

Abstract
Equol is a metabolite produced from daidzein by enteric microflora, and it has attracted a great deal of attention because of its protective or ameliorative ability against several sex hormone-dependent diseases (e.g., menopausal disorder and lower bone density), which is more potent than that of other isoflavonoids. We purified a novel NADP(H)-dependent daidzein reductase (L-DZNR) from Lactococcus strain 20-92 (Lactococcus 20-92; S. Uchiyama, T. Ueno, and T. Suzuki, international patent WO2005/000042) that is involved in the metabolism of soy isoflavones and equol production and converts daidzein to dihydrodaidzein. Partial amino acid sequences were determined from purified L-DZNR, and the gene encoding L-DZNR was cloned. The nucleotide sequence of this gene consists of an open reading frame of 1,935 nucleotides, and the deduced amino acid sequence consists of 644 amino acids. L-DZNR contains two cofactor binding motifs and an 4Fe-4S cluster. It was further suggested that L-DZNR was an NAD(H)/NADP(H):flavin oxidoreductase belonging to the old yellow enzyme (OYE) family. Recombinant histidine-tagged L-DZNR was expressed in Escherichia coli. The recombinant protein converted daidzein to (S)-dihydrodaidzein with enantioselectivity. This is the first report of the isolation of an enzyme related to daidzein metabolism and equol production in enteric bacteria.
Isoflavones are flavonoids present in various plants and are known to be abundant in soybeans and legumes. These compounds have been called phytoestrogens because their chemical structure is similar to that of the female sex hormone, estrogen. Isoflavones have an ability to bind to estrogen receptors and show protection against or improvement in several sex hormone-dependent diseases, such as breast cancer, prostate cancer, menopausal disorder, lower bone density, and hypertension, due to their weak agonistic or antagonistic effects (1, 19, 27).
Daidzein is one of the main soy isoflavonoids produced from daidzin by the glucosidase of intestinal bacteria (17). Equol is a metabolite produced from daidzein by the enterobacterial microflora (5). Recently, equol has attracted a great deal of attention because its estrogenic activity is more potent than that of other isoflavonoids, including daidzein (27). It is well known that individual variation exists in the ability of these enteric microflora to produce equol and that less than half the human population is capable of producing equol after ingesting soy isoflavones (3). Therefore, to increase the production of equol in the enteric environment of each individual, the development of probiotics using safe bacteria which have the ability to produce equol from daidzein is ongoing.
Lactococcus strain 20-92 (Lactococcus 20-92; 30a) is an equol-producing lactic acid bacterium isolated from the feces of healthy humans by Uchiyama et al. (30). This bacterium is spherical and Gram positive and is a strain of L. garvieae. The application of Lactococcus 20-92 in probiotics is advantageous because L. garvieae is not pathogenic or toxic to humans.
To date, other bacterial strains that are capable of transforming daidzein to dihydrodaidzein or equol have been isolated (9, 21, 22, 23, 29, 32, 36, 37). Daidzein is thought to be metabolized by human intestinal bacteria to equol or to O-desmethylangolensin via dihydrodaidzein and tetrahydrodaidzein (14, 15, 22, 32); however, neither the enzymes involved in the metabolism of daidzein to equol nor even the metabolic pathway has been clarified fully for equol-producing bacteria.
In this study, we purified an enzyme from Lactococcus 20-92 that assisted in the conversion of daidzein to dihydrodaidzein. Furthermore, we cloned the L-DZNR gene and expressed the active recombinant enzyme in E. coli.
MATERIALS AND METHODS
Bacteria and culture conditions.
Lactococcus 20-92 was used for enzyme purification and isolation of genomic DNA. It was cultured statically and anaerobically in GAM broth (Nissui Pharmaceutical, Tokyo, Japan) containing 10 μg/ml of daidzein (Fujicco, Kobe, Japan) at 37°C for 16 to 18 h. An anaerobic atmosphere was generated with the use of a BBL Gaspak plus system (Becton Dickinson and Company, Baltimore, MD). Escherichia coli JM109 (Takara Bio, Inc., Otsu, Japan) was used as a host for standard cloning experiments, and the resulting transformants were grown in Luria-Bertani (LB) broth containing ampicillin (50 μg/ml) at 37°C. E. coli BL21(DE3) (Novagen, Inc., Madison, WI) was used as a host for expression of the recombinant protein and was grown in LB broth supplemented with ampicillin (50 μg/ml) at 30°C.
Quantitation of daidzein metabolites by HPLC.
Quantitation of daidzein metabolites by high-performance liquid chromatography (HPLC) was performed according to the method of Lundh et al. (13) with slight modifications and using daidzein, dihydrodaidzein (Toronto Research Chemicals, Toronto, Canada), and equol (LC Laboratories, Woburn, MA) as standards. For quantitation of daidzein metabolites, samples (1 ml) were extracted with 3 ml of ethyl acetate. The organic solvent was vacuum dried at 40°C, and the dried extract was dissolved in 0.5 ml of a 1:1 mixture of solvent A (1.8% [vol/vol] ethyl acetate, 18% [vol/vol] methanol in 0.04% [vol/vol] phosphoric acid, 8 μg/ml EDTA-Na) and solvent B (2% [vol/vol] ethyl acetate in methanol). A 50-μl aliquot of the dissolved extract was then analyzed with an HPLC system (Shimadzu, Kyoto, Japan) equipped with a reversed-phase Capcell-pack UG120 column (5-μm particle size, 250-mm by 4.6-mm internal diameter; Shiseido, Tokyo, Japan). Detection of the daidzein metabolites was performed by increasing the concentration of solvent B in solvent A under the following conditions: a linear gradient for 15 min with 20 to 35% solvent B, an isocratic elution for 5 min with 35% solvent B, and a linear gradient for 5 min with 35 to 70% solvent B at a flow rate of 1 ml/min at 40°C, with monitoring at 254 and 280 nm.
Enzyme assay for L-DZNR.
An enzyme source was added to the ice-cooled substrate solution and incubated at 37°C for 2 h. The final reaction mixture (1 ml) contained 10 μg/ml of daidzein as a substrate, 2 mM NADPH (Wako Pure Chemicals, Osaka, Japan), and 2 mM NADH (Wako Pure Chemicals) in buffer A, which was composed of 0.1 M potassium phosphate buffer (KPB), pH 7.0, containing 2 mM dithiothreitol (DTT; Wako Pure Chemicals), 5 mM sodium hydrosulfite (Wako Pure Chemicals), and 1 mM phenylmethylsulfonyl fluoride (PMSF; Sigma-Aldrich Corp., St. Louis, MO). Following the 2-h incubation, extraction with ethyl acetate was carried out and daidzein metabolites were analyzed by HPLC as described above.
