoside | 89.52 ± 4.43 |
| PNP-β-d-glucosaminide | 0 |
| PNP-β-d-mannopyranoside | 0 |
| PNP-β-d-xylopyranoside | 0 |
| PNP-β-l-arabinopyranoside | 0 |
| ONP-α-d-galactopyranoside | 0 |
| ONP-β-d-fucopyranoside | 15.88 ± 0.54 |
| ONP-β-d-galactopyranoside | 2.33 ± 0.08 |
| ONP-β-d-glucopyranoside | 100.00 ± 3.54 |
Activity toward ONP-β-d-glucopyranoside was set as 100% and corresponded to a specific activity of 44.3 U/mg.
Biotransformation of ginsenoside Rb1 and structural identification of reaction products.
The products of BgpA hydrolysis of ginsenoside Rb1 were determined at regular intervals by TLC and HPLC analysis. As shown in Fig. 5 (and see Fig. S2 in the supplemental material), the enzyme catalyzed the production of three different metabolites from ginsenoside Rb1. Based on a comparison of Rf values (determined by TLC) and metabolite retention times (determined by HPLC) with those of standard ginsenosides, metabolite 3 was identified as C-K, while metabolites 1 and 2 appeared to be unknown compounds, as their Rf values and retention times were different from the standard ginsenosides (Fig. 5; see Fig. S2 in the supplemental material). Therefore, metabolites 1 and 2 were examined by NMR and MS. Metabolite 1 was obtained as a white powder and displayed a pseudomolecular ion peak [M+H]+ at m/z 947.5581 in HR-ESIMS, corresponding to the elemental formula C48H83O18 (calculated molecular weight, 947.5579). The 1H- and 13C-NMR spectroscopic data of metabolite 1 (see the supplemental material) closely resembled Gyp XVII (4). Heteronuclear multiple bond correlations (HMBCs) of δH 5.14 (1H, d, J = 7.2 Hz, H-1") to δC 83.9 (C-20) and δH 4.95 (1H, d, J = 7.2 Hz, H-1′) to δC 89.3 (C-3) enabled the assignment of a glucose residue to both C-20 and C-3. The remaining glucose was affixed to C-6", as determined by the long-range correlation between δH 5.11 (1H, d, J = 7.2 Hz, H-1‴) and δC 70.7 (C-6") in the HMBC spectrum. Thus, metabolite 1 was confirmed as Gyp XVII (4).
FIG. 5.

Time course of ginsenoside Rb1 bioconversion by BgpA at enzyme concentrations of 0.1 mg/ml (A), 0.5 mgl/ml (B), and 1.0 mg/ml (C). Metabolites were analyzed by TLC. Lanes: Rb1 to C-K, standards; 5 min to 24 h, reaction times.
The molecular formula of metabolite 2 was determined to be C42H72O13 based on the protonated molecular ion peak at m/z 785.5089 (calculated molecular weight for C42H73O13, 785.5051) in HR-ESIMS. The 1H- and 13C-NMR spectroscopic data of metabolite 2 (see the supplemental material) were similar to those of metabolite 1, but lacked the proton and carbon signals of 1 glucose unit. Only two anomeric proton signals were observed at δH 5.09 (1H, d, J = 7.8 Hz, H-1") and 5.06 (1H, d, J = 7.6 Hz, H-1′), and the corresponding carbon signals were detected at δC 98.3 (C-1") and 105.5 (C-1′) in the 1H- and 13C-NMR spectra, respectively, implying the presence of two glucose residues in metabolite 2. In the 13C-NMR spectrum, the chemical shift of C-3 (δC 78.5) of metabolite 2 appeared more pronounced than that of metabolite 1 (δC 89.3), suggesting that there was a free hydroxyl group at C-3 in metabolite 2. These data were consistent with values reported in the literature (47). Accordingly, the structure of metabolite 2 was established as Gyp LXXV.
