

Abstract
ɛ-Poly-l-lysine (ɛ-PL) is produced by Streptomyces albulus NBRC14147 as a secondary metabolite and can be detected only when the fermentation broth has an acidic pH during the stationary growth phase. Since strain NBRC14147 produces ɛ-PL-degrading enzymes, the original chain length of the ɛ-PL polymer product synthesized by ɛ-PL synthetase (Pls) is unclear. Here, we report on the identification of the gene encoding the ɛ-PL-degrading enzyme (PldII), which plays a central role in ɛ-PL degradation in this strain. A knockout mutant of the pldII gene was found to produce an ɛ-PL of unchanged polymer chain length, demonstrating that the length is not determined by ɛ-PL-degrading enzymes but rather by Pls itself and that the 25 to 35 l-lysine residues of ɛ-PL represent the original chain length of the polymer product synthesized by Pls in vivo. Transcriptional analysis of pls and a kinetic study of Pls further suggested that the Pls catalytic function is regulated by intracellular ATP, high levels of which are required for full enzymatic activity. Furthermore, it was found that acidic pH conditions during ɛ-PL fermentation, rather than the inhibition of the ɛ-PL-degrading enzyme, are necessary for the accumulation of intracellular ATP.
Two amino acid homopolymers comprising a single type of amino acid are known in nature (13): poly-γ-glutamic acid (γ-PGA) and ɛ-poly-l-lysine (ɛ-PL). The latter consists of 25 to 35 l-lysine residues with linkages between α-carboxyl groups and ɛ-amino groups (Fig. 1). ɛ-PL exhibits antimicrobial activity against a spectrum of microorganisms including bacteria and fungi (16-20). Because of its high levels of safety and biodegradability (7), ɛ-PL is used as a food preservative in several countries.
FIG. 1.

Chemical structure of ɛ-PL.
The biological activity of ɛ-PL is dependent on its molecular size. Shima and coworkers investigated this relationship in Escherichia coli K-12 (20) and showed that ɛ-PL with more than nine l-lysine residues severely inhibited microbial growth; however, the l-lysine octamer demonstrated negligible antimicrobial activity. In contrast, chemically synthesized α-poly-l-lysine with a chain of 50 l-lysine residues and linkages between the α-carboxyl and α-amino groups demonstrated a lower level of activity than ɛ-PL. Thus, polymerization of l-lysine via an isopeptide bond is required to exert its biological activity, and the polymerization mechanisms involved in the chain length and the diversity of ɛ-PL are of particular interest.
We recently reported on the purification of an ɛ-PL synthetase (Pls) and the cloning of its gene (accession number AB385841) from Streptomyces albulus NBRC14147, which is an ɛ-PL-producing strain (22). Pls was found to be a membrane protein with adenylation and thiolation domains characteristic of the nonribosomal peptide synthetases (NRPSs) (11, 12, 15, 21). In vitro, Pls produced ɛ-PL with chain lengths ranging from 3 to 17 residues, demonstrating that the chain length diversity of ɛ-PL is directly generated by the synthetase rather than via the differential degradation of a uniform polymer by ɛ-PL-degrading enzymes (22). However, in vivo, it is still unclear whether the 25 to 35 l-lysine residues of ɛ-PL represent the original chain length of the polymer product synthesized by Pls as strain NBRC14147 produces ɛ-PL-degrading enzymes (9).
During ɛ-PL fermentation, its production and accumulation are detected when the pH value of the fermentation broth decreases from approximately 7.0 to the self-stabilized final value of approximately 3.2 (9). After fermentation is complete, ɛ-PL is enzymatically degraded by increasing the pH value to approximately 7.0 (9). Such degradation is also observed using washed mycelium at pH 7.0. In fact, Kito and coworkers successfully purified the ɛ-PL-degrading enzyme Pld from S. albulus (10). Pld was classified as a type of Zn2+-containing aminopeptidase that is tightly bound to the cell membrane and that releases amino (N)-terminal l-lysines one at a time; the mode of ɛ-PL degradation with Pld is an exo-type action. Its highest activity was observed at pH 7.0, which was in good agreement with the observation that the ɛ-PL produced was degraded at a neutral pH.
Furthermore, we previously reported that the pld gene (accession number AB243405) plays a partial role in self-resistance (5). We expected increased productivity or a change in the molecular size of the ɛ-PL (25- to 35-mer) produced on inactivation of the pld gene; however, contrary to this, no alterations were observed. In addition, a high level of ɛ-PL-degrading activity was still detected in the pld gene disruptant while its aminopeptidase activity was significantly lower than that of the wild type due to the inactivation of pld. ɛ-PL degradation was shown to be catalyzed by endo-type peptidases as no l-lysine monomers were released.
Here, we report on the identification and inactivation of the gene encoding the unidentified ɛ-PL-degrading enzyme with endo-type peptidase activity in S. albulus NBRC14147. Our study provides insight into the original chain length of ɛ-PL synthesized by Pls in vivo. Moreover, it reveals that acidic pH conditions during ɛ-PL fermentation, rather than the inhibition of the ɛ-PL-degrading enzyme, are necessary for the accumulation of intracellular ATP.
MATERIALS AND METHODS
Chemicals.
ɛ-PL is commercially available and was a gift from Chisso Corp. (Tokyo, Japan). All other chemicals were of analytical grade.
Bacterial strains, plasmids, and media.
The S. albulus strains and plasmids used in this study are listed in Table 1. S. albulus CRM001, in which the pld gene is inactivated (5), was used as the parent strain for the pldII gene inactivation experiment. The SLB medium and growth conditions used for S. albulus strains have been previously described (3). E. coli NovaBlue (Novagen) was used for routine cloning, and E. coli S17-1 was used for intergeneric conjugation. E. coli strains were grown in Luria-Bertani (LB) medium (14) supplemented with neomycin (25 μg/ml). A transposon carrying the apramycin resistance gene [aac(3)IV] was prepared as described previously (4). The E. coli-S. albulus shuttle vector pLAE003 (Table 1) was used for the pldII gene inactivation experiment in S. albulus CRM001. Recombinant DNA procedures for E. coli were performed using standard techniques as described by Sambrook and Russell (14).
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Descriptiona | Reference or source |
|---|---|---|
| S. albulus strains | ||
| CR1 | Derivative of strain NBRC14147 (wild type); cryptic plasmid (pNO33) free | 3 |
| CRM001 | CR1 pld::tsr (Tspr) | 5 |
| CRM003 | CR1 pls::aac(3)IV (Apmr) | 22 |
| CRM004 | CRM001 derivative; pld::tsr (Tspr)pldII::aac(3)IV (Apmr) | This study |
| E. coli strains | ||
| NovaBlue | Cloning host; endA1 hsdR17(rK12− mK12+) supE44 thi-1 recA1 gyrA96 relA1 lac [F′proA+B+lacIqZΔM15::Tn10] (Tetr) | TaKaRa |
| S17-1 | Donor strain used for intergeneric conjugation | 3 |
| Plasmids | ||
| pLAE003 | E. coli-S. albulus shuttle vector harboring pNO33 replicon;oriT of plasmid RK2; Neor (aphII) | 3 |
| pLAE3800 | pLAE003 derivative with 3.8-kbp PCR fragment containing the pldII gene; Neor (aphII) | This study |
| pLAE3800-apr | pLAE3800 derivative in which the pldII gene was inactivated by the insertion of the aac(3)IV gene; Neor Aprr | This study |
| pLAE006 | pLAE003 harboring ermE* promoter; oriT of plasmid RK2; Neor (aphII) | 6 |
| pLAE006-pldII | pLAE006 harboring the pldII gene; oriT of plasmid RK2; Neor (aphII) | This study |
Tspr, thiostrepton resistant; Apmr, apramycin resistant; Tetr, tetracycline resistant; Neor, neomycin resistant.
Inactivation of the pldII gene.
A 3.8-kb fragment carrying the pldII gene was amplified with the primers frp3200-F (5′-GCTCTAGACTGACCCTGCTGGGCCTG-3′) and frp7000-R (5′-ACCAAGCTTCAACGACGTCGTCGCCC-3′) and inserted into the XbaI and HindIII sites of pLAE003 to give pLAE3800. To inactivate the pldII gene, the transposon carrying the apramycin resistance gene aac(3)IV was randomly inserted into pLAE3800 by in vitro transposon insertion with EZ-Tn5 transposase (Epicentre). The pLAE3800-apr plasmid, with the transposon inserted into the pldII gene, was selected by restriction enzyme digests and sequencing analysis. It was subsequently introduced into S. albulus CRM001 (a pld gene disruptant) by intergeneric conjugation with E. coli S17-1, as described previously (3), and exconjugants showing resistance against both neomycin and apramycin were isolated. After the exconjugants were subjected to protoplasting and regeneration as previously described (3), colonies showing apramycin resistance and neomycin sensitivity were selected. Insertional inactivation via double crossing over induced by protoplasting and regeneration was confirmed by Southern hybridization using an AlkPhos direct labeling and detection system (GE Healthcare) (see Fig. 4). Strain CRM004, in which both the pld and the pldII genes were inactivated, was used for further experiments.
FIG. 4.

Construction of the pldII gene disruptant. Gene replacement via double crossing over was carried out as shown in the schematic diagram (a) and confirmed by Southern blot analysis (b). Genomic DNA from the CRM001 strain (lane 1) and the CRM004 strain (lane 2) was digested by SphI and subjected to Southern hybridization using the pldII gene as a probe.
Complementation experiment with the pldII gene and strain CRM003.
To construct pLAE006-pldII, in which the pldII gene was expressed under the control of the ermE* constitutive promoter, the following set of primers was designed to amplify the plsII gene region from 60 bp upstream of the start codon (containing the ribosome binding site) to the pldII gene stop codon: 5′-CAACCGGATCCCCGGCGAACGCCCCGCCCG-3′ (forward) and 5′-ACCAAGCTTTCAGGCCGCGAGCGCGGGCAGCGGCTT-3′ (reverse). Restriction enzyme sites (shown in italics) of BamHI and HindIII were introduced using these primers. PCR was carried out under standard conditions using S. albulus CR1 genomic DNA as a template. After sequence confirmation of amplified fragments, the pldII gene was ligated into pLAE006 (6) carrying the ermE* promoter and neomycin resistance gene. The resulting plasmid, pLAE006-pldII, was introduced into the S. albulus CRM003 strain by intergeneric conjugation from E. coli, as described previously (3).
ɛ-PL-degrading activity in S. albulus strains.
S. albulus strains were cultured in the ɛ-PL-production medium M3G containing 5% glucose, 0.5% yeast extract, 1% (NH4)2SO4, 0.16% Na2HPO4, 0.14% KH2PO4, 0.05% MgSO4·7H2O, 0.004% ZnSO4·7H2O, and 0.003% FeSO4·7H2O (pH 6.8) for 20 h at 30°C. Mycelia were harvested by centrifugation and then washed twice with 100 mM 2-morpholinoethanesulfonic acid (MES) buffer (pH 6.0). To initiate the degrading reaction, washed mycelia were added to the buffer containing 1.0 mg ml−1 (100 mg of mycelium [wet weight] ml−1) ɛ-PL. After incubation for 3 h, mycelia were removed by centrifugation, and the supernatants containing ɛ-PL degradation products were analyzed by reverse-phase high-performance liquid chromatography (HPLC) using an ion pair reagent, as described previously (5, 10).
ɛ-PL fermentation with S. albulus strains.
S. albulus CR1 and CRM004 strains were inoculated into 5 ml of SLB medium and cultivated until the stationary phase was reached (20 h at 30°C). Aliquots (0.5 ml) of the culture were transferred to 50 ml of M3G medium and grown at 30°C. After the fermentation broths were centrifuged, the amount of ɛ-PL produced in the supernatant was determined according to the method reported by Itzhaki (8).
A two-stage fermentation method was also employed for the analysis of ɛ-PL productivity. Mycelia of S. albulus strains cultured with 50 ml of M3G medium for 24 h at 30°C were resuspended in the same volume of 100 mM citrate buffer (pH 3.8 to 5.5) containing 5% glucose and 1% (NH4)2SO4. After incubation with shaking at 30°C for 20 h, the ɛ-PL concentrations were measured.
ɛ-PL fermentation in a 3-liter bench-scale jar fermentor with strict pH control was performed according to the method reported by Kahar and coworkers (9). S. albulus NBRC14147 was inoculated into 5 ml of SLB medium and cultivated until the stationary phase was reached (for 20 h at 30°C). Aliquots (1.0 ml) of the culture were transferred to 500-ml Erlenmeyer flasks containing 100 ml of M3G medium and precultured at 30°C for 20 h with rotary shaking. A 100-ml sample of the preculture was inoculated into 2 liters of M3G medium in 3-liter jar fermentors and then cultured aerobically at 30°C with agitation. The pH of the medium was maintained at 4.2 by feeding 10% aqueous ammonia in the stationary phase.
Gene expression analysis by quantitative RT-PCR.
S. albulus NBRC14147 was cultivated in M3G medium in a 3-liter bench-scale jar fermentor. Total RNA was isolated from strain NBRC14147 grown at 30°C for 9, 11, 13, 15, and 24 h, as described previously (2). RNA samples were treated with DNase I (TaKaRa), and 5 μg was used as a template for reverse transcription (RT) at 42°C with ReverTra Ace (Toyobo) and oligonucleotides. The resulting cDNAs were used for PCR amplifications. The 5′ primer 5′-GGGGGATCCTCGTCGCCCCTTCTCGAATCG-3′ (pls-N) and the 3′ primer 5′-ACCAAGCTTTCACGCGGCCGCACCTCCCTC-3′ (pls-C) were used to amplify the pls gene. The gene encoding aspartokinase (Ask) (5) was amplified with the primer pair 5′-GGGGGATCCGGCCTTGTCGTGCAGAAGTAC-3′ (ask-N) and 5′-ACCAAGCTTTCATCGCCCGGTGCCGCCGTA-3′ (ask-C). The primers pls-C and ask-C were also used in the RT reactions. RNA samples that had not been subjected to RT were used as negative controls.
Steady-state kinetics of Pls.
Pls was purified from S. albulus NBRC14147 and assayed as described previously (22), with the exception that the enzyme concentration and reaction time were reduced to enable measurement of steady-state kinetic parameters. All assays were carried out under linear conditions. A Lineweaver-Burk plot and a Hill plot were used to estimate the apparent kinetic constants.
Measurement of intracellular ATP levels in S. albulus NBRC14147.
Quantitative analysis of intracellular ATP levels in S. albulus NBRC14147 was performed according to the method reported by Bagnara et al. (1). Culture broth (0.5 ml) of S. albulus NBRC14147 was added into 0.6 ml of ice-cold extraction reagent consisting of 510 mM trichloroacetic acid and 17 mM EDTA. The samples were mixed thoroughly using a Voltex mixer for 10 s and were placed on ice for 15 min. After centrifugation, supernatant ATP concentrations were analyzed by reverse-phase HPLC. The analytical conditions were as follows: a Develosil RP-Aqueous-AR-5 column (250 by 4.6 mm; Nomura Chemical, Japan) was used at a temperature of 30°C; detection was at 260 nm with an isocratic flow (1 ml min−1) of 100 mM KH2PO4 (pH 4.0).
MIC values of ɛ-PL for the S. albulus strains.
S. albulus strains were grown in M3G medium for 20 h, and aliquots (5 μl) were transferred to 1 ml of 0.8% nutrient broth (Difco) containing ɛ-PL, in which the pH was adjusted to 4.5 (20 mM citrate buffer) or 7.0 (20 mM HEPES-NaOH). After cultivation for 48 h at 30°C, the MIC values were obtained.
RESULTS
Identification and sequence analysis of the pldII gene.
During in silico analysis of a 33-kb DNA fragment containing the pls gene (open reading frame 13 [ORF13]) (22), we found a metalloproteinase gene (ORF14) with which it was translationally coupled (Fig. 2 a). The deduced amino acid sequence of this gene product consisted of 467 amino acid residues with a calculated molecular mass of 51,945 Da, which showed similarity to the ɛ-PL-degrading enzyme Pld (36% identity and 51% similarity) (Fig. 2b). It was therefore predicted to be the gene encoding an unidentified ɛ-PL-degrading enzyme of our target.
FIG. 2.

The pldII gene located immediately downstream of the pls gene. (a) Schematic organization of the pls and pldII genes (accession number AB385841). (b) Alignment of Pld (accession number AB243405) with PldII (accession number AB385841). Areas shaded in dark gray and light gray indicate identical and similar amino acid residues, respectively. The conserved motif involved in Zn2+ binding is indicated by plus signs.
To clarify this, we initially investigated the ɛ-PL-degrading activity in the pls gene knockout mutant strain CRM003 (Table 1), in which the metalloproteinase gene should also be inactivated due to downstream polar effects (22). HPLC analysis revealed that the ɛ-PL-degrading activity of this mutant was remarkably decreased (Fig. 3 d), which suggested that the gene indeed encodes the major ɛ-PL-degrading enzyme PldII. We initially attempted to construct a recombinant PldII enzyme to investigate its catalytic function. However, soluble recombinant enzymes could not be obtained using heterologous expression systems because of the tendency to form inclusion bodies (data not shown). Hence, we investigated the biological function of pldII using the knockout mutant strain CRM004, in which the pldII gene was inactivated by homologous recombination (Fig. 4). Southern hybridization using the pldII gene as a probe confirmed that strain CRM004 carried the pldII gene that had been inactivated by the insertion of the apramycin resistance gene (pldII::apr).
FIG. 3.
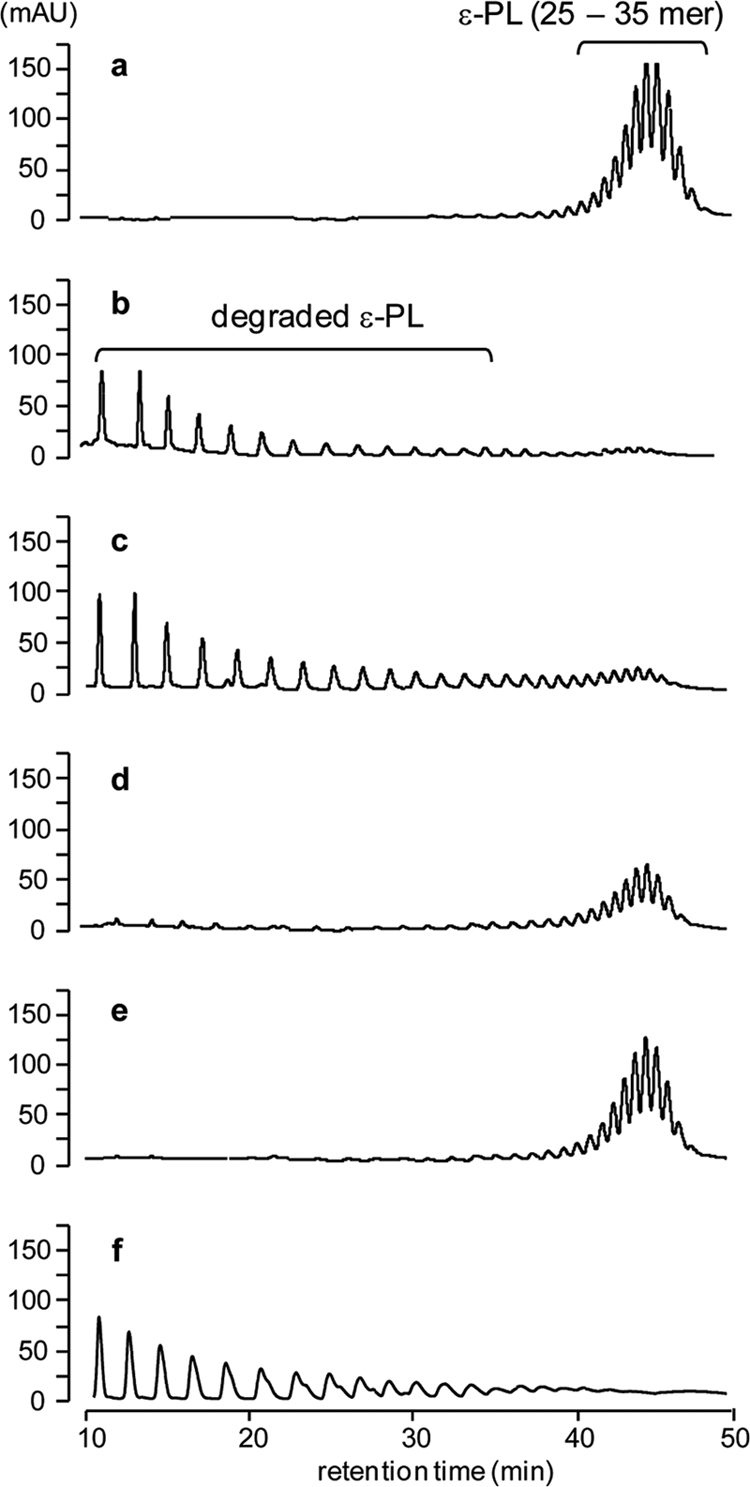
ɛ-PL-degrading activities in S. albulus strains. Washed mycelia of CR1 (b), CRM001 (c), CRM003 (d), CRM004 (e), and CRM003 harboring the pLAE006-pldII plasmid (f) were incubated with 100 mM MES buffer (pH 6.0) containing ɛ-PL (1.0 mg ml−1) for 3 h, and the supernatants were analyzed by reverse-phase HPLC. The authentic ɛ-PL (1.0 mg ml−1) was also analyzed (a).
ɛ-PL-degrading activity in the S. albulus strains.
ɛ-PL-degrading activities in S. albulus CR1 and its derivatives including the CRM004 strain were investigated by incubating mycelia with ɛ-PL (1 mg ml−1) in 100 mM MES buffer (pH 6.0). The CR1 strain degraded ɛ-PL (25- to 35-mer) by the catalytic functions of Pld and PldII, and its complete digestion was observed in a 3-h incubation (Fig. 3b). Similarly, the pld-inactivated CRM001 also showed ɛ-PL-degrading activity (Fig. 3c), which was in good agreement with a previous study (5). In contrast, the degrading profiles of the PldII-null mutants CRM003 and CRM004 differed (Fig. 3d and e). In particular, pld- and pldII-inactivated CRM004 showed negligible ɛ-PL-degrading activity, whereas CRM003 only slightly degraded ɛ-PL. The strain CRM003 harboring the pLAE006-pldII plasmid, in which the pldII gene was expressed under the control of the ermE* constitutive promoter, showed a restored ɛ-PL-degrading activity (Fig. 3f). This complementation experiment demonstrated that the pldII gene on the genome of strain CRM003 was inactivated due to downstream polar effects. These results clearly demonstrated that PldII is the major ɛ-PL-degrading enzyme produced by S. albulus NBRC14147.
ɛ-PL productivities of the mutant strain.
It was expected that the productivity and/or the chain length of ɛ-PL produced by CRM004 would be altered because this strain lacks ɛ-PL-degrading activity. To confirm this, the CR1 and CRM004 strains were cultivated in M3G medium. However, contrary to expectations, these strains showed identical ɛ-PL productivities and chain lengths (data not shown). As described earlier, the production and accumulation of ɛ-PL are detected when the pH value of the fermentation broth in which it is growing decreases from its initial pH value (approximately 7.0) to its self-stabilized final value (approximately 3.2), and ɛ-PL-degrading activity is mainly detected on the cell surface at neutral pH (5). The self-stabilized acidic pH condition was believed to be an important fermentation parameter inhibiting ɛ-PL degradation on the cell surface, thereby resulting in its production and accumulation. However, considering the fact that the optimum pH for l-lysine polymerization catalyzed by Pls is 8.5 (22), the productivity and/or chain length of ɛ-PL in strain CRM004 would be altered in a medium with a pH higher than 3.2.
To examine the pH effect directly, strain CRM004 was incubated in citrate buffer at various pH values (3.8 to 5.5) (Fig. 5) because S. albulus NBRC14147 can produce ɛ-PL in citrate buffer containing only glucose and ammonium sulfate as a carbon and nitrogen source, respectively (using the two-stage fermentation method as described in Materials and Methods). Similar ɛ-PL productivity levels were again observed in the CRM004 and CR1 strains although the former did show negligible ɛ-PL-degrading activity (Fig. 5). The chain length diversity of ɛ-PL was also unchanged (data not shown). In addition, the ɛ-PL productivity of CRM004 was decreased at pH values above 4.7, demonstrating that S. albulus NBRC14147 does not produce ɛ-PL under neutral pH conditions. In general, fermentation by microorganisms is characterized by the accumulation of organic acids and the accompanying reduction in pH. However, organic acids are not involved in the Pls catalytic machinery. This suggests another reason why the production and accumulation of ɛ-PL occurred under acidic pH conditions.
FIG. 5.
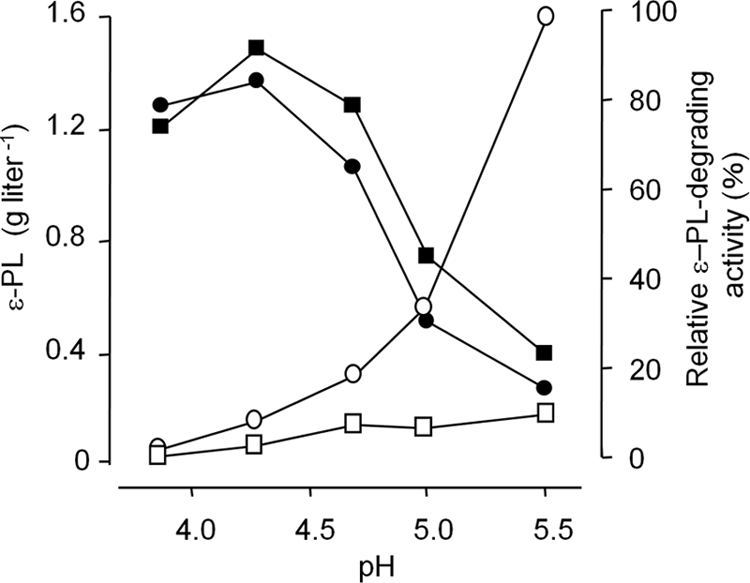
ɛ-PL productivities and ɛ-PL-degrading activities of the CR1 and CRM004 strains. Strains CR1 (circles) and CRM004 (squares) were incubated with 100 mM citrate buffer (pH 3.8 to 5.5) containing 5% glucose and 1% (NH4)2SO4. After incubation with shaking at 30°C for 20 h, the supernatants and mycelia were obtained by centrifugation. The supernatants were used for the measurements of the ɛ-PL concentrations (closed symbols). The mycelia were washed and resuspended with 100 mM citrate buffer (pH 3.8 to 5.5) without glucose and ammonium sulfate, and then ɛ-PL (1 mg/ml) was added and incubated at 30°C for 3 h to investigate ɛ-PL-degrading activities (open symbols).
Gene expression analysis by quantitative RT-PCR in S. albulus NBRC14147.
Strict culture broth pH control is essential to obtain high ɛ-PL productivity (9). Optimized fermentation was divided into two phases: in the first, cell growth was accelerated by maintaining the pH at values above 5.0; in the second, the pH was maintained at around 4.0 to achieve the highest ɛ-PL productivity. In the present study, we demonstrated that acidic pH conditions are not required to inhibit ɛ-PL-degrading enzymes, as described earlier. To investigate the regulation mechanisms of ɛ-PL biosynthesis, S. albulus NBRC14147 was cultivated in M3G medium under strict pH control (Fig. 6 a), and total RNA was isolated after 9, 11, 13, 15, and 24 h (Fig. 6b). Quantitative RT-PCR (Fig. 6c) revealed that the ask gene (6), which encodes the rate-limiting enzyme of l-lysine biosynthesis, Ask, was transcribed throughout the cultivation (Fig. 6c). pls transcription was not detected in the early growth phase (∼9 h) but appeared at 11 h, showing that pls was transcriptionally regulated to operate during the mid-log and stationary phases of growth. In this experiment, ɛ-PL production was initiated at 20 h, revealing that Pls was expressed 9 h before the ɛ-PL production occurred (Fig. 6a). In addition, accumulated intracellular ATP levels were observed during the stationary phase of growth.
FIG. 6.

Transcriptional analysis of the pls gene. (a) S. albulus NBRC14147 was cultivated in M3G medium in a 3-liter bench-scale jar fermentor under strict pH control. (b) Total RNA was isolated from mycelia grown at 30°C for 9, 11, 13, 15, and 24 h and analyzed in formaldehyde-agarose gels (2.5 μg per lane). (c) Quantitative RT-PCR was performed.
Steady-state kinetics of Pls.
Pls showed a typical hyperbolic saturation curve with respect to l-lysine concentration at a fixed ATP concentration, and Km and Vmax values for l-lysine at 30°C were calculated to be 70.9 ± 10.7 μM and 6,607 ± 324 pmol−1 s−1 mg−1, respectively (Fig. 7 a). A double-sigmoid curve was obtained for ATP at a fixed l-lysine concentration (Fig. 7b), suggesting that Pls is allosterically regulated by ATP. In fact, two Hill coefficients (nH) were calculated from the graph: 0.72 (0.25 to 2 mM ATP) and 3.26 (3 to 5 mM ATP). Although the regulation mechanism was complicated, the affinity of Pls for ATP represented by the substrate concentration that yield half-maximal velocity (S0.5), which is synonymous with Km on allosteric enzymes, was low compared with that of l-lysine (estimated to be approximately 2 mM).
FIG. 7.

Kinetic analysis of Pls for l-lysine (a) and ATP (b). Insets of show the Lineweaver-Burk plot (a) and the Hill plot (b). Each value represents the mean of three experiments. Error bars are not shown because relative standard deviations of less than 5% were commonly calculated.
MIC values of ɛ-PL for the S. albulus strains.
To confirm the biological function of the pldII gene, we measured the MICs of ɛ-PL for the CR1, CRM001, CRM003, and CRM004 strains under acidic and neutral pH conditions. All strains showed resistance to ɛ-PL (>10 mg/ml) at pH 4.5. In contrast, they were sensitive to ɛ-PL at pH 7.0, and the MIC values for the CR1, CRM001, CRM003, and CRM004 strains were determined as 313, 156, 78, and 39.6 μg/ml, respectively.
DISCUSSION
The present study identified the pldII gene encoding the major ɛ-PL-degrading enzyme and showed it to be translationally coupled with the pls gene (accession number AB385841) (Fig. 2a). The primary structure of the pldII gene product showed similarity to Pld (Fig. 2b). PldII activity was detected on the cell surface but not in the supernatant of the culture broth, suggesting that PldII bound to the outside of cytoplasmic membranes. Furthermore, like Pld (accession number AB243405), the presence of the Zn2+-binding motif HEXXH and of the N-terminal signal peptide in the gene product was indicated by a database search using PROSITE (http://www.expasy.org/prosite) and the SignalP, version 3.0, server (http://www.cbs.dtu.dk/services/SignalP), respectively.
The pldII-null mutants, CRM003 and CRM004, did not produce any short-chain oligomers (Fig. 3). In particular, CRM004, in which the pld and pldII genes were both inactivated, showed negligible ɛ-PL-degrading activity and released no l-lysine monomers during degradation (data not shown), whereas CRM003 released l-lysine monomers through exo-type degrading activity caused by Pld (data not shown). These observations clearly demonstrated that the mode of ɛ-PL degradation with PldII was endo-type activity, in contrast to the mode of Pld. Our results further revealed that S. albulus NBRC14147 produces no ɛ-PL-degrading enzyme(s) other than Pld and PldII.
To date, it has been unclear whether the 25 to 35 l-lysine residues of ɛ-PL represent the original chain length of the polymer product synthesized by Pls in vivo because it can be generated via differential degradation of a longer-chain ɛ-PL. However, our findings clearly show that Pls indeed produces the ɛ-PL consisting of 25 to 35 l-lysine residues in vivo.
Pls was found to be a membrane protein with an adenylation domain and thiolation domains characteristic of NRPSs (22). It has no traditional condensation or thioesterase domain; instead, its six transmembrane domains surround three tandem soluble domains. These tandem domains iteratively catalyzed l-lysine polymerization using free l-lysine polymer (or monomer in the initial reaction) as the acceptor and T-domain-bound l-lysine as the donor, directly yielding chains of diverse lengths. Using this machinery, l-lysine molecules are polymerized via multiple enzymatic reactions: adenylation, thiolation, and peptide formations. However, Pls showed a typical hyperbolic saturation curve with respect to l-lysine concentration at a fixed ATP concentration (Fig. 7a).
Based on the kinetics studies, the affinity of ATP for Pls was found to be much lower than that of l-lysine. Moreover, considering the calculated nH values, a low concentration (0.25 to 2 mM) and a high concentration (3 to 5 mM) of ATP showed negative and positive cooperative allosteric interactions, respectively, implying that Pls requires a large amount of ATP for full activity. This was strongly supported by the fermentation profiles. Although S. albulus NBRC14147 grew slowly during the stationary phase (at an acidic pH), large amounts of glucose and dissolved oxygen in the medium were still consumed (Fig. 6). These results indicated that the glycolytic pathway and electron transport chain were still active and that almost all ATP generated was not used for cell growth but, instead, accumulated to be used as a cofactor of Pls in ɛ-PL biosynthesis. In fact, the accumulated ATP level was observed when ɛ-PL was produced (Fig. 6a).
Taken together, these findings indicate that expressed Pls in the early growth phase (Pls was expressed 9 h earlier than ɛ-PL) is negatively regulated due to low intracellular ATP levels, thereby producing no ɛ-PL; in other words, the allosteric regulation of Pls avoids excess consumption of ATP for primary metabolism in the early growth phase. Thus, the concentration of intracellular ATP is a key regulatory factor for ɛ-PL biosynthesis. In contrast, considering the fact that the l-lysine affinity for Pls is relatively high, the l-lysine biosynthetic pathway should provide sufficient l-lysine for ɛ-PL biosynthesis (Fig. 6).
We further investigated the biological function of the pldII gene. Most self-resistant genes for antibiotic compounds produced by actinomycetes exist in a cluster with biosynthetic genes, and this was also expected for pldII and ɛ-PL. We therefore measured the MIC values of ɛ-PL for strains CR1, CRM001, CRM003, and CRM004. The antibiotic activities of ɛ-PL were observed under the neutral pH condition (pH 7.0), and pldII gene inactivation led to diminished resistance to ɛ-PL. In contrast, ɛ-PL exhibited no antibiotic activity against all strains at pH 4.5, revealing that ɛ-PL is inhibited by an acidic pH. This was in good agreement with the fact that the ɛ-PL-sensitive strain CRM004 can produce ɛ-PL. Considering that S. albulus NBRC14147 produces no ɛ-PL under neutral pH conditions in nature, ɛ-PL-degrading enzymes (Pld and PldII) should be involved in the resistance to an exogenous ɛ-PL rather than to its own ɛ-PL. In particular, the biological role of PldII is much more important.
In conclusion, our results reveal that the chain length of ɛ-PL is determined not by ɛ-PL-degrading enzymes but by Pls itself and that the catalytic function of Pls is regulated by intracellular ATP levels. S. albulus NBRC14147 appears to be an intriguing and high-ATP-producing microorganism.
Acknowledgments
This work was supported, in part, by KAKENHI (20780057), a Grant-in-Aid for Young Scientists (B) from the Japan Society for the Promotion of Science (to Y.H.), and by the Industrial Technology Research Grant Program in 2008 from the New Energy and Industrial Technology Development Organization of Japan (to Y.H.).
Footnotes
Published ahead of print on 2 July 2010.
REFERENCES
- 1.Bagnara, A. S., and Finch, L. R. 1972. Quantitative extraction and estimation of intracellular nucleoside triphosphates of Escherichia coli. Anal. Biochem. 45:24-34. [DOI] [PubMed] [Google Scholar]
- 2.Hamano, Y., T. Dairi, M. Yamamoto, T. Kuzuyama, N. Itoh, and H. Seto. 2002. Growth-phase dependent expression of the mevalonate pathway in a terpenoid antibiotic-producing Streptomyces strain. Biosci. Biotechnol. Biochem. 66:808-819. [DOI] [PubMed] [Google Scholar]
- 3.Hamano, Y., I. Nicchu, Y. Hoshino, T. Kawai, S. Nakamori, and H. Takagi. 2005. Development of gene delivery systems for the ɛ-poly-L-lysine producer, Streptomyces albulus. J. Biosci. Bioeng. 99:636-641. [DOI] [PubMed] [Google Scholar]
- 4.Hamano, Y., C. Maruyama, and H. Kimoto. 2006. Construction of a knockout mutant of the streptothricin-resistance gene in Streptomyces albulus by electroporation. Actinomycetologica 20:35-41. [Google Scholar]
- 5.Hamano, Y., T. Yoshida, M. Kito, S. Nakamori, T. Nagasawa, and H. Takagi. 2006. Biological function of the pld gene product that degrades ɛ-poly-l-lysine in Streptomyces albulus. Appl. Microbiol. Biotechnol. 72:173-181. [DOI] [PubMed] [Google Scholar]
- 6.Hamano, Y., I. Nicchu, T. Shimizu, Y. Onji, J. Hiraki, and H. Takagi. 2007. ɛ-Poly-l-lysine producer, Streptomyces albulus, has feedback-inhibition resistant aspartokinase. Appl. Microbiol. Biotechnol. 76:873-882. [DOI] [PubMed] [Google Scholar]
- 7.Hiraki, J., M. Hatakeyama, H. Morita, and Y. Izumi. 1998. Improved ɛ-poly-l-lysine production of an S-(2-aminoethyl)-l-cysteine resistant mutant of Streptomyces albulus. Seibutu Kougaku Kaishi 76:487-493. (In Japanese.) [Google Scholar]
- 8.Itzhaki, R. F. 1972. Colorimetric method for estimating polylysine and polyarginine. Anal. Biochem. 50:569-574. [DOI] [PubMed] [Google Scholar]
- 9.Kahar, P., T. Iwata, J. Hiraki, E. Y. Park, and M. Okabe. 2001. Enhancement of ɛ-polylysine production by Streptomyces albulus strain 410 using pH control. J. Biosci. Bioeng. 91:190-194. [DOI] [PubMed] [Google Scholar]
- 10.Kito, M., R. Takimoto, T. Yoshida, and T. Nagasawa. 2002. Purification and characterization of an ɛ-poly-l-lysine-degrading enzyme from an ɛ-poly-l-lysine-producing strain of Streptomyces albulus. Arch. Microbiol. 178:325-330. [DOI] [PubMed] [Google Scholar]
- 11.Marahiel, M. A., T. Stachelhaus, and H. D. Mootz. 1997. Modular peptide synthases involved in nonribosomal peptide synthesis. Chem. Rev. 97:2651-2673. [DOI] [PubMed] [Google Scholar]
- 12.Mootz, H. D., D. Schwarzer, and M. A. Marahiel. 2002. Ways of assembling complex natural products on modular nonribosomal peptide synthetases. Chembiochem 3:490-504. [DOI] [PubMed] [Google Scholar]
- 13.Oppermann-Sanio, F. B., and A. Steinbüchel. 2002. Occurrence, functions and biosynthesis of polyamides in microorganisms and biotechnological production. Naturwissenschaften 89:11-22. [DOI] [PubMed] [Google Scholar]
- 14.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 15.Schwarzer, D., R. Finking, and M. A. Marahiel. 2003. Nonribosomal peptides: from genes to products. Nat. Prod. Rep. 20:275-287. [DOI] [PubMed] [Google Scholar]
- 16.Shima, S., and H. Sakai. 1977. Polylysine produced by Streptomyces. Agric. Biol. Chem. 41:1807-1809. [Google Scholar]
- 17.Shima, S., and H. Sakai. 1981. Poly-l-lysine produced by Streptomyces. II. Taxonomy and fermentation studies. Agric. Biol. Chem. 45:2497-2502. [Google Scholar]
- 18.Shima, S., and H. Sakai. 1981. Poly-l-lysine produced by Streptomyces. III. Chemical studies. Agric. Biol. Chem. 45:2503-2508. [Google Scholar]
- 19.Shima, S., S. Oshima, and H. Sakai. 1983. Biosynthesis of ɛ-poly-l-lysine by washed mycelium of Streptomyces albulus no. 346. Nippon Nogeikagaku Kaishi 57:221-226. [Google Scholar]
- 20.Shima, S., H. Matsuoka, T. Iwamoto, and H. Sakai. 1984. Antimicrobial action of ɛ-poly-l-lysine. J. Antibiot. (Tokyo) 37:1449-1455. [DOI] [PubMed] [Google Scholar]
- 21.Walsh, C. 2003. Antibiotics: action, origins, resistance. ASM Press, Washington, DC.
- 22.Yamanaka, K., C. Maruyama, H. Takagi, and Y. Hamano. 2008. Epsilon-poly-l-lysine dispersity is controlled by a highly unusual nonribosomal peptide synthetase. Nat. Chem. Biol. 4:766-772. [DOI] [PubMed] [Google Scholar]