

Abstract
Harnessing endogenous cardioprotectants is a novel therapeutic strategy to combat ischemia/reperfusion (I/R) injury. Thrombin causes I/R injury, whereas exogenous adenosine prevents I/R injury. We hypothesized that blocking thrombin receptor activation with a protease-activated receptor (PAR) 4 antagonist would unmask the cardioprotective effects of endogenous adenosine. The protective role of two structurally unrelated PAR4 antagonists, trans-cinnamoyl-YPGKF-amide (tc-Y-NH2) and palmitoyl-SGRRYGHALR-amide (P4pal10), were evaluated in two rat models of myocardial I/R injury. P4pal10 (10 μg/kg) treatment before ischemia significantly decreased infarct size (IS) by 31, 21, and 19% when given before, during, and after ischemia in the in vivo model. tc-Y-NH2 (5 μM) treatment before ischemia decreased IS by 51% in the in vitro model and increased recovery of ventricular function by 26%. To assess whether the cardioprotective effects of PAR4 blockade were due to endogenous adenosine, isolated hearts were treated with a nonselective adenosine receptor blocker, 8-sulfaphenyltheophylline (8-SPT), and tc-Y-NH2 before ischemia. 8-SPT abolished the protective effects of tc-Y-NH2 but did not affect IS when given alone. Adenosine-mediated survival pathways were then explored. The cardioprotective effects of tc-Y-NH2 were abolished by inhibition of Akt (wortmannin), extracellular signal-regulated kinase 1/2 [PD98059 (2′-amino-3′-methoxyflavone)], nitric-oxide synthase [NG-monomethyl-L-arginine (L-NMA)], and KATP channels (glibenclamide). PD98059, L-NMA, and glibenclamide alone had no effect on cardioprotection in vitro. Furthermore, inhibition of mitochondrial KATP channels [5-hydroxydecanoic acid (5-HD)] and sarcolemmal KATP channels (sodium (5-(2-(5-chloro-2-methoxybenzamido)ethyl)-2-methoxyphenylsulfonyl)(methylcarbamothioyl)amide; HMR 1098) abolished P4pal10-induced cardioprotection in vivo. Thrombin receptor blockade by PAR4 inhibition provides protection against injury from myocardial I/R by unmasking adenosine receptor signaling and supports the hypothesis of a coupling between thrombin receptors and adenosine receptors.
Limiting infarct size by timely reperfusion is critical to improve outcomes in patients with myocardial infarction. In contrast, reperfusion may increase infarct size, a phenomenon known as ischemia/reperfusion (I/R) injury. Exogenously delivered pharmacological agents, such as adenosine, have been shown to limit infarct size resulting from I/R in experimental studies. However translating these studies into clinical practice has been disappointing. Therefore, harnessing endogenous cardioprotectants may be a novel therapeutic strategy to limit I/R injury.
Adenosine is an endogenous nucleoside present in normal conditions; however, during ischemia, the production of adenosine increases severalfold (Shryock and Belardinelli, 1997). Adenosine activates four well characterized G protein-coupled receptors (GPCRs) to produce various physiological effects that attenuate many of the proposed mechanisms of I/R injury (Headrick et al., 2003a). However, the specific adenosine receptor (AR) subtype (A1AR, A2aAR, A2bAR, or A3AR) responsible for adenosine-mediated cardioprotective effects remains unclear. The signaling pathway for adenosine-mediated cardioprotection involves the activation of PI3K/Akt, ERK1/2, nitric-oxide synthase (NOS), and the sarcolemmal (sarc) or mitochondrial (mito) ATP-sensitive potassium channels (KATP channels) (Headrick et al., 2003b). In experimental models as well as in small clinical studies, administration of adenosine with reperfusion results in a marked reduction in infarct size and improvement in ventricular function. In the larger AMISTAD and AMISTAD II trials, a 3-h infusion of adenosine as an adjunct to reperfusion resulted in a striking reduction in infarct size (55–65%) but did not significantly affect clinical outcome (Mahaffey et al., 1999; Ross et al., 2005). Despite these potential cardioprotective actions, adenosine treatment has not been incorporated into standard clinical practice. Adenosine has a very short half-life, necessitating a continuous infusion. Not only is this inconvenient, but it increases the chance of side effects, such as bradycardia and hypotension. Harnessing endogenous adenosine provides a novel treatment strategy to reduce I/R, which would eliminate the side effects and expense of adenosine infusions.
Thrombin contributes to I/R injury independently of its effects on platelets and fibrinogen (Chong et al., 2003; Strande et al., 2007). Thrombin activates the seven trans-membrane G protein-coupled protease-activated receptors (PARs) 1 and 4 by proteolytic cleavage of the N terminus to expose a tethered ligand, which then transactivates the receptor (Ossovskaya and Bunnett, 2004). Thrombin is a non-selective activator of PARs; therefore, in experimental systems, synthetic activating peptides (APs) containing 5- to 14-amino acid residues that correspond to the tethered ligand are used to selectively activate each receptor. The peptide AYPGKF-amide is a PAR4 AP. PAR1 and PAR4 were originally detected in platelets where their activation causes platelet activation and aggregation. As a result, they have been identified in a variety of cell types and tissues and have been shown to modulate a variety of physiological processes. We have recently shown that PAR1 inhibition protects against myocardial I/R injury by recruiting cardioprotective pathways including PI3K/Akt, NOS, and KATP channels (Strande et al., 2007). However, the role of PAR4 in determining the extent of myocardial I/R injury is not known. Therefore, we focused on PAR4 in this study to determine whether PAR4 inhibition is cardioprotective.
Adenosine has been shown to modulate thrombin signaling in various models. For instance, in vein endothelial cells, adenosine is able to down-regulate tissue factor induction by thrombin (Deguchi et al., 1998). Additionally in corneal endothelium, adenosine opposes thrombin-mediated inhibition of calcium wave propagation (D’Hondt et al., 2007). However, the mechanism of this adenosine and thrombin receptor “cross-talk” is poorly understood. Furthermore, it is not known whether this receptor cross-talk occurs in the myocardium. Therefore, we proposed that by blocking thrombin activation pathways with a PAR4 antagonist, endogenous adenosine would be allowed to exert its cardioprotective effects.
We used both an in vivo rat model and an in vitro isolated rat heart model of I/R injury to assess the effects of PAR4 blockade on infarct size. The absence of blood in the isolated heart model eliminated the possible interference of coagulation as well as platelet activation in mediating injury. We studied the ability of two structurally different PAR4 inhibitors, trans-cinnamoyl-YPGKF-amide (tc-Y-NH2) and palmitoyl-SGRRY-GHALR-amide (P4pal10), to protect the heart against injury from I/R. We then assessed the role of adenosine by blocking its effects with a nonselective adenosine receptor antagonist. Furthermore, based on the assumption that antagonizing PAR4 signaling would unmask AR signaling, we confirmed the signaling pathway leading to protection was similar to that previously reported for AR activation. Therefore, we explored the adenosine-mediated cardioprotective pathways including Akt, ERK1/2, NOS, sarc, and mitoKATP channels.