In reaction mixtures containing Rb1 at 1 mg/ml and BgpA at 0.1, 0.5, or 1.0 mg/ml, Rb1 was completely hydrolyzed within 5 min. Only one product, metabolite 1 (Gyp XVII), was detected at enzyme concentrations of 0.1 and 0.5 mg/ml. At longer incubation times, the levels of Gyp XVII gradually decreased. At enzyme concentrations of 0.1 and 0.5 mg/ml, Gyp XVII was completely absent after 2 h; at an enzyme concentration of 1.0 mg/ml, the metabolite was undetectable after 24 h. Metabolite 2 (Gyp LXVV) started to appear at 2 h at an enzyme concentration of 0.1 mg/ml and reached a maximum level at 12 h, after which it gradually decreased. At enzyme concentrations of 0.1 and 0.5 mg/ml, Gyp LXXV started to appear at 10 min and reached a maximum level at 2 h, before gradually decreasing to an almost undetectable level at 24 h. Metabolite 3 (C-K) continuously increased throughout the incubation time at all enzyme concentrations. These data indicated that Gyp XVII and Gyp LXXV are intermediate metabolites and that C-K is the final product. The observation that the highest concentration of Gyp LXXV occurred after the peak of Gyp XVII production confirmed that Rb1 was first converted into Gyp XVII, then into Gyp LXXV, and finally into C-K. Therefore, the putative conversion pathway of Rb1 by BgpA was Rb1 → Gyp XVII → Gyp LXXV → C-K by stepwise hydrolysis of the outer and inner glucose of C-3, followed by hydrolysis of the outer glucose of C-20 of Rb1.
Substrate specificity of BgpA.
Theoretically, four types of glucose moieties attached to ginsenoside Rb1 could be available for hydrolysis by BgpA, namely, the outer and inner glucose moieties attached at positions C-3 and C-20. Based on an analysis of the hydrolysis products of ginsenoside Rb1, BgpA hydrolyzed three of the four possible types of glucose moieties: the outer and inner glucose moieties at position C-3 and the outer glucose at position C-20. The preferential reactivity of BgpA toward these three glucose moieties of Rb1 could be deduced from the Rb1 biotransformation pathway: i.e., Rb1 → Gyp XVII → Gyp LXXV → C-K. BgpA preferred the outer glucose at position C-3, followed by the inner glucose at position C-3, and finally the outer glucose at position C-20. By comparison, family 1 ginsenoside-hydrolyzing glycosidases preferentially hydrolyze the outer glucose at position C-20 (16, 28).
To investigate whether BgpA showed the same specificity and selectivity for glycone residues attached at the C-3 and C-20 positions of other PPD-type ginsenosides, ginsenosides Rd and Rg3 were also used as substrates. Ginsenoside Rd has one glucose residue at the C-20 position and two glucose residues at the C-3 position, respectively, while Rg3 has only two glucose residues at the C-3 position (Fig. 1). Ginsenoside Rd was converted to F2 by cleavage of the terminal glucose at the C-3 position; F2 was further converted to C-K by cleavage of the inner glucose at the C-3 position of F2. When ginsenoside Rg3 was used as a substrate, BgpA hydrolyzed the terminal glucose at the C-3 position of Rg3 to form ginsenoside Rh2, which was subsequently converted to PPD by cleavage of the inner glucose at the C-3 position of Rh2 (Fig. 6). These results indicated that BgpA shows substrate specificity for PPD-type ginsenosides that have one or two glucose moieties at the C-3 or C-20 position and prefers the outer glucose at position C-3, followed by the inner glucose at position C-3, and finally the outer glucose at position C-20.
FIG. 6.
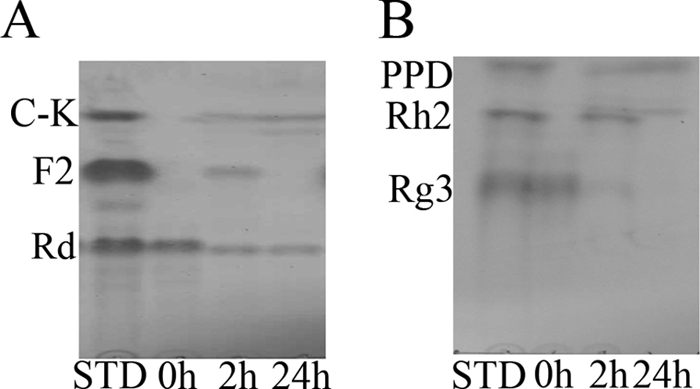
Time course of ginsenoside Rd (A) and Rg3 (B) bioconversion by BgpA at an enzyme concentration of 0.1 mg/ml and ginsenoside concentration of 0.5 mg/ml. Metabolites were analyzed by TLC. Lanes: STD, standard; 0 to 24 h, reaction times.
In summary, we have cloned and characterized a family 3 β-glucosidase, termed BgpA, from a novel bacterium isolated from the soil of a ginseng farm in Korea. The enzyme was able to convert ginsenoside Rb1 into rare ginsenosides through selective hydrolysis of glucose moieties, yielding Gyp XVII, Gyp LXXV, and C-K. This is the first report of the cloning of an enzyme that produces Gyp XVII and Gyp LXXV through the biotransformation of ginsenosides.
Supplementary Material
Acknowledgments
This work was supported by KRIBB research initiatives and the 21C Frontier Microbial Genomics and Applications Center Program, Ministry of Education, Science & Technology (grant MG08-0101-2-0), Republic of Korea.
We are grateful to Eun-Hee Kim, Korea Basic Science Institute, for the NMR spectral data.
Footnotes
Published ahead of print on 9 July 2010.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Akao, T., H. Kida, M. Kanaoka, M. Hattori, and K. Kobashi. 1998. Intestinal bacterial hydrolysis is required for the appearance of compound K in rat plasma after oral administration of ginsenoside Rb1 from Panax ginseng. J. Pharm. Pharmacol. 50:1155-1160. [DOI] [PubMed] [Google Scholar]
- 2.Bae, E. A., M. J. Han, E. J. Kim, and D. H. Kim. 2004. Transformation of ginseng saponins to ginsenoside Rh2 by acids and human intestinal bacteria and biological activities of their transformants. Arch. Pharm. Res. 27:61-67. [DOI] [PubMed] [Google Scholar]
- 3.Buck, J. D. 1982. Nonstaining (KOH) method for determination of Gram reactions of marine bacteria. Appl. Environ. Microbiol. 44:992-993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng, L. Q., J. R. Na, M. K. Kim, M. H. Bang, and D. C. Yang. 2007. Microbial conversion of ginsenoside Rb1 to minor ginsenoside F2 and gypenoside XVII by Intrasporangium sp. GS603 isolated from soil. J. Microbiol. Biotechnol. 17:1937-1943. [PubMed] [Google Scholar]
- 5.Christensen, L. P. 2009. Ginsenosides: chemistry, biosynthesis, analysis, and potential health effects. Adv. Food Nutr. Res. 55:1-99. [DOI] [PubMed] [Google Scholar]
- 6.Cleland, W. W. 1979. Statistical analysis of enzyme kinetic data. Methods Enzymol. 63:103-138. [DOI] [PubMed] [Google Scholar]
- 7.Collins, M. D. 1985. Analysis of isoprenoid quinones. Methods Microbiol. 18:329-366. [Google Scholar]
- 8.Ezaki, T., Y. Hashimoto, and E. Yabuuchi. 1989. Fluorometric deoxyribonucleic acid-deoxyribonucleic acid hybridization in microdilution wells as an alternative to membrane filter hybridization in which radioisotopes are used to determine genetic relatedness among bacterial strains. Int. J. Syst. Bacteriol. 39:224-229. [Google Scholar]
- 9.Felsenstein, J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783-791. [DOI] [PubMed] [Google Scholar]
- 10.Fitch, W. M. 1971. Toward defining the course of evolution: minimum change for a specific tree topology. Syst. Zool. 20:406-416. [Google Scholar]
- 11.Hall, T. A. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41:95-98. [Google Scholar]
- 12.Harvey, A. J., M. Hrmova, R. De Gori, J. N. Varghese, and G. B. Fincher. 2000. Comparative modeling of the three-dimensional structures of family 3 glycoside hydrolases. Proteins 41:257-269. [DOI] [PubMed] [Google Scholar]
- 13.Henrissat, B., and G. Davies. 1997. Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 7:637-644. [DOI] [PubMed] [Google Scholar]
- 14.Kim, B. H., S. Y. Lee, H. J. Cho, S. N. You, Y. J. Kim, Y. M. Park, J. K. Lee, M. Y. Baik, C. S. Park, and S. C. Ahn. 2006. Biotransformation of Korean Panax ginseng by Pectinex. Biol. Pharm. Bull. 29:2472-2478. [DOI] [PubMed] [Google Scholar]
- 15.Kim, S., S. Y. Nah, and H. Rhim. 2008. Neuroprotective effects of ginseng saponins against L-type Ca2+ channel-mediated cell death in rat cortical neurons. Biochem. Biophys. Res. Commun. 365:399-405. [DOI] [PubMed] [Google Scholar]
- 16.Kim, S. J., C. M. Lee, M. Y. Kim, Y. S. Yeo, S. H. Yoon, H. C. Kang, and B. S. Koo. 2007. Screening and characterization of an enzyme with β-glucosidase activity from environmental DNA. J. Microbiol. Biotechnol. 17:905-912. [PubMed] [Google Scholar]
- 17.Kim, W. Y., J. M. Kim, S. B. Han, S. K. Lee, N. D. Kim, M. K. Park, C. K. Kim, and J. H. Park. 2000. Steaming of ginseng at high temperature enhances biological activity. J. Nat. Prod. 63:1702-1704. [DOI] [PubMed] [Google Scholar]
- 18.Kim, Y. S., C. S. Park, and D. K. Oh. 2006. Lactulose production from lactose and fructose by a thermostable β-galactosidase from Sulfolobus solfataricus. Enzyme Microb. Technol. 39:903-908. [Google Scholar]
- 19.Kimura, M. 1983. The neutral theory of molecular evolution. Cambridge University Press, Cambridge, United Kingdom.
- 20.Ko, S. R., Y. Suzuki, K. J. Choi, and Y. H. Kim. 2000. Enzymatic preparation of genuine prosapogenin, 20(S)-ginsenoside Rh1, from ginsenosides Re and Rg1. Biosci. Biotechnol. Biochem. 64:2739-2743. [DOI] [PubMed] [Google Scholar]
- 21.Komagata, K., and K. Suzuki. 1987. Lipid and cell-wall analysis in bacterial systematics. Methods Microbiol. 19:161-207. [Google Scholar]
- 22.Lee, J.-E., J. P. Seo, D. W. Lee, Y. H. Ko, and S. D. Lee. 2008. Terrabacter lapilli sp. nov., a novel actinomycete isolated from stone. Int. J. Syst. Evol. Microbiol. 58:1084-1088. [DOI] [PubMed] [Google Scholar]
- 23.Lee, S. J., J. H. Sung, C. K. Moon, and B. H. Lee. 1999. Antitumor activity of a novel ginseng saponin metabolite in human pulmonary adenocarcinoma cells resistant to cisplatin. Cancer Lett. 144:39-43. [DOI] [PubMed] [Google Scholar]
- 24.Margolles, A., and C. G. de Los Reyes-Gavilán. 2003. Purification and functional characterization of a novel α-l-arabinofuranosidase from Bifidobacterium longum B667. Appl. Environ. Microbiol. 69:5096-5103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mesbah, M., U. Premachandran, and W. B. Whitman. 1989. Precise measurement of the G+C content of deoxyribonucleic acid by high-performance liquid chromatography. Int. J. Syst. Bacteriol. 39:159-167. [Google Scholar]
- 26.Minnikin, D. E., A. G. O'Donnell, M. Goodfellow, G. Alderson, M. Athalye, K. Schaal, and J. H. Parlett. 1984. An integrated procedure for the extraction of isoprenoid quinones and polar lipids. J. Microbiol. Methods 2:233-241. [Google Scholar]
- 27.Nagai, M., T. Ando, N. Tanaka, O. Tanaka, and S. Shibata. 1972. Chemical studies on the oriental plant drugs. XXVIII. Saponins and sapogenins of ginseng: stereochemistry of the sapogenin of ginsenoside Rb1, Rb2 and Rc. Chem. Pharm. Bull. 20:1212-1216. [Google Scholar]
- 28.Noh, K. H., J. W. Son, H. J. Kim, and D. K. Oh. 2009. Ginsenoside compound K production from ginseng root extract by a thermostable beta-glycosidase from Sulfolobus solfataricus. Biosci. Biotechnol. Biochem. 73:316-321. [DOI] [PubMed] [Google Scholar]
- 29.Park, J. D., D. K. Rhee, and Y. H. Lee. 2005. Biological activities and chemistry of saponins from Panax ginseng C. A. Meyer. Phytochem. Rev. 4:159-175. [Google Scholar]
- 30.Park, J. H. 2004. Sun ginseng—a new processed ginseng with fortified activity. Food Ind. Nutr. 9:23-27. [Google Scholar]
- 31.Rhule, A., B. Rase, J. R. Smith, and D. M. Shepherd. 2008. Toll-like receptor ligand-induced activation of murine DC2.4 cells is attenuated by Panax notoginseng. J. Ethnopharmacol. 116:179-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saitou, N., and M. Nei. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4:406-425. [DOI] [PubMed] [Google Scholar]
- 33.Sasser, M. 1990. Identification of bacteria by gas chromatography of cellular fatty acids. Technical note 101. Microbial ID, Newark, DE.
- 34.Shin, H. Y., S. Y. Park, J. H. Sung, and D. H. Kim. 2003. Purification and characterization of α-l-arabinopyranosidase and α-l-arabinofuranosidase from Bifidobacterium breve K-110, a human intestinal anaerobic bacterium metabolizing ginsenoside Rb2 and Rc. Appl. Environ. Microbiol. 69:7116-7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smibert, R. M., and N. R. Krieg. 1994. Phenotypic characterization, p. 607-654. In P. Gerhardt, R. G. E. Murray, W. A. Wood, and N. R. Krieg (ed.), Methods for general and molecular microbiology. ASM Press, Washington, DC.
- 36.Tamura, K., J. Dudley, M. Nei, and S. Kumar. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596-1599. [DOI] [PubMed] [Google Scholar]
- 37.Tawab, M. A., U. Bahr, M. Karas, M. Wurglics, and M. Schubert-Zsilavecz. 2003. Degradation of ginsenosides in humans after oral administration. Drug Metab. Dispos. 31:1065-1071. [DOI] [PubMed] [Google Scholar]
- 38.Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G. Higgins. 1997. The CLUSTAL X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang, W., E. R. Rayburn, M. Hao, Y. Zhao, D. L. Hill, R. Zhang, and H. Wang. 2008. Experimental therapy of prostate cancer with novel natural product anti-cancer ginsenosides. Prostate 68:809-819. [DOI] [PubMed] [Google Scholar]
- 40.Wayne, L. G., D. J. Brenner, and R. R. Colwell. 1987. Report of the ad hoc committee on reconciliation of approaches to bacterial systematics. Int. J. Syst. Bacteriol. 37:463-464. [Google Scholar]
- 41.Weon, H.-Y., P. Schumann, R. M. Kroppenstedt, B.-Y. Kim, J. Song, S.-W. Kwon, S.-J. Go, and E. Stackebrandt. 2007. Terrabacter aerolatus sp. nov., isolated from an air sample. Int. J. Syst. Evol. Microbiol. 57:2106-2109. [DOI] [PubMed] [Google Scholar]
- 42.Weon, H. Y., J. A. Son, S. H. Yoo, B. Y. Kim, S. W. Kwon, P. Schumann, R. Kroppenstedt, and E. Stackebrandt. 2010. Terrabacter aeriphilus sp. nov., isolated from an air sample. Int. J. Syst. Evol. Microbiol. 60:1130-1134. [DOI] [PubMed] [Google Scholar]
- 43.Wu, J. Y., B. H. Gardner, C. I. Murphy, J. R. Seals, C. R. Kensil, and J. Recchia. 1992. Saponin adjuvant enhancement of antigen-specific immune responses to an experimental HIV-1 vaccine. J. Immunol. 148:1519-1525. [PubMed] [Google Scholar]
- 44.Xu, Q. F., X. L. Fang, and D. F. Che. 2003. Pharmacokinetics and bioavailability of ginsenoside Rb1 and Rg1 from Panax notoginseng in rats. J. Ethnopharmacol. 84:187-192. [DOI] [PubMed] [Google Scholar]
- 45.Yan, Q., X. W. Zhou, W. Zhou, X. W. Li, M. Q. Feng, and P. Zhou. 2008. Purification and properties of a novel beta-glucosidase, hydrolyzing ginsenoside Rb1 to CK, from Paecilomyces Bainier. J. Microbiol. Biotechnol. 18:1081-1089. [PubMed] [Google Scholar]
- 46.Yoon, J. H., S. Park, S. J. Kang, Y. T. Jung, and W. Kim. 2009. Terrabacter terrigena sp. nov., isolated from soil. Int. J. Syst. Evol. Microbiol. 59:2798-2802. [DOI] [PubMed] [Google Scholar]
- 47.Yoshikawa, K., M. Arimitsu, K. Kishi, T. Takemoto, and S. Arihara. 1987. Studies on the constituents of Cucurbitaceae plants. XVIII. On the saponin constituents of Gynostemma pentaphyllum Makino. Yakugaku Zasshi 107:361-366. (In Japanese.) [Google Scholar]
- 48.Yun, T.-K., Y.-S. Lee, Y. H. Lee, S. I. Kim, and H. Y. Yun. 2001. Cancer chemopreventive compounds of red ginseng produced from Panax ginseng C. A. Meyer. J. Ginseng Res. 25:107-111. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.