Preparation of supernatant and precipitate from cell lysates.
Lactococcus 20-92 cells were centrifuged at 5,000 × g for 20 min. The cell pellets were suspended in 0.1 M KPB containing 2 mM DTT, 5 mM sodium hydrosulfite, and 1 mM PMSF, with a 200-ml pellet resuspended in 1 ml of buffer. The suspension was dispensed onto 0.7 ml of 0.1-mm zirconia/silica beads (BioSpec Products, Inc., Bartlesville, OK) in 2-ml tubes, and cells were disrupted four times using a FastPrep-24 instrument (MP Biomedical, Inc., Solon, OH) at 6,500 rpm for 20 s. Between each disruption cycle, the samples were cooled on ice. After centrifugation at 15,000 × g for 10 min, the supernatant and precipitate were separated.
Purification of L-DZNR from Lactococcus 20-92.
The Lactococcus 20-92 cells were harvested, and the supernatant from the cell lysate was prepared as described above. The supernatant was loaded onto a butyl Sepharose 4 Fast Flow column (GE Healthcare, Uppsala, Sweden) preequilibrated with buffer A containing 1 M ammonium sulfate. After the column was washed, the bound proteins were eluted with buffer A containing 0.5 M ammonium sulfate. The eluate was then applied to an HPLC system equipped with a TSK gel ether 5-PW column (Tosoh, Tokyo, Japan) equilibrated with buffer B (0.1 M KPB, pH 7.0, 2 mM DTT, 2.5 mM sodium hydrosulfite, 1% [vol/vol] 2-propanol) containing 1 M ammonium sulfate. Elution was performed along a 15-min linear gradient of 1 to 0 M ammonium sulfate in buffer B at a flow rate of 0.1 ml/min. The active fractions were diluted with buffer A and then applied to a spin column packed with 2′,5′-ADP-Sepharose 4B (GE Healthcare) and eluted with 20 mM NADPH in buffer A. The resultant eluate was then loaded onto a Mono-Q column (GE Healthcare) preequilibrated with buffer B at pH 7.5. The column was washed and eluted using a linear gradient of 0 to 0.65 M NaCl in buffer B over 32.5 min at a flow rate of 0.1 ml/min. Activity was observed in fractions 28 through 30, corresponding to 0.4 to 0.47 M NaCl. The activity and purity of the purified enzyme were analyzed by the HPLC method as described above and by SDS-PAGE (10) on a 5-to-20% (wt/vol) gradient polyacrylamide gel (SuperSep, Wako Pure Chemicals), respectively. The SDS-PAGE gels were stained with the Flamingo fluorescent gel stain system (Bio-Rad Laboratories, Inc., Hercules, CA).
Determination of amino acid sequences.
For N-terminal sequence analysis, 70 μl of the purified L-DZNR from the Mono-Q HPLC was added to 30 μl of 0.1% (vol/vol) trifluoroacetic acid (TFA) and transferred to a polyvinylidene difluoride (PVDF) membrane (ProSorb cartridge; Applied Biosystems, Foster City, CA). After washing with 20% (vol/vol) methanol and drying, the membrane was applied to a Procise 494cLC protein sequencer (Applied Biosystems).
The internal amino acid sequences were determined by partially purifying L-DZNR from a 600-ml cell culture by serial chromatography with butyl Sepharose 4 Fast Flow and 2′,5′-ADP-Sepharose 4B. The active fraction from the 2′,5′-ADP-Sepharose 4B column was concentrated to 5 μl and subjected to SDS-PAGE using a 10-to-20% gradient gel (Super Sep HG, Wako Pure Chemicals). After staining with colloidal blue (Invitrogen, Carlsbad, CA), a band at about 70 kDa corresponding to L-DZNR was excised, crushed, and destained in a 50% acetonitrile solution. After dehydration and drying, the resultant gel pieces were reduced with 55 mM DTT, alkylated with 100 mM iodoacetamide, and further digested with 2 μg Achromobacter protease I (Wako Pure Chemicals) for 7 h. The digested peptides were extracted with 60% (vol/vol) acetonitrile in 0.1% TFA and then, after concentration, applied to reversed-phase HPLC using a micro RPC C2/C18 SC2.1/10 column (GE Healthcare) equilibrated with eluent A (0.05% TFA) and eluted over a 40-min linear gradient of 5 to 65% eluent B (90% acetonitrile, 0.04% TFA) in eluent A at a flow rate of 0.1 ml/min. To facilitate the determination of L-DZNR-derived peptides, an area of equal size corresponding to where a 70-kDa band should have migrated was also excised from a control, sample-free lane and treated in the same way. Consequently, three peaks, eluted at 20.6 min (peptide 1), 22.1 min (peptide 2), and 26.6 min (peptide 3), were selected and subjected to internal amino acid analysis using the Procise 494cLC protein sequencer.
Isolation of Lactococcus 20-92 strain genomic DNA.
Genomic DNA was extracted from Lactococcus 20-92 cells cultured for 18 h using the genomic DNA buffer set and genome chip 500 (Qiagen, Hilden, Germany) according to the manufacturer's instructions, with slight modifications. Briefly, 50 ml of cultured cells were harvested by centrifugation at 5,000 × g for 20 min. The collected cells were resuspended in 11 ml of buffer B1 (from the genomic DNA buffer set) containing 200 ng/ml RNase A, 300 μl of a 100-mg/ml lysozyme solution (Sigma-Aldrich), and 500 μl of Qiagen proteinase K solution (Qiagen). After incubation at 37°C overnight, 4 ml of buffer B2 (from the genomic DNA buffer set) was added, and the solution was mixed slowly and incubated at 50°C for 2 h. This lysate was centrifuged at 5,000 × g and 4°C for 10 min. The supernatant obtained was loaded onto a Qiagen genomic-tip 500/G, and genomic DNA was purified according to the manufacturer's instructions.
Amplification of the L-DZNR gene from Lactococcus 20-92.
To clone the L-DZNR gene from Lactococcus 20-92 genomic DNA, we designed four degenerate primers, E1-N-terminal-F31, E1-N-terminal-F32, E1-N-terminal-F37, and E1-internal-RP1, based on corresponding segments of the N-terminal (MKNKFYPKTFERGYIGNLEVEN) and internal (ASRMVMDAVHEGYIAG) amino acid sequences of the purified enzyme (Table 1). We carried out degenerate PCR amplification using Ex-Taq polymerase (Takara Bio, Inc.) with an initial denaturation step at 95°C for 2 min, followed by 50 cycles of 95°C for 45 s, 54°C for 30 s, and 72°C for 2 min, with a final extension step at 72°C for 3 min. The resultant DNA fragment was purified with a gel extraction kit (Qiagen) and then subcloned into a pT7Blue T-vector (Novagen, Inc.). We obtained the DNA clone, pT7-E1-PCR, and sequenced it. Determination of the nucleotide sequence was carried out using a BigDye terminator v3.1 cycle sequence kit (Applied Biosystems) on an Applied Biosystems 3130xl genetic analyzer.
TABLE 1.
Sequences of primers employed in this study
| Primer | Sequence |
|---|---|
| E1-N-terminal-F31 | 5′-TGAAGAATAANTTNTAYCCNAARACNTTYGA-3′ |
| E1-N-terminal-F32 | 5′-ATGAAGAATAAGTTTTAYCCNAARACNTTYGA-3′ |
| E1-N-terminal-F37 | 5′-TGAAGAATAAGTTTTAYCCNAARACNTTYGARRGNGG-3′ |
| E1-internal-RP1 | 5′-CCTGCAATATAACCTTCATGTACNGCRTCCATNACCAT-3′ |
| E1-RACE-N-P1 | 5′-ATGCGGATCGCTCGGTTCTCGACCTCTAGGTTAC-3′ |
| E1-RACE-N-P2 | 5′-TTCTCGACCTCTAGGTTACCGATGTAGCCGC-3′ |
| E1-RACE-RP2-1 | 5′-ATCGAGGAGAAGTGCGAGGACGTCAGGGTCATC-3′ |
| E1-RACE-RP2-2 | 5′-ACGTCAGGGTCATCGGCATCGGCGACTGCAAG-3′ |
| pUC19-FP-1 | 5′-ACACAGGAAACAGCTATGACCATGATTACG-3′ |
| pUC19-FP-2 | 5′-ATGATTACGCCAAGCTTGCATGCCTGCAGG-3′ |
| exp.E1 pet F Nde | 5′-AGCTCATATGAAGAACAAGTTCTATCCGAA-3′ |
| exp.E1 pet R His | 5′-AATCGAATTCGTACAGGTTGCAGCCAGCGATGT-3′ |
Construction of genomic DNA libraries.
Genomic DNA libraries were constructed by inserting DNA fragments, obtained by digesting the Lactococcus 20-92 genomic DNA with several restriction enzymes (Takara Bio, Inc.), into pUC19 plasmids which were linearized with the corresponding restriction enzymes and dephosphorylated with shrimp alkaline phosphatase (Takara Bio, Inc.). The ligation of DNA fragments with linearized pUC19 was done with a DNA ligation kit, version 2.1 (Takara Bio, Inc.). The final reaction solutions were diluted 10-fold with water, and these diluents used as templates for rapid amplification of cDNA ends (RACE) (7).
Isolation and sequencing of the L-DZNR gene.
For isolation of the upstream and downstream regions of the degenerate-PCR-amplified DNA fragment, RACE-like procedures were performed with genomic DNA libraries as templates. The upstream and downstream DNA portions were amplified by a first round of RACE, followed by a nested RACE using gene-specific primers which were designed based on the sequence of the degenerate PCR product (primers E1-RACE-N-P1 and E1-RACE-N-P2 for the upstream portion and E1-RACE-RP2-1 and E1-RACE-RP2-2 for the downstream region) and the pUC19-specific primers pUC19-FP-1 and pUC19-FP-2 (Table 1). Using Ex-Taq DNA polymerase (Takara Bio, Inc.), the RACE involved an initial denaturation step at 95°C for 2 min, followed by 30 cycles of 95°C for 45 s, 60°C for 30 s, and 72°C for 1 min and a final extension at 72°C for 3 min. Amplified DNA from the nested RACEs was purified with a gel extraction kit (Qiagen) and directly sequenced. The entire nucleotide sequence of the L-DZNR gene was determined by overlapping the clone pT7-E1-PCR and three DNA fragments amplified by RACE.
Construction of the L-DZNR expression vector plasmid.
A pair of primers, exp.E1 pet F Nde and exp.E1 pet R His, incorporating NdeI and EcoRI restriction sites, respectively, were designed to allow amplification of the entire coding region of the L-DZNR gene (Table 1). An L-DZNR-His (N-terminally 6×His-tagged L-DZNR) expression plasmid, pET-E1-His, was constructed by digesting the resulting PCR product, amplified with primers exp.E1 pet F Nde and exp.E1 pet R His, with NdeI and EcoRI and ligating the liberated fragment into pET-21a(+) (Novagen, Inc.) between the NdeI and EcoRI sites. The recombinant plasmid was transformed into E. coli JM109 by conventional methods, and the transformants were grown on LB plates containing ampicillin at 37°C for 15 h. A single colony was picked and cultured in LB medium containing ampicillin. The isolation of pET-E1-His from E. coli JM109 transformants was carried out using a PI200 automatic genomic DNA isolation system (Kurabo Industries, Osaka, Japan).
Expression of recombinant L-DZNR-His in E. coli.
The expression plasmid pET-E1-His was introduced into E. coli BL21(DE3). Cultures of the pET-E1-His transformant and E. coli BL21(DE3) containing pET-21a(+) were grown at 30°C until the optical density at 600 nm was 0.6 in LB medium containing ampicillin. The expression of recombinant L-DZNR-His was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG; Wako Pure Chemicals), and the culture was continued for a further 3 h at 30°C. The expression of recombinant L-DZNR-His in the E. coli cells was confirmed by SDS-PAGE analysis (10). The IPTG-induced E. coli cells were harvested by centrifugation, and the collected cells were suspended in 1 ml buffer B and disrupted using a FastPrep-24 instrument as described above. From the cell lysates, 5 μl was analyzed by SDS-PAGE and visualized with a quick CBB gel staining kit (Wako Pure Chemicals) and 50 μl was used for the enzyme assay.
Enantioselectivity analysis.
The enantioselectivity of L-DZNR was determined using HPLC, with a 50-μl aliquot of the dissolved extract injected into an optically active Sumichiral-OA7000 column (5 μm, 250-mm by 4.6-mm internal diameter; Sumika Chemical Analysis Service, Osaka, Japan). The elution was isocratic, with a 50% (vol/vol) methanol-water mobile phase for 30 min at 40°C and a flow rate of 1.0 ml/min. The judgment of whether it was (R)- or (S)-dihydrodaidzein was made based on the report of Wang et al. (33). Optical resolution of a dihydrodaidzein racemate (purchased from Toronto Research Chemicals, Toronto, Canada) was performed by Daicel Chemical Industries (Osaka, Japan), and we obtained two enantiomers.
Nucleotide sequence accession number.
The sequence of the L-DZNR gene reported in this paper has been deposited in the DDBJ database under the accession number AB558141.
RESULTS
Validation of the conversion activity of daidzein to dihydrodaidzein in Lactococcus 20-92.
Lactococcus 20-92 can produce equol from daidzein in anaerobic culture, and we examined whether or not cell lysates possessed daidzein-metabolizing activity in vitro. We confirmed the presence of conversion activity from daidzein to dihydrodaidzein in the centrifuged supernatant and precipitate of the disrupted cell material in the presence but not the absence of coenzymes NADH and NADPH. The centrifuged supernatant was used as a material for the purification of L-DZNR, although the majority of enzymatic activity existed in the precipitate. This activity was only obvious in the presence of NADPH and not in the presence of NADH, revealing that the coenzyme NADPH was essential for this activity (Fig. 1).
FIG. 1.

The necessity of cofactors for biotransformation in the cell extract. (A to C) Conversion of daidzein to dihydrodaidzein in the cell extract with NADPH (A) or NADH (B) and without a cofactor (C). Elution profiles of the reference standards daidzein, dihydrodaidzein, and equol are shown in the panel labeled STD.
Purification of L-DZNR from Lactococcus 20-92.
We purified L-DZNR from the centrifuged supernatant of the disrupted cell material using a sequential four-step purification procedure consisting of hydrophobic chromatography, affinity chromatography, and two rounds of anion-exchange chromatography. The active fractions of the final Mono-Q anion-exchange chromatography exhibited a single band with an apparent molecular mass of 70 kDa (Fig. 2a), and the staining intensity of the bands from each fraction correlated with the conversion activity from daidzein to dihydrodaidzein (Fig. 2b). As a result, we concluded that this purified protein was L-DZNR from Lactococcus 20-92.
FIG. 2.

Purification of L-DZNR from cell extracts of Lactococcus 20-92. (a) SDS-PAGE analysis of Mono-Q HPLC fractions 27 to 31. Purified L-DZNR protein (approximately 70 kDa) is indicated by an arrow. (b) Dihydrodaidzein-producing activity in Mono-Q HPLC fractions 27 to 31. MW, molecular weight in thousands.
Cloning and sequencing of L-DZNR and its gene.
For the determination of the primary structure of L-DZNR, we initially obtained the N-terminal and internal amino acid sequences of the purified L-DZNR protein. The 22 amino acids at the N terminus were MKNKFYPKTFERGYIGNLEVEN. The three internal portions of L-DZNR were 10, 16, and 17 amino acids long, with the sequences FDEPVYPQAE (peptide 1), ASRMVMDAVHEGYIAG (peptide 2), and GYIGNLEVENRAIRMPM (peptide 3), respectively. Given that the 10 amino acids at the N-terminal end of peptide 3 coincided with 10 residues of the identified N-terminal sequence, these two identified sequences were thought to be continuous at the N-terminal region of this enzyme.
Degenerate primers were designed on the basis of the N-terminal amino acid sequence and the sequence of peptide 2. We tried to amplify the gene encoding L-DZNR with the genomic DNA from Lactococcus 20-92 and obtained a 1.9-kb DNA fragment by degenerate PCR when an annealing temperature of 54°C was used (Fig. 3). The DNA fragment was cloned and sequenced. The deduced amino acid sequence from the nucleotide sequence contained the same sequence that was observed in peptide 1 at amino acids 372 to 381 (Fig. 4). Therefore, we concluded that this DNA fragment was derived from the L-DZNR gene. This DNA fragment was incomplete; therefore, we performed RACE to search for the missing segment and obtained three new DNA fragments containing portions of the L-DZNR gene. These were designated RACE-fragment-N-SacI (1.2 kb), RACE-fragment-N-Sau3AI (1.0 kb), and RACE-fragment-C-SacI (0.6 kb). We determined the entire nucleotide sequence of the L-DZNR gene by overlapping the sequences of the initially amplified 1.9-kb DNA fragment and these three DNA fragments. The assembled sequence consisted of 3,553 nucleotides and contained an open reading frame of 1,935 nucleotides encoding L-DZNR. The open reading frame of this gene encoded 644 amino acids with a calculated molecular mass of 68,943 Da, demonstrating good agreement with the apparent molecular mass of 70 kDa obtained from the purified L-DZNR. Figure 4 shows the nucleotide and deduced amino acid sequences of L-DZNR. A putative Shine-Dalgarno sequence and a putative terminator sequence were identified in 5′- and 3′-untranslated regions, respectively; however, −35 and −10 boxes were not found in the 5′-untranslated regions.
FIG. 3.

Agarose gel electrophoresis of degenerate PCR products obtained using primer sets 1, 2, and 3, indicated above the gel. Amplification of a 1.9-kb DNA fragment from the L-DZNR gene with primer set 3 is indicated by an arrow. The combinations of forward and reverse primers were as follows: 1, E1-N-terminal-31 and E1-internal-RP1; 2, E1-N-terminal-37 and E1-internal-RP1; and 3, E1-N-terminal-F32 and E1-internal-RP1. Ma, λ/StyI DNA size marker; Mb, 100-bp DNA ladder size marker.
FIG. 4.

Nucleotide sequence of the L-DZNR gene from Lactococcus 20-92 and predicted amino acid sequence of its product. Nucleotides and translated amino acids are numbered on the right side of the figure. Peptide sequences obtained by Edoman degradation are boxed. The 4Fe-4S iron-sulfur cluster motif and two cofactor binding sites are indicated by the shaded areas. Consensus amino acid sequences of each motif are shown in bold. Putative terminator sequences are indicated by the arrows.
Gene characteristics.
Analysis of the deduced amino acid sequence of L-DZNR has indicated that this sequence contains a putative 4Fe-4S iron-sulfur cluster motif (CXXCX3CX12C) (20) at amino acids 343 to 363 and two cofactor binding sites (GXGXXG) (11, 31) at amino acids 390 to 395 and 512 to 517 (Fig. 4 and 5). A BlastP homology search (2) revealed that the deduced amino acid sequence of L-DZNR was similar to archaeal or bacterial open reading frames which are annotated as NADH:flavin oxidoreductase/NADH oxidase (28 to 36% identity). An old yellow enzyme (OYE)-like FMN binding domain sequence was also conserved at the N terminus of this protein (Fig. 5). The majority of organisms which had a sequence similar to that of L-DZNR were obligate or facultative anaerobes. Genes which were similar to L-DZNR were not found when a Blastn search with default parameters was conducted. Three characterized enzymes, NADH oxidase from Thermoanaerobacter brockii (GenBank accession number P32382, 35.4% identity) (12), 2-enoate reductase from Moorella thermoacetica (GenBank accession number Y16136, 32.9% identity) (26), and 2,4-dienoyl coenzyme A reductase from E. coli (GenBank accession number AAB82738, 28.6% identity) (8), were found to be similar to L-DZNR. Figure 5 shows the alignment of the deduced amino acid sequences for L-DZNR and these three characterized enzymes. The two nucleotide binding motifs (GXGXXG) and a putative 4Fe-4S iron-sulfur cluster motif (CXXCX3CX11-12C) were conserved in all of the aligned enzymes (Fig. 5). Analysis of the protein structure and localization with PSORT (http://psort.ims.u-tokyo.ac.jp/) (24, 25) demonstrated three transmembrane regions at amino acids 460 to 477, 507 to 525, and 546 to 562 in the C-terminal region of L-DZNR; therefore, L-DZNR is likely to be a transmembrane protein.
FIG. 5.

Sequence alignment of L-DZNR from Lactococcus 20-92 with the NADH oxidase from T. brockii (GenBank accession no. P32382), 2-enoate reductase from M. thermoacetica (GenBank accession no. Y16136), and 2,4-dienoyl coenzyme A (CoA) reductase from E. coli (GenBank accession no. AAB82738). Identical amino acid residues are indicated in black, and 3/4 matched amino acid residues are in gray. Box A, OYE-like FMN binding domain; box B, 4Fe-4S cluster motif; box C, coenzyme binding motifs. Consensus amino acid residues of 4Fe-4S cluster motif and coenzyme binding motifs are indicated in bold letters under boxes B and C, respectively.
Expression of recombinant L-DZNR-His in E. coli.
The expression of recombinant L-DZNR-His in E. coli harboring pET-E1-His encoding L-DZNR-His was attempted under different conditions. Using SDS-PAGE, we analyzed the expression of recombinant L-DZNR-His. A protein with a molecular mass of approximately 70 kDa, consistent with the size expected from pET-E1-His, was observed in cell lysates of E. coli harboring pET-E1-His but not in the controls (Fig. 6). An enzyme assay of the expressed recombinant L-DZNR-His was performed using E. coli cell extracts as an enzyme source, and the cell extracts displayed conversion activity from daidzein to dihydrodaidzein by HPLC analysis, proving that this recombinant protein had daidzein reductase activity (Fig. 7).
FIG. 6.
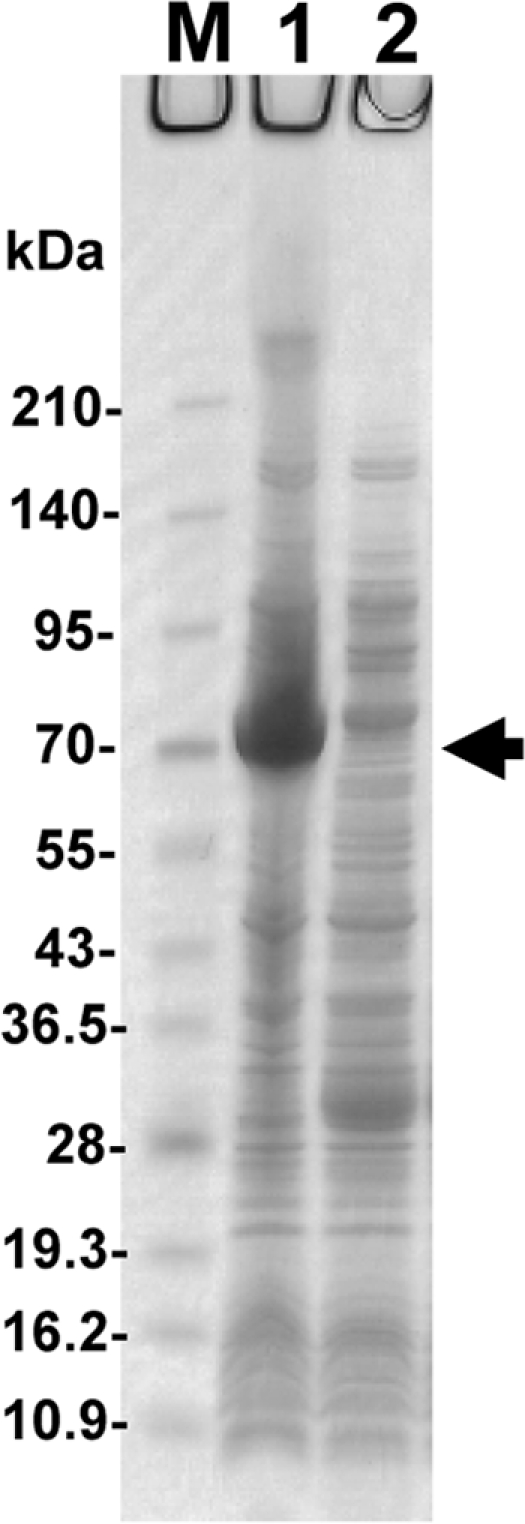
SDS-PAGE analysis of the expressed recombinant L-DZNR-His. M, molecular mass standards (kDa); lane 1, crude cell extract from E. coli BL21(DE3)/pET-E1-His; lane 2, crude cell extract from E. coli BL21(DE3)/pET21a. Recombinant L-DZNR-His (approximately 70 kDa) is indicated by the arrow.
FIG. 7.

(a and b) HPLC elution profiles of daidzein converted to dihydrodaidzein in crude cell extracts from E. coli BL21(DE3)/pET-E1-His (a) and E. coli BL21(DE3)/pET21a (b). Elution profiles of reference standards (daidzein, dihydrodaidzein, and equol) are shown in the panel labeled STD.
The L-DZNR-His activities in E. coli cell extracts were vastly different depending on the culture conditions, with the cell extracts exhibiting lower enzymatic activities in high-aeration than in low-aeration cultures. Moreover, the recombinant proteins demonstrated a tendency to aggregate into inclusion bodies (data not shown). Therefore, the recombinant protein was finally expressed under very-low-aeration culture conditions at 30°C.
Enantioselectivity of recombinant L-DZNR-His.
Two enantiomers of dihydrodaidzein exist. We examined the enantioselectivity of recombinant L-DZNR-His during the conversion of daidzein to dihydrodaidzein by chiral HPLC analysis. The results are shown in Fig. 8. This recombinant protein possessed enantioselectivity, with high production of (S)-dihydrodaidzein rather than (R)-dihydrodaidzein from daidzein evident.
FIG. 8.

Chiral HPLC analysis of the products in enantioselective reduction of dihydrodaidzein. (a) Reaction mixture with Lactococcus 20-92 cell extract. (b) Reaction mixture with recombinant L-DZNR-His expressed in E. coli. The enantiomers of dihydrodaidzein, (R)-dihydrodaidzein and (S)-dihydrodaidzein, are indicated as (R) and (S), respectively. The elution profile of the reference standard (dihydrodaidzein racemate) is shown in the panel labeled STD.
DISCUSSION
Metabolic pathways leading to the formation of equol from daidzein have been proposed previously (6, 15, 18, 23, 32). We observed dihydrodaidzein and tetrahydrodaidzein as reaction products in the conversion of daidzein to equol using homogenates of Lactococcus 20-92 cells as a enzyme source; however, dehydroequol, a metabolite which has been detected in human urine and is thought to be a metabolite produced directly from tetrahydrodaidzein (18, 32), was not observed (data not shown). Accordingly, we assumed that equol was produced from daidzein in this bacterium following three stages of enzymatic reactions. Those stages were the conversion of daidzein to dihydrodaidzein (first stage), of dihydrodaidzein to tetrahydrodaidzein (second stage), and of tetrahydrodaidzein to equol (third stage). We called the enzymes responsible for each stage E1, E2, and E3, respectively (Fig. 9). For the verification of our hypothetical metabolic pathway, we attempted to purify these enzymes from the homogenates of Lactococcus 20-92 cells and succeeded in isolating E1, also known as L-DZNR.
FIG. 9.
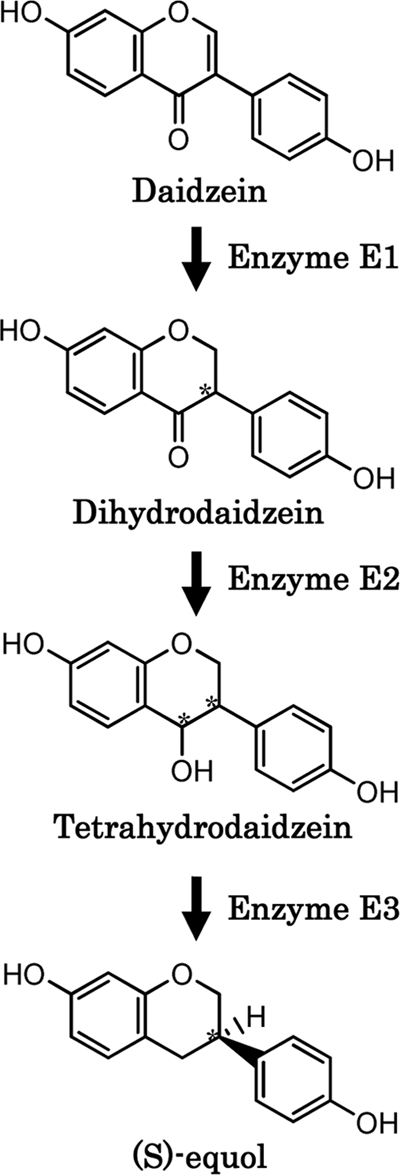
Postulated metabolic pathway for conversion of daidzein to equol in Lactococcus 20-92. Asymmetric carbons of the molecules are indicated by asterisks.
The conversion from daidzein to dihydrodaidzein is a reducing reaction. In general, a reductase often needs a coenzyme, such as NAD(H), NADP(H), or FAD(H), for its catalytic activity. We found that the conversion of daidzein to dihydrodaidzein by the cell extract of Lactococcus 20-92 is dependent on the coenzyme NADPH. Therefore, we examined the affinity of this enzyme for blue Sepharose, red Sepharose, and 2′,5′-ADP-Sepharose, which are the affinity resins generally used for purification of enzymes that use NAD(H) or NADP(H) as a coenzyme. We found that this enzyme had a higher affinity to 2′,5′-ADP-Sepharose (data not shown). Consequently, 2′,5′-ADP-Sepharose was adopted as the affinity resin for purification of this enzyme.
We purified L-DZNR using hydrophobic, affinity, and anion-exchange chromatography. We also succeeded in cloning the L-DZNR gene by using the information provided by the N-terminal and partial internal amino acid sequences obtained from the purified protein. The molecular mass (68,943 Da) calculated from the sequence of the L-DZNR gene was in strong agreement with molecular mass of the protein analyzed by SDS-PAGE (70 kDa).
The nucleotide sequence of the L-DZNR gene was unique, but the primary structure of the L-DZNR protein contained an 4Fe-4S cluster and two cofactor binding motifs. An OYE-like FMN binding domain near the N terminus strongly suggested that this enzyme belongs to the OYE family. Homology searches in public databases revealed numerous genes encoding proteins that have motifs similar to those seen in L-DZNR that have been cloned from several bacteria or identified in bacterial genomic DNA, and almost all the products of these genes are annotated as putative flavin oxidoreductases or hypothetical proteins. Such a gene was found frequently in obligate and facultative anaerobic organisms, including Lactococcus 20-92. One of the features of such gene products is that they have an 4Fe-4S cluster at the center of the putative primary sequence.
The protein containing the Fe-S cluster is generally called an iron-sulfur protein. Iron-sulfur proteins are widely found in many organisms and have essential physiological roles in electron transport and oxidation-reduction catalysis. An iron-sulfur cluster is thought to facilitate electron transfer in oxidation-reduction reactions (4, 28). These properties suggest that the electron transfer function of the 4Fe-4S cluster in the L-DZNR molecule contributes to the reduction of daidzein to dihydrodaidzein under anaerobic conditions.
The predicted putative structure of L-DZNR is similar to those of bacterial enoate reductases. Enoate reductases catalyze the reduction of carbon-carbon double bonds of nonactivated 2-enoate, α,β-unsaturated aldehydes, cyclic ketones, and methylketones by using NADH as an electron donor (26). It was reported that the recombinant enoate reductase of Clostridium thermoaceticum expressed in E. coli under aerobic conditions was insoluble and enzymatically inactive but that, when expressed under anaerobic conditions, the enzyme was soluble and enzymatically active (26).
Lactococcus 20-92 is able to grow both aerobically and anaerobically; however, Lactococcus 20-92 does not exhibit the ability to convert daidzein to equol when grown aerobically. Moreover, the enzyme activity of the recombinant L-DZNR tends to be lower when expressed in highly aerated E. coli cultures. Therefore, these facts suggest that L-DZNR has characteristics similar to those of the enoate reductase of C. thermoaceticum, with both enzymes being unstable under aerobic conditions.
As described above, the existence of an OYE-like FMN binding domain in the determined putative primary structure of L-DZNR reveals that this enzyme could be classified in the OYE family. The OYE member NAD(P)H dehydrogenase was the first flavoprotein purified from brewer's yeast and was named so because a solution of this protein is yellow in color (34, 35). A solution of the purified recombinant L-DZNR-His from E. coli was light yellow in color (data not shown). Kataoka et al. (16) reported that the OYE from Candida macedoniensis could catalyze the enantioselective hydrogenation of the carbon double bond of ketoisophorone (KIP; 2,6,6-trimethyl-2-cyclohexen-1,4-dione). We examined whether L-DZNR catalyzes a reduction of daidzein with enantioselectivity or not and showed that recombinant L-DZNR-His participated in the enantioselective hydrogenation of daidzein, with the dihydrodaidzein produced almost always being (S)-dihydrodaidzein.
The conversion of daidzein was performed using the cell extract of Lactococcus 20-92 as an enzyme source, and the produced dihydrodaidzein was detected as a racemate. Recently, Kim et al. (18) reported that (R)- and (S)-dihydrodaidzeins are rapidly converted to racemates in the cell extract and the whole cell of Eggerthella sp. Julong 732 and proposed the existence of a dihydrodaidzein racemase in this bacterium. Taken together, our results from this study strongly support the existence of a dihydrodaidzein racemase as predicted by Kim et al. (18).
This is the first report elucidating the sequence of the gene encoding L-DZNR, one of the enzymes involved in the metabolism of daidzein to equol. Our study contributes to the identification of other enzymes involved in the equol production pathway and to the elucidation of metabolic mechanisms involved in converting daidzein to equol in enteric bacteria.
Acknowledgments
We thank the staff of the Institute of Biomedical Innovation, Saga Nutraceuticals Research Institute, and Microbiological Institute of Otsuka Pharmaceutical Co., Ltd., for technical assistance. We also thank Kuniaki Natsume and Yasuo Irie for their continuous support of this study.
Footnotes
Published ahead of print on 16 July 2010.
REFERENCES
- 1.Adlercreutz, H. 2002. Phyto-oestrogens and cancer. Lancet Oncol. 3:364-373. [DOI] [PubMed] [Google Scholar]
- 2.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 3.Arai, Y., M. Uehara, Y. Sato, M. Kimira, A. Eboshida, H. Adlercreutz, and S. Watanabe. 2000. Comparison of isoflavones among dietary intake, plasma concentration and urinary excretion for estimation of phytoestrogen intake. J. Epidermiol. 10:127-135. [DOI] [PubMed] [Google Scholar]
- 4.Beinert, H., R. H. Holm, and E. Münck. 1997. Iron-sulfur clusters: nature's modular, multipurpose structures. Science 277:653-659. [DOI] [PubMed] [Google Scholar]
- 5.Bowey, E., H. Adlercreutz, and I. Rowland. 2003. Metabolism of isoflavones and lignans by the gut microflora: a study in germ-free and human flora associated rats. Food Chem. Toxicol. 41:631-636. [DOI] [PubMed] [Google Scholar]
- 6.Decroos, K., S. Vanhemmems, S. Cattoir, and W. Verstraete. 2005. Isolation and characterization of an equol-producing mixed microbial culture from a human faecal sample and its activity under gastrointestinal conditions. Arch. Microbiol. 183:45-55. [DOI] [PubMed] [Google Scholar]
- 7.Frohman, M. A., M. K. Dush, and G. R. Martin. 1988. Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proc. Natl. Acad. Sci. U. S. A. 85:8998-9002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He, X. Y., S. Y. Yang, and H. Schulz. 1997. Cloning and expression of the fadH gene and characterization of the gene product 2,4-dienoyl coenzyme A reductase from Escherichia coli. Eur. J. Biochem. 248:516-520. [DOI] [PubMed] [Google Scholar]
- 9.Hur, H. G., J. O. Lay, R. D. Beger, J. P. Freeman, and F. Rafii. 2000. Isolation of human intestinal bacteria metabolizing the natural isoflavone glycosides daidzin and genistin. Arch. Microbiol. 174:422-428. [DOI] [PubMed] [Google Scholar]
- 10.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 11.Liu, Z. J., Y. J. Sun, J. Rose, Y. J. Chung, C. D. Hsiao, W. R. Chang, I. Kuo, J. Perozich, R. Lindahl, J. Hemple, and B. C. Wang. 1997. The first structure of an aldehyde dehydrogenase reveals novel interactions between NAD and the Rossmann fold. Nat. Struct. Biol. 4:317-326. [DOI] [PubMed] [Google Scholar]
- 12.Liu, X. L., and R. K. Scopes. 1993. Cloning, sequencing and expression of the gene encoding NADH oxidase from the extreme anaerobic thermophile Thermoanaerobium brockii. Biochim. Biophys. Acta 1174:187-190. [DOI] [PubMed] [Google Scholar]
- 13.Lundh, T. J., H. Pettersson, and K. H. Kiessling. 1988. Liquid chromatographic determination of the estrogens daidzein, formononetin, coumestrol, and equol in bovine blood plasma and urine. J. Assoc. Off. Anal. Chem. 71:938-941. [PubMed] [Google Scholar]
- 14.Jin, J. S., T. Nishihata, N. Kakiuchi, and M. Hattori. 2008. Biotransformation of C-glucosylisoflavone puerarin to estrogenic (3S)-equol in co-culture of two human intestinal bacteria. Biol. Pharm. Bull. 31:1621-1625. [DOI] [PubMed] [Google Scholar]
- 15.Joannou, G. E., G. E. Kelly, A. Y. Reeder, M. Waring, and C. Nelson. 1995. A urinary profile study of dietary phytoestrogens. The identification and mode of metabolism of new isoflavonoids. J. Steroid Biochem. Mol. Biol. 54:167-184. [DOI] [PubMed] [Google Scholar]
- 16.Kataoka, M., A. Kotaka, A. Hasegawa, M. Wada, A. Yoshizumi, S. Nakamori, and S. Shimizu. 2002. Old yellow enzyme from Candida macedoniensis catalyzes the stereospecific reduction of the C=C bond of ketoisophorone. Biosci. Biotechnol. Biochem. 66:2651-2657. [DOI] [PubMed] [Google Scholar]
- 17.Kim, D. H., E. A. Jung, I. S. Sohng, J. A. Han, T. H. Kim, and M. J. Han. 1998. Intestinal bacterial metabolism of flavonoids and its relation to some biological activities. Arch. Pharm. Res. 21:17-23. [DOI] [PubMed] [Google Scholar]
- 18.Kim, M., S. I. Kim, J. Han, L. Wang, D. G. Song, and S. U. Kim. 2009. Stereospecific biotransformation of dihydrodaidzein into (3S)-equol by the human intestinal bacterium Eggerthella strain Julong 732. Appl. Environ. Microbiol. 75:3062-3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kostelac, D., G. Rechkemmer, and K. Briviba. 2003. Phytoestrogens modulate binding response of estrogen receptors alpha and beta to the estrogen response element. J. Agric. Food. Chem. 51:7632-7635. [DOI] [PubMed] [Google Scholar]
- 20.Krauth-Siegel, R. L., R. Blatterspiel, M. Saleh, E. Schiltz, R. H. Schirmer, and R. Untucht-Grau. 1982. Glutathione reductase from human erythrocytes. The sequences of the NADPH domain and of the interface domain. Eur. J. Biochem. 121:259-267. [DOI] [PubMed] [Google Scholar]
- 21.Maruo, T., M. Sakamoto, C. Ito, T. Toda, and Y. Benno. 2008. Adlercreutzia equolifaciens gen. nov., sp. nov., an equol-producing bacterium isolated from human faeces, and emended description of the genus Eggerthella. Int. J. Syst. Evol. Microbiol. 58:1221-1227. [DOI] [PubMed] [Google Scholar]
- 22.Minamida, K., K. Ota, M. Nishimukai, M. Tanaka, A. Abe, T. Sone, F. Tomita, H. Hara, and K. Asano. 2008. Asaccharobacter celatus gen. nov., sp. nov., isolated from rat caecum. Int. J. Syst. Evol. Microbiol. 58:1238-1240. [DOI] [PubMed] [Google Scholar]
- 23.Minamida, K., M. Tanaka, A. Abe, T. Sone, F. Tomita, H. Hara, and K. Asano. 2006. Production of equol from daidzein by gram-positive rod-shaped bacterium isolated from rat intestine. J. Biosci. Bioeng. 102:247-250. [DOI] [PubMed] [Google Scholar]
- 24.Nakai, K. 1991. Predicting various targeting signals in amino acid sequences. Bull. Inst. Chem. Res. 69:269-291. [Google Scholar]
- 25.Nakai, K., and M. Kanehisa. 1992. A knowledge base for predicting protein localization sites in eukaryotic cells. Genomics 14:897-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rohdich, F., A. Wiese, R. Feicht, H. Simon, and A. Bacher. 2001. Enoate reductases of Clostridia. Cloning, sequencing, and expression. J. Biol. Chem. 276:5779-5787. [DOI] [PubMed] [Google Scholar]
- 27.Setchell, K. D. R., N. M. Brown, and E. L. Olsen. 2002. The clinical importance of the metabolite equol—a clue to the effectiveness of soy and its isoflavone. J. Nutr. 132:3577-3584. [DOI] [PubMed] [Google Scholar]
- 28.Singh, B. B., I. Curdt, D. Shomburg, P. S. Bisen, and H. Böhme. 2001. Valine 77 of heterocystous ferredoxin FdxH2 in Anabaena variabilis strain ATCC 29413 is critical for its oxygen sensitivity. Mol. Cell. Biochem. 217:137-142. [DOI] [PubMed] [Google Scholar]
- 29.Tamura, M., T. Tsushida, and K. Shinohara. 2007. Isolation of an isoflavone-metabolizing, Clostridium-like bacterium, strain TM-40, from human faeces. Anaerobe 13:32-35. [DOI] [PubMed] [Google Scholar]
- 30.Uchiyama, S., T. Ueno, and T. Suzuki. 2007. Identification of a newly isolated equol-producing lactic acid bacterium from the human feces. J. Intest. Microbiol. 21:217-220. [Google Scholar]
- 30a.Uchiyama, S., T. Ueno, and T. Suzuki. 2005. Composition containing lactic acid bacterium producing equol. International patent WO2005/000042.
- 31.Vasiliou, V., A. Bairoch, K. F. Tipton, and D. W. Nebert. 1999. Eukaryotic aldehyde dehydrogenase (ALDH) genes: human polymorphisms, and recommended nomenclature based on divergent evolution and chromosomal mapping. Pharmacogenetics 9:421-434. [PubMed] [Google Scholar]
- 32.Wang, X. L., H. G. Hur, J. H. Lee, K. T. Kim, and S. I. Kim. 2005. Enantioselective synthesis of S-equol from dihydrodaidzein by a newly isolated anaerobic human intestinal bacterium. Appl. Environ. Microbiol. 71:214-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang, X. L., K. H. Shin, H. G. Hur, and S. I. Kim. 2005. Enhanced biosynthesis of dihydrodaidzein and dihydrogenistein by a newly isolated bovine rumen anaerobic bacterium. J. Biotechnol. 115:261-269. [DOI] [PubMed] [Google Scholar]
- 34.Warburg, O., and W. Christian. 1932. Ein zweites sauerstoffübertragendes Ferment und sein Absorptionsepktrum. Naturwissenschafte 20:688. [Google Scholar]
- 35.Warburg, O., and W. Christian. 1933. Über das gelbe Ferment und seine Wirkungen. Biochem. Z. 266:377-411. [Google Scholar]
- 36.Yokoyama, S., and T. Suzuki. 2008. Isolation and characterization of a novel equol-producing bacterium from human feces. Biosci. Biotechnol. Biochem. 72:2660-2666. [DOI] [PubMed] [Google Scholar]
- 37.Yu, Z. T., W. Yao, and W. Y. Zhu. 2008. Isolation and identification of equol-producing bacterial strains from cultures of pig faeces. FEMS Microbiol. Lett. 282:73-80. [DOI] [PubMed] [Google Scholar]