Materials and Methods
Male Sprague-Dawley (SD) rats at 8 weeks of age used in this study received humane care in compliance with the Guide for the Care and Use of Laboratory Animals formulated by the National Research Council (1996). tc-Y-NH2 was obtained from Peptides International (Louisville, KY), AYPGKF-amide was obtained from Bachem (King of Prussia, PA), and P4pal10 was synthesized by GenScript (Piscataway, NJ). Lepirudin (recombinant hirudin; Bayer HealthCare Pharmaceuticals, West Haven, CT) was obtained through the Froedtert Hospital Pharmacy (Milwaukee, WI). All other chemicals and reagents were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise specified.
PCR
Rat heart PCR Ready First Strand cDNA was purchased from BioChain Institute, Inc. (Hayward, CA). This consists of cDNA reverse transcribed from mRNA pooled from three rat hearts. Forward and reverse primers used for amplifying rat PAR4 were prepared commercially, based on published data (Rohatgi et al., 2004): sense, 5′-GGA ATG CCA GAC GCC CAG CAT C-3′; and antisense, 5′-GGT GAG GCG TTG ACC ACG CA-3′ (PCR product, 559 bp). The conditions for amplification were 95°C for 10 min for one cycle, 94°C for 30 s, 64°C for 90 s, 72°C for 60 s for 40 cycles, and 72°C for 10 min for 1 cycle. One microliter of cDNA was used in each reaction along with iTaq DNA Polymerase and dNTP mix (Bio-Rad, Hercules, CA). Electrophoresis was conducted on a 1.2% agarose gel stained with SYBR Green.
Isolation of Rat Cardiac Fibroblasts and Cardiomyocytes
Cardiomyocytes from SD rats were a generous gift from Eugene Konorev (Medical College of Wisconsin, Milwaukee, WI). They were isolated according to previously published methods (Piper et al., 1990). Cardiac fibroblasts from SD rats were isolated as described previously (Dubey et al., 1997).
Immunoblot Analysis
For PAR4 protein detection, heart homogenates and immunoblot analysis were performed using methods described previously (Strande et al., 2007), and immunoblots were incubated with a 1:200 dilution of the primary antibody [PAR4 (C-20), catalog number sc-8464; Santa Cruz Biotechnology, Inc., Santa Cruz, CA]. For phosphorylated Akt and ERK1/2 detection, the following alterations were made. The free wall of the left ventricle from each group (control, tc-Y-NH2, wortmannin, and PD98059) were harvested for protein extraction either immediately after perfusion with the compound or after 30-min ischemia followed by 5 min of reperfusion. After the proteins were separated by SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes, the membrane was blocked in 5% bovine serum albumin in Tris-buffered saline/0.1% Tween 20. Immunoblots were then incubated with a 1:1000 dilution of either phospho-Akt (Ser473) or phospho-p44/42 mitogen-activated protein kinase (Thr202/Tyr204) antibodies from Cell Signaling Technology (Danvers, MA). An antibody against β-actin (Sigma-Aldrich) or glycer-aldehyde-3-phosphate dehydrogenase (Cell Signaling Technology) was used to standardize protein loading of each of the blots. A secondary antibody was then applied, followed with enhanced chemiluminescence solution (Amersham Life Science, Arlington Heights, IL), and exposed to film for 1 min.
P4pal10 and Cardioprotection Studies in Vivo
An in vivo anesthetized rat model was used for these experiments with the general surgical protocol and determination of infarct size as described previously (Peart and Gross, 2005). For infarct size studies, rats (n = 6/group) underwent 30 min of regional ischemia followed by 120 min of reperfusion. P4pal10 was administered i.v. over 1 min starting 15 min before ischemia, 15 min after the onset of ischemia, and 10 s after the onset of reperfusion in a separate series of experiments (n = 6 rats/group).
tc-Y-NH2 and Cardioprotection Studies in Vitro
Rats were anesthetized with a mixture containing pentobarbital sodium (50 mg/kg) and heparin (1000 IU/kg) i.p. Excised hearts were retrograde perfused through the aorta with a modified Krebs’ buffer and instrumented as described previously (Strande et al., 2007). In brief, coronary flow rate was determined by timed collection of the coronary effluent. A saline-filled latex balloon connected to a pressure transducer was inserted into the left ventricle, and baseline end-diastolic pressure was set at 5 to 10 mm Hg. Heart rate, left ventricle end-diastolic pressure, and left ventricular developed pressures (LVDPs) were recorded continuously. The measurements for postischemic recovery of LVDP used for comparison were taken at 180 min of reperfusion. After stabilization for 15 to 20 min, the hearts (n = 6/group) were subjected to 30 min of regional ischemia, followed by 180 min of reperfusion. Group 1 received no treatment (Fig. 1A). The hearts in group 2 were perfused continuously with different concentrations of tc-Y-NH2 from 15 min before coronary occlusion until occlusion (Fig. 1B). The hearts in group 3 were continuously perfused with an inhibitor (PAR4 AP, wortmannin, PD98059, L-NMA, or glibenclamide) starting 15 min before the commencement of tc-Y-NH2 perfusion (i.e., from 15 min before ischemia) for 30 min (Fig. 1C). The hearts in group 4 were continuously perfused with an inhibitor (PAR4 AP, wortmannin, PD98059, L-NMA, or glibenclamide) starting at 30 min before occlusion (Fig. 1D). The hearts in group 5 were continuously perfused for 15 min with tc-Y-NH2 and a nonspecific AR antagonist, 8-sulfaphenyltheophylline (8-SPT) (Fig. 1E). The results from groups 1 and 2 were used for comparison in Figs. 4 to 8.
Fig. 1.

Experimental protocols. All hearts were subjected to 30 min of regional ischemia after a 50-min stabilization period and followed by 180 min of reperfusion. The control group (A) received no treatment. The tc-Y-NH2 group (B) was continuously perfused with different concentrations of tc-Y-NH2 15 min before the onset of ischemia. The inhibitor groups were continuously perfused with an inhibitor (PAR4 AP, wortmannin, PD98059, L-NMA, or glibenclamide) for 30 min before ischemia with (C) or without (D) the addition of tc-Y-NH2 15 min before ischemia. E, 8-SPT was perfused with tc-Y-NH2 for the 15 min before ischemia.
Fig. 4.

Analysis of the cardioprotective effects of PAR4 inhibition in vitro. Concentration-response curve of tc-Y-NH2, a PAR4 antagonist. Rat hearts were perfused with buffer with increasing concentrations of tc-Y-NH2 (0, 1, 5, and 10 μM) for 15 min before 30-min regional ischemia and 180-min reperfusion. A, infarct size expressed as a percentage of AAR. B, recovery of LVDP. The protective effects of tc-Y-NH2 were abolished by a PAR4-activating peptide. Rat hearts were perfused with PAR4 AP (20 μM) for 15 min before the addition of tc-Y-NH2 (5 μM) and perfused an additional 15 min before 30 min of regional ischemia and 180-min reperfusion. C, infarct size expressed as a percentage of AAR. D, recovery of LVDP. Data are mean ± S.D., n = 6/group.*, P < 0.05, treated versus control.
Fig. 8.
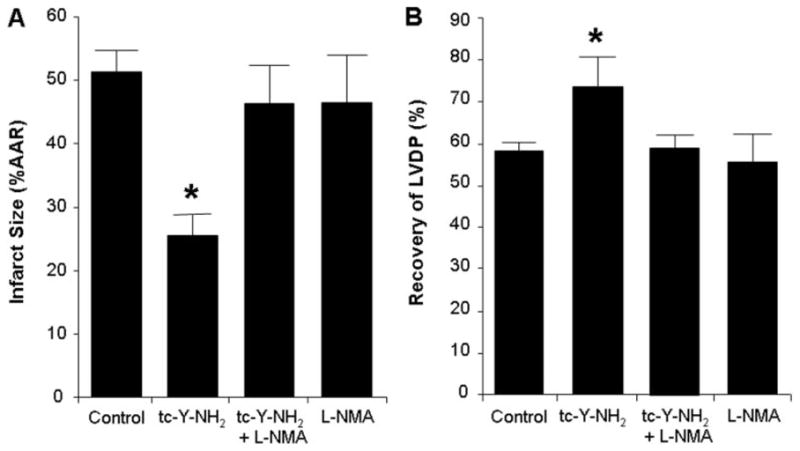
Nitric oxide is required for the cardioprotective effects of PAR4 inhibition. Inhibition of nitric oxide synthase (L-NMA) abolished the cardioprotective effect of the PAR4 antagonist tc-Y-NH2. Hearts were perfused with L-NMA (100 μM) for 15 min before the addition of tc-Y-NH2 (5 μM) and perfused an additional 15 min before 30-min regional ischemia and 180-min reperfusion. A, infarct size expressed as a percentage of AAR. B, percentage of recovery of LVDP. Data are mean ± S.D., n = 6/group. *, P < 0.05, treated versus control.
Infarct Induction and Measurement
To induce ischemia, a ligature was positioned around the left main coronary artery and threaded through a plastic snare to permit reversible occlusion of the coronary artery. Coronary occlusion was induced for 30 min by clamping the snare onto the heart, and reperfusion was achieved by releasing the snare. At the end of 180-min reperfusion, the coronary artery was reoccluded, and the risk zone was delineated by perfusion of 0.5% Evans blue into the aortic cannula. Hearts were sectioned and incubated in 1% triphenyltetrazolium chloride in phosphate buffer, pH 7.4 (37°C) for 15 min to define white necrotic tissue when fixed in 10% formalin for 24 h. Area at risk (AAR) and infarct-to-risk ratios were determined by computerized planimetry using J-Image version 1.6 software (National Institutes of Health, Bethesda, MD) (Strande et al., 2007).
Statistical Analysis
Six rats/hearts were included in each group. Data reported are mean ±S.D. Statistical analysis was performed by use of repeated measures analysis of variance with the Greenhouse-Geisser adjustment used to correct for the inflated risk of a type I error (Strande et al., 2007). If significant, the Mann-Whitney test was used as a second step to identify which groups were significantly different (Strande et al., 2007). Significance was set at P < 0.05.
Results
Presence of PAR4 in the Rat Heart
cDNA reverse transcribed from pooled rat heart mRNA was amplified using primers specific for the rat PAR4 sequence. PAR4 mRNA is constitutively expressed in the rat heart (Fig. 2A, lane 1). The presence of PAR4 protein in whole-heart homogenates obtained from SD rats was then detected by immunoblotting (Fig. 2B, lanes 2–3). PAR4 was also expressed in isolated cardiomyocytes but not cardiac fibroblasts (Fig. 2B, lanes 1 and 5).
Fig. 2.

Analysis of PAR4 expression by reverse transcription-PCR and immunoblotting rat heart tissue. A, PCR of rat heart cDNA with PAR4-specific primers. Lane 1, 100-bp ladder. PAR4 (559 bp, lane 2) was detected in pooled cDNA from three rat hearts. Lane 3, negative control that contains the PAR4 primers but no cDNA. β-Actin (838 bp, lane 4) is an internal control. B, determination of PAR4 protein by immunoblot analysis of rat heart ventricles (lanes 2 and 3) and isolated cardiomyocytes (lane 5) and cardiac fibroblasts (lane 1). Glyceraldehyde-3-phosphate dehydrogenase is the loading control.
PAR4 Inhibition and Cardioprotection in Vivo
We used P4pal10, a synthetic peptide that blocks the intracellular signaling of PAR4 (Covic et al., 2002), to determine whether PAR4 inhibition limited I/R injury in an in vivo rat model. We first performed a dose-response analysis (0.1–100 μg/kg) to determine the optimal protective dose. The principal endpoint of this study was infarct size, expressed as a percentage of the AAR. Infarct size was 64 ± 2% of the AAR in the control group. No significant change in infarct size was detected with the 0.1, 30, and 100 μg/kg doses of P4pal10. However, a graded reduction was seen with the 1 to 10 μg/kg doses, and 10 μg/kg was the optimally effective dose (Fig. 3A). These hearts had an infarct size of 43 ± 5% of the AAR, which is a 31% reduction in infarct size compared with the control. Heart rate and blood pressures were monitored throughout the procedure, and there were no significant differences between baseline hemodynamics between groups. Mean arterial pressure decreased during ischemia and reperfusion in all groups, but there was no significant difference between groups (data not shown).
Fig. 3.

Analysis of the cardioprotective effects of PAR4 inhibition in vivo. A, dose-response curve of P4pal10, a PAR4 antagonist. Rats were treated with either saline or increasing doses of P4pal10 (0.1, 1, 3, 10, 30, and 100 μg/kg) administered as an i.v. bolus 15 min before ischemia. B, phase action of P4pal10 on cardioprotection in the rat. Rats were treated with either saline or increasing doses of P4pal10 (10 μg/kg) 15 min before ischemia, 15 min after onset of ischemia, or 10 s after onset of reperfusion. Data are mean ± S.D., n = 6/group. *, P < 0.05, treated versus control.
Clinically, patients generally receive medical treatment for acute myocardial infarction after the onset of symptoms. Thus, we determined whether P4pal10 reduces infarct size when given during ischemia or at reperfusion. Rats were treated with an i.v. bolus of 10 μg/kg P4pal10 15 min after the onset of ischemia or 5 min after initiation of reperfusion. P4pal10 was able to reduce infarct size when administered during ischemia by 21% and at reperfusion by 19% compared with the control (Fig. 3B).
PAR4 Inhibition and Cardioprotection in Vitro
To eliminate the potential role of platelets contributing to the cardioprotective effect of PAR4 inhibition, we studied the effects of tc-Y-NH2 on I/R injury in an isolated heart model. Even though this model is buffer-perfused to wash out blood cells and components, we have recently shown that thrombin is present and active in the isolated heart and thus able to activate PAR4 (Strande et al., 2007). We first performed a concentration-response analysis, using 1, 5, or 10 μM, to determine the optimal protective dose for tc-Y-NH2. It has been shown previously that tc-Y-NH2 acts as a PAR4 antagonist by competing with the binding site of the tethered ligand (Hollenberg and Saifeddine, 2001).
Control hearts produced an infarct size of 51 ± 3% of the AAR (Fig. 4A). Continuous administration of tc-Y-NH2 for 15 min immediately before ischemia resulted in a concentration-dependent reduction of infarct size. Five micromolar tc-Y-NH2 led to the largest reduction in infarct size (25 ± 3%), a 49% decrease. Other concentrations of tc-Y-NH2 had no significant effect on infarct size.
Likewise, tc-Y-NH2 increased recovery of LVDP in a concentration-dependent manner. Again, the optimal concentration was 5 μM (Fig. 4B). There were no differences in hemodynamics at any time points measured between control and tc-Y-NH2-treated groups (data not shown).
The infarct-limiting effect of tc-Y-NH2 was abolished by cotreatment with 20 μM PAR4 AP (49 ± 4%; Fig. 4C). PAR4 AP did not affect infarct size (48 ± 5%) when given alone. Likewise, cotreatment with PAR4 AP also blocked the enhanced recovery of LVDP produced by tc-Y-NH2 (54 ± 4% preischemic LVDP; Fig. 4D) and did not affect LVDP when given alone (52 ± 4%).
P4pal10 was also used to inhibit PAR4 activation in this model. Although we saw a trend favoring a decrease in infarct size, the compound markedly reduced LVDP and coronary flow rate even before ischemia (data not shown). Therefore, we continued to use tc-Y-NH2 (5 μM) instead of P4pal10 for the remaining in vitro studies.
Role of Adenosine in PAR4 Antagonist-Mediated Cardioprotection in Vitro
We considered the possibility of adenosine being a trigger of the infarct-limiting effects observed with PAR4 blockade; therefore, we investigated the role of ARs with the use of 8-SPT (1 μM), a nonspecific antagonist of ARs. Perfusion of hearts with 8-SPT and tc-Y-NH2 for 15 min before ischemia resulted in an infarct size and LVDP similar to the values observed with the control hearts (Fig. 5, A and B). In other words, 8-SPT abolished the cardioprotective effects of tc-Y-NH2. 8-SPT alone did not affect infarct size or LVDP when given alone.
Fig. 5.
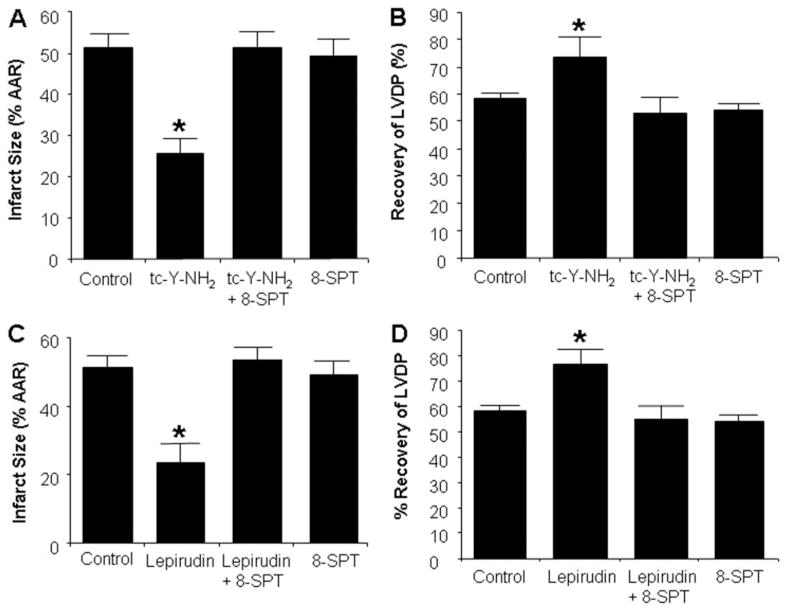
The cardioprotective effect of PAR4 and thrombin inhibition is dependent on endogenous adenosine. Inhibition of adenosine receptors (8-SPT) abolished the cardioprotective effects of the PAR4 antagonist tc-Y-NH2. Hearts were perfused with 8-SPT (1 μM) with or without tc-Y-NH2 (5 μM) for 15 min before 30-min regional ischemia and 180-min reperfusion. A, infarct size expressed as a percentage of AAR. B, percentage of recovery of LVDP. Data are mean ± S.D., n = 6/group. *, P < 0.05, treated versus control. The protective effect of thrombin inhibition is abolished by 8-SPT. Rat hearts were perfused with 8-SPT (1 μM) with or without lepirudin (1 U/ml) for 15 min before 30 min of regional ischemia and 180-min reperfusion. C, infarct size expressed as a percentage of AAR. D, percentage recovery of LVDP. Data are mean ± S.D., n = 6/group. *, P < 0.05, treated versus control.
We also investigated whether the cardioprotective effects of direct thrombin inhibition with lepirudin could be abrogated by 8-SPT. We have recently shown that lepirudin decreased I/R injury in rat hearts in vitro (Strande et al., 2007). Perfusion of rat hearts with 8-SPT and lepirudin (1 U/ml) for 15 min before ischemia abolished the protective effects of lepirudin (Fig. 5, C and D). Because adenosine protects the heart by activating survival pathways that include PI3K/Akt, ERK1/2, NOS, and KATP channels, we examined these pathways in PAR4 antagonist-induced cardioprotection.
Role of the PI3K/Akt Pathway in PAR4 Antagonist-Mediated Cardioprotection in Vitro
We used wortmannin, a PI3K inhibitor, to examine the potential role of the PI3K/Akt pathway in mediating the protection after PAR4 inhibition. Wortmannin (100 nM) when given alone had no effect on infarct size or LVDP. However, it abolished the cardioprotective effects of tc-Y-NH2 (Fig. 6, A and B). tc-Y-NH2 had no effect on phosphorylation of Akt before ischemia (data not shown). Akt phosphorylation was increased in the control and tc-Y-NH2-treated groups at reperfusion, but tc-Y-NH2 did not further increase the level of phosphorylation of Akt over the control levels during reperfusion. However, wortmannin significantly decreased Akt phosphorylation at reperfusion (Fig. 6, C and D).
Fig. 6.

The cardioprotective effects of PAR4 inhibition are dependent on PI3K/Akt activation. Inhibition of PI3K/Akt [wortmannin (Wort)] abolished the cardioprotective effects of the PAR4 antagonist tc-Y-NH2. Hearts were perfused with Wort (100 nM) for 15 min before the addition of tc-Y-NH2 (5 μM) and perfused an additional 15 min before 30-min regional ischemia and 180-min reperfusion. A, infarct size expressed as a percentage of AAR. B, percentage of recovery of LVDP. Data are mean ± S.D., n = 6/group. *, P < 0.05, treated versus control. PAR4 inhibition did not increase activation of Akt after 5 min of reperfusion as measured by phosphorylation of Akt. Rat hearts were perfused with Wort (100 nM) or -tc-Y-NH2 (5 μM) for 15 min before 30min regional ischemia. The free wall of the left ventricle was harvested for protein extraction after 5 min of reperfusion. C, immunoblot for phosphorylated Akt. β-actin is the loading control. D, quantitation of phosphorylated Akt when normalized to β-actin. Data are mean ± S.D., n = 3/group. *, P < 0.05, treated versus control.
Role of the ERK Pathway in PAR4 Antagonist-Mediated Cardioprotection in Vitro
Isolated hearts were pre-treated with the ERK1/2 inhibitor, PD98059 (10 μM), alone or in combination with tc-Y-NH2. PD98059 abrogated the cardioprotective effect of tc-Y-NH2 (Fig. 7, A and B). PD98059 alone had no effect on cardioprotection.
Fig. 7.

ERK1/2 is required for the cardioprotective effects of PAR4 inhibition, but it is not activated by tc-Y-NH2. Inhibition of ERK1/2 (PD98059) abolished the cardioprotective effects of the PAR4 antagonist tc-Y-NH2. Hearts were perfused with PD98059 (10 μ M) for 15 min before the addition of tc-Y-NH2 (5 μM) and perfused an additional 15 min before 30-min regional ischemia and 180-min reperfusion. A, infarct size expressed as a percentage of AAR. B, percentage of recovery of LVDP. Data are mean ± S.D., n = 6/group. *, P < 0.05, treated versus control. PAR4 inhibition did not increase activation of ERK1/2 after 5 min of reperfusion as measured by phosphorylation of ERK1/2. Rat hearts were perfused with PD98059 (10 μM) or tc-Y-NH2 (5 μM) for 15 min before 30 min of regional ischemia. The free wall of the left ventricle was harvested for protein extraction after 5 min of reperfusion. C, immunoblot for phosphorylated ERK1/2. β-actin is the loading control. D, quantitation of phosphorylated ERK2 when normalized to β-actin. E, quantitation of phosphorylated ERK1 when normalized to β-actin. Data are mean ± S.D., n = 3/group. *, P < 0.05, treated versus control.
tc-Y-NH2 had no effect on phosphorylation of ERK1/2 before ischemia (data not shown) and did not further increase phosphorylation over control values after reperfusion. PD98059 significantly decreased ERK2 phosphorylation at reperfusion but did not significantly change ERK1 phosphorylation (Fig. 7, C and D).
Role of Nitric-Oxide Synthase in PAR4 Antagonist-Mediated Cardioprotection in Vitro
Blockade of NOS by L-NMA (100 μM) before ischemia had no effect on infarct size or LVDP; however, L-NMA abolished the cardioprotective effect of tc-Y-NH2 (Fig. 8, A and B). Nitric oxide (NO) levels were assessed by measuring tissue nitrite and nitrate levels after 1 h of reperfusion. No differences in nitrite or nitrate levels were detected in the tc-Y-NH2-treated hearts compared with the control hearts, although L-NMA decreased NO levels below control values (data not shown).
Role of KATP Channels in PAR4 Antagonist-Mediated Cardioprotection in Vivo and in Vitro
We examined the role of KATP channels in mediating the cardioprotective effects of both tc-Y-NH2 and P4pal10. Isolated hearts were perfused with the nonselective KATP channel blocker, glibenclamide (3 μM), alone for 15 min or in combination with tc-Y-NH2 for 15 min before ischemia. Glibenclamide completely abolished the cardioprotective effect of tc-Y-NH2 (Fig. 9, A and B). Glibenclamide alone had no effect on infarct size.
Fig. 9.
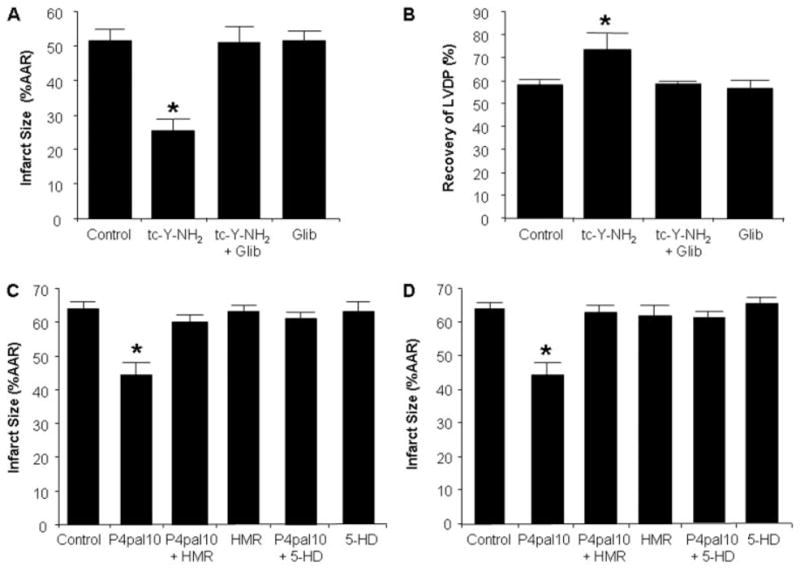
The cardioprotective effects of PAR4 inhibition are mediated through opening of KATP channels. Inhibition of KATP channels [glibenclamide (Glib)] abolished the cardioprotective effect of the PAR4 antagonist tc-Y-NH2. Hearts were perfused with Glib (3 μM) for 15 min before the addition of tc-Y-NH2 (5 μM) and perfused an additional 15 min before 30-min regional ischemia and 180-min reperfusion. A, infarct size expressed as a percentage of AAR. B, percentage of recovery of LVDP. Data are mean ± S.D., n = 6/group. *, P < 0.05, treated versus control. The protective effects of P4pal10 was abolished by blocking either sarcolemmal KATP channels (HMR; 3 mg/kg) or mitochondrial KATP channels (5-HD; 10 mg/kg) before ischemia (C) and before reperfusion (D). In these studies, infarct size was determined after 30 min of regional ischemia and 120-min reperfusion. Data are mean ± S.D., n = 6/group. *, P < 0.05, treated versus control.
We further examined the relative roles of the sarc and mitoKATP channels in the in vivo rat model of I/R injury. The selective sarcKATP channel inhibitor, HMR 1098 (3 mg/kg), did not alter infarct size when given alone 20 min before regional ischemia. When administered 5 min before P4pal10, it completely abrogated the infarct-limiting effects of P4pal10 (Fig. 9, C and D). Similar results were seen with the selective mitoKATP channel inhibitor, 5-hydroxydecanoic acid (5-HD; 10 mg/kg) (Fig. 9, C and D). Furthermore, HMR 1098 and 5-HD eliminated the protective effects of P4pal10 when after P4pal10 infusion and 5 min before reperfusion but had no effect on infarct size when administered alone (Fig. 9, C and D).
Discussion
The results of our study show, for the first time, that PAR4 inhibition reduces infarct size and increases LVDP in rat hearts after I/R. In addition, P4pal10 protects the heart against infarction when given after the onset of ischemia and immediately at the time of reperfusion. We also demonstrate that this effect relies on the endogenous activation of cell survival pathways including PI3K/Akt, ERK1/2, NOS, and KATP channels. More importantly, we demonstrate that the infarct-limiting effects of PAR4 antagonists involve ARs. PAR4 inhibition results in a similar reduction in infarct size as adenosine receptor agonists or ischemic preconditioning (35–50%) (Yellon et al., 1992; Lasley et al., 2007). This is also the first study that has shown cross-talk between PAR4 and adenosine receptors during myocardial ischemia and reperfusion.
PAR4-Adenosine Receptor Cross-Talk
The results of this study clearly suggest there is cross-talk between the thrombin and adenosine signaling pathways in the ischemic-reperfused rat myocardium. However, the basis of receptor cross-talk remains to be elucidated and may be occurring through receptor heterodimerization, G protein uncoupling from the inhibited receptor, distal signal regulation, or the regulation of other receptor agonist release. Adenosine receptors are known to couple with other GPCRs whose activation is required for the infarct-limiting effects of adenosine. In fact, the interaction between adenosine and opioids has been well documented (Peart and Gross, 2003, 2005). Studies have also shown that different GPCRs heterodimerize, resulting in altered ligand-binding properties, modified signaling and trafficking properties (George et al., 2000; Gomes et al., 2001; Minneman, 2007). The different adenosine receptors have also been shown to form heterodimers with other GCPRs such as purinergic receptor P2Y1 and dopamine D1 receptors (Ginés et al., 2000; Yoshioka et al., 2002). In addition, the PARs are known to heterodimerize with other members of their family (Leger et al., 2006; McLaughlin et al., 2007). In theory, PARs may oligomerize with ARs altering signaling of the ARs. For instance, PAR4 inhibition with tc-Y-NH2 may uncouple the AR and allow adenosine signaling, ultimately conferring a cardioprotective action. However, receptor heterodimerization is not likely to explain the cross-talk between PAR4 and AR because direct thrombin inhibition has also been shown to mediate its cardioprotective effects through the AR. Therefore, receptor cross-talk probably occurs further down in the signaling pathway. For instance, thrombin and PAR4 activation may result in signaling pathways that down-regulate adenosine signaling pathways, and the removal of PAR4’s signaling pathways may then allow the protective effects of adenosine signaling to take precedence. This is the first report that protection provided by PAR4 inhibition may be mediated by endogenous adenosine in a rat model of myocardial I/R injury.
PI3K/Akt
PI3K/Akt is an important mediator of cardioprotection (Hausenloy et al., 2005). Inhibition of this pathway has been shown to abolish the infarct-limiting effects of several pharmaceuticals (Li and Sato, 2001; Qin et al., 2003). However, thrombin and its receptor PAR4 have both been found to increase phosphorylation of Akt in isolated rat cardiomyocytes (Sabri et al., 2003; Snabaitis et al., 2005), so as confirmed by our results, we did not expect tc-Y-NH2 to up-regulate Akt phosphorylation. Nonetheless, Akt phosphorylation was increased in control and treatment groups at reperfusion and significantly reduced in the wortmannin-treated groups. This indicates that a basal level of activation of Akt is necessary for cardioprotection. This basal level of activation may result from adenosine release at reperfusion and stimulation of its receptors.
In contrast, other studies have questioned the role of PI3K/Akt in adenosine-mediated cardioprotection (Qin et al., 2003; Yang et al., 2004). This is probably due to the different signaling patterns of the various ARs important in cardioprotection. For instance, A1ARs do not depend on activation of PI3K/Akt for their infarct-limiting effects, whereas A2AR and A3AR do signal through this kinase pathway to produce their cardioprotective effects (Germack and Dickenson, 2005). Our data would suggest that the cardioprotective effects of PAR4 inhibition are due to the activation of A2AR and/or A3AR.
ERK1/2
ERK1/2 is also an important mediator of cardioprotection (Hausenloy et al., 2005). ERK1/2 phosphorylation is increased after thrombin or PAR4 AP treatment of isolated rat cardiomyocytes (Sabri et al., 2003; Snabaitis et al., 2005). Therefore, we did not expect tc-Y-NH2 to induce phosphorylation of ERK1/2 as supported by our results. On the other hand, adenosine has been shown to signal through ERK1/2 to produce its infarct-sparing effects (Headrick et al., 2003a). We found that ERK2 phosphorylation was increased at reperfusion in the control and tc-Y-NH2-treated groups but that PD98059 significantly decreased phosphorylation at reperfusion. This would suggest that a basal level of ERK2 activation is necessary for cardioprotection. It is presumed that this basal activation is at least in part caused by adenosine.
NOS
Nitric-oxide synthase produces NO to act as a mediator against myocardial stunning and infarction. Blocking NOS with Nω-nitro-L-arginine methyl ester abrogates pharmacological preconditioning with AR agonists in whole hearts (Krieg et al., 2004; Oldenburg et al., 2004). Thrombin and PAR4 activation have been associated with increasing NO production in endothelium and intact vessels (Momota et al., 2006; Touyz, 2007); therefore, it is not surprising that PAR4 inhibition with tc-Y-NH2 did not change NO production over control values. However, L-NMA alone did decrease NO production, indicating that a basal level of NO is required for cardioprotection in this system. Adenosine has been shown to mediate cardioprotection by up-regulating NO production (Headrick et al., 2003a; Yang et al., 2004). It is possible that the basal NO production that is required for cardioprotection is at least in part caused by adenosine receptor activation.
KATP Channels
Activation of KATP channels are both integral triggers and end-effectors of cardioprotection. They are highly expressed in myocardial sarcolemma and thought to be expressed in myocardial mitochondria. Both ischemic and pharmacologic preconditioning are dependent on the opening of these channels (Much-Ellingsen et al., 1996; Peart and Gross, 2002). Furthermore, both sarc and mitoKATP channels have been implicated in adenosine-mediated cardioprotection (Peart and Gross, 2002). The current study describes a role for both sarc and mitoKATP channels in the infarct-limiting effects of the PAR4 antagonist. Blockade of either channel 5 min before reperfusion abolishes the cardioprotective effects of P4pal10, suggesting that the channels are end-effectors of cardioprotection. A similar effect is observed when the KATP channel inhibitors are administered before ischemia. This may suggest that the sarc and mitoKATP channels are also triggers of cardioprotection induced by PAR4 blockade.
Study Limitations
This study is based on the assumption that endogenous adenosine is mediating cardioprotection by acting on its receptors. A limitation of this study is the lack of direct measurement of adenosine concentration in the presence and absence of the PAR4 antagonist. However, increased production of adenosine during myocardial ischemia has been well documented (Sommerschild and Kirkeboen, 2000), and we do not expect tc-Y-NH2 to affect production of adenosine in treated hearts. Moreover, the cardioprotective effects of PAR4 inhibition were verified with two structurally different compounds, and it is unlikely that both of them would have the same effects on endogenous adenosine production. Furthermore, if the levels of endogenous adenosine differed between the control and treated hearts, we would expect a difference in hemodynamic parameters such as a decreased heart rate and increase in coronary flow. We did not observe any differences between the tc-Y-NH2 treatment and control groups in vitro or P4pal10 treatment and control groups in vivo. P4pal10 treatment in vitro did result in a marked reduction of coronary flow in these hearts and was not associated with cardioprotection. It is possible the PAR4 antagonists, tc-Y-NH2 and P4pal10, have effects other than blocking PAR4 signaling. Although these potential effects should be considered, the reversal of the infarct-limiting effects of tc-Y-NH2 at the receptor level with a PAR4 agonist, PAR4 AP, which would compete for the ligand binding site would suggest that the protection seen is indeed due to the blockade of PAR4 coupled with the activation of the AR by endogenous adenosine.
In summary, the data presented demonstrate that inhibition of PAR4 affords marked protection against infarct development in an in vivo and in vitro model of myocardial I/R injury, probably due to activation of ARs by endogenously produced adenosine. The protection observed appears to be dependent upon AR activation, Akt, ERK1/2, and NOS, along with opening of the sarc and mitoKATP channels. Inhibition of PAR4 presents a novel approach to achieve potent adenosine-mediated cardioprotection after production of endogenous adenosine.
Acknowledgments
The secretarial assistance of Mary Lynne Koenig is gratefully acknowledged.
This study was supported in part by National Institutes of Health Grants HL54075 (to J.E.B.) and HL0831 (to G.J.G.). J.L.S. is supported by National Institutes of Health Training Grant HL07792 (awarded to the Medical College of Wisconsin).
ABBREVIATIONS
- I/R
ischemia/reperfusion
- GPCR
G protein-coupled receptor
- AR
adenosine receptor
- ERK
extracellular signal-regulated kinase
- NOS
nitric-oxide synthase
- sarc
sarcolemmal
- mito
mitochondrial
- PAR
protease-activated receptor
- AP
activating peptide
- tc-Y-NH2
trans-cinnamoyl-YPGKF-amide
- P4pal10
palmitoyl-SGRRYGHALR-amide
- SD
Sprague-Dawley
- PCR
polymerase chain reaction
- PD98059
2′-amino-3′-methoxyflavone
- LVDP
left ventricular developed pressure
- L-NMA
NG-monomethyl-L-arginine
- 8-SPT
8-sulfaphenyltheophylline
- AAR
area at risk
- NO
nitric oxide
- 5-HD
5-hydroxydecanoic acid
- PI3K
phosphoinositide-3 kinase
- HMR 1098
sodium (5-(2-(5-chloro-2-methoxybenzamido)ethyl)-2-methoxyphenylsulfonyl)(methylcarbamothioyl)amide
References
- Chong AJ, Pohlman TH, Hampton CR, Shimamoto A, Mackman N, Verrier ED. Tissue factor and thrombin mediate myocardial ischemia-reperfusion injury. Ann Thorac Surg. 2003;75:S649–S655. doi: 10.1016/s0003-4975(02)04691-x. [DOI] [PubMed] [Google Scholar]
- Covic L, Misra M, Badar J, Singh C, Kuliopulos A. Pepducin-based intervention of thrombin-receptor signaling and systemic platelet activation. Nat Med. 2002;8:1161–1165. doi: 10.1038/nm760. [DOI] [PubMed] [Google Scholar]
- Deguchi H, Takeya H, Urano H, Gabazza EC, Zhou H, Suzuki K. Adenosine regulates tissue factor expression on endothelial cells. Thromb Res. 1998;91:57–64. doi: 10.1016/s0049-3848(98)00045-0. [DOI] [PubMed] [Google Scholar]
- D’Hondt C, Srinivas SP, Vereecke J, Himpens B. Adenosine opposes thrombin-induced inhibition of intercellular calcium wave in corneal endothelial cells. Invest Ophthalmol Vis Sci. 2007;48:1518–1527. doi: 10.1167/iovs.06-1062. [DOI] [PubMed] [Google Scholar]
- Dubey RK, Gillespie DG, Mi Z, Jackson EK. Exogenous and endogenous adenosine inhibits fetal calf serum-induced growth of rat cardiac fibroblasts: role of A2B receptors. Circulation. 1997;96:2656–2666. doi: 10.1161/01.cir.96.8.2656. [DOI] [PubMed] [Google Scholar]
- George SR, Fan T, Xie Z, Tse R, Tam V, Varghese G, O’Dowd BF. Oligomerization of μ- and δ-opioid receptors: generation of novel functional properties. J Biol Chem. 2000;275:26128–26135. doi: 10.1074/jbc.M000345200. [DOI] [PubMed] [Google Scholar]
- Germack R, Dickenson JM. Adenosine triggers preconditioning through MEK/ERK1/2 signalling pathway during hypoxia/reoxygenation in neonatal rat cardiomyocytes. J Mol Cell Cardiol. 2005;39:429–442. doi: 10.1016/j.yjmcc.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Ginés S, Hillion J, Torvinen M, Le Crom S, Casado V, Canela EI, Rondin S, Lew JY, Watson S, Zoli M, et al. Dopamine D1 and adenosine A1 receptors form functionally interacting heteromeric complexes. Proc Natl Acad Sci U S A. 2000;97:8606–8611. doi: 10.1073/pnas.150241097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Jordan BA, Gupta A, Rios C, Trapaidze N, Devi LA. G protein coupled receptor dimerization: implications in modulating receptor function. J Mol Med. 2001;79:226–242. doi: 10.1007/s001090100219. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Tsang A, Yellon DM. The reperfusion injury salvage kinase pathway: a common target for both ischemic preconditioning and postconditioning. Trends Cardiovasc Med. 2005;15:69–75. doi: 10.1016/j.tcm.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Headrick JP, Hack B, Ashton KJ. Acute adenosinergic cardioprotection in ischemic-reperfused hearts. Am J Physiol Heart Circ Physiol. 2003a;285:H1797–H1818. doi: 10.1152/ajpheart.00407.2003. [DOI] [PubMed] [Google Scholar]
- Headrick JP, Hack B, Ashton KJ. Acute adenosinergic cardioprotection in ischemic-reperfused hearts. Am J Physiol Heart Circ Physiol. 2003b;285:H1797–H1818. doi: 10.1152/ajpheart.00407.2003. [DOI] [PubMed] [Google Scholar]
- Hollenberg MD, Saifeddine M. Proteinase-activated receptor 4 (PAR4): activation and inhibition of rat platelet aggregation by PAR4-derived peptides. Can J Physiol Pharmacol. 2001;79:439–442. [PubMed] [Google Scholar]
- Krieg T, Qin Q, Philipp S, Alexeyev MF, Cohen MV, Downey JM. Acetylcholine and bradykinin trigger preconditioning in the heart through a pathway that includes Akt and NOS. Am J Physiol Heart Circ Physiol. 2004;287:H2606–H2611. doi: 10.1152/ajpheart.00600.2004. [DOI] [PubMed] [Google Scholar]
- Lasley RD, Kristo G, Keith BJ, Mentzer RM., Jr The A2a/A2b receptor antagonist ZM-241385 blocks the cardioprotective effect of adenosine agonist pretreatment in in vivo rat myocardium. Am J Physiol Heart Circ Physiol. 2007;292:H426–H431. doi: 10.1152/ajpheart.00675.2006. [DOI] [PubMed] [Google Scholar]
- Leger AJ, Jacques SL, Badar J, Kaneider NC, Derian CK, Andrade-Gordon P, Covic L, Kuliopulos A. Blocking the protease-activated receptor 1–4 heterodimer in platelet-mediated thrombosis. Circulation. 2006;113:1244–1254. doi: 10.1161/CIRCULATIONAHA.105.587758. [DOI] [PubMed] [Google Scholar]
- Li Y, Sato T. Dual signaling via protein kinase C and phosphatidylinositol 3′-kinase/Akt contributes to bradykinin B2 receptor-induced cardioprotection in guinea pig hearts. J Mol Cell Cardiol. 2001;33:2047–2053. doi: 10.1006/jmcc.2001.1455. [DOI] [PubMed] [Google Scholar]
- Mahaffey KW, Puma JA, Barbagelata NA, DiCarli MF, Leesar MA, Browne KF, Eisenberg PR, Bolli R, Casas AC, Molina-Viamonte V, et al. Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction: results of a multicenter, randomized, placebo-controlled trial: the Acute Myocardial Infarction STudy of ADenosine (AMISTAD) trial. J Am Coll Cardiol. 1999;34:1711–1720. doi: 10.1016/s0735-1097(99)00418-0. [DOI] [PubMed] [Google Scholar]
- McLaughlin JN, Patterson MM, Malik AB. Protease-activated receptor-3 (PAR3) regulates PAR1 signaling by receptor dimerization. Proc Natl Acad Sci U S A. 2007;104:5662–5667. doi: 10.1073/pnas.0700763104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minneman KP. Heterodimerization and surface localization of G protein coupled receptors. Biochem Pharmacol. 2007;73:1043–1050. doi: 10.1016/j.bcp.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momota F, Hirano K, Hirano M, Nishimura J, Kanaide H. Involvement of Gi/o in the PAR-4-induced NO production in endothelial cells. Biochem Biophys Res Commun. 2006;342:365–371. doi: 10.1016/j.bbrc.2006.01.165. [DOI] [PubMed] [Google Scholar]
- Much-Ellingsen J, Bugge E, Ytrehus K. Blockade of the KATP-channel by glibenclamide aggravates ischemic injury, and counteracts ischemic preconditioning. Basic Res Cardiol. 1996;91:382–388. doi: 10.1007/BF00788718. [DOI] [PubMed] [Google Scholar]
- Oldenburg O, Qin Q, Krieg T, Yang XM, Philipp S, Critz SD, Cohen MV, Downey JM. Bradykinin induces mitochondrial ROS generation via NO, cGMP, PKG, and mitoKATP channel opening and leads to cardioprotection. Am J Physiol Heart Circ Physiol. 2004;286:H468–H476. doi: 10.1152/ajpheart.00360.2003. [DOI] [PubMed] [Google Scholar]
- Ossovskaya VS, Bunnett NW. Protease-activated receptors: contribution to physiology and disease. Physiol Rev. 2004;84:579–621. doi: 10.1152/physrev.00028.2003. [DOI] [PubMed] [Google Scholar]
- Peart JN, Gross GJ. Sarcolemmal and mitochondrial KATP channels and myocardial ischemic preconditioning. J Cell Mol Med. 2002;6:453–464. doi: 10.1111/j.1582-4934.2002.tb00449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peart JN, Gross GJ. Adenosine and opioid receptor-mediated cardioprotection in the rat: evidence for cross-talk between receptors. Am J Physiol Heart Circ Physiol. 2003;285:H81–H89. doi: 10.1152/ajpheart.00985.2002. [DOI] [PubMed] [Google Scholar]
- Peart JN, Gross GJ. Cardioprotection following adenosine kinase inhibition in rat hearts. Basic Res Cardiol. 2005;100:328–336. doi: 10.1007/s00395-005-0526-7. [DOI] [PubMed] [Google Scholar]
- Piper HM, Volz A, Schwartz P. Adult ventricular rat heart muscle cells. In: Piper HM, editor. Cell Culture Techniques in Heart and Vessel Research. Springer Verlag; Berlin, Germany: 1990. pp. 36–60. [Google Scholar]
- Qin Q, Downey JM, Cohen MV. Acetylcholine but not adenosine triggers preconditioning through PI3-kinase and a tyrosine kinase. Am J Physiol Heart Circ Physiol. 2003;284:H727–H734. doi: 10.1152/ajpheart.00476.2002. [DOI] [PubMed] [Google Scholar]
- Rohatgi T, Henrich-Noack P, Sedehizade F, Goertler M, Wallesch CW, Reymann KG, Reiser G. Transient focal ischemia in rat brain differentially regulates mRNA expression of protease-activated receptors 1 to 4. J Neurosci Res. 2004;75:273–279. doi: 10.1002/jnr.10847. [DOI] [PubMed] [Google Scholar]
- Ross AM, Gibbons RJ, Stone GW, Kloner RA, Alexander RW. A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II) J Am Coll Cardiol. 2005;45:1775–1780. doi: 10.1016/j.jacc.2005.02.061. [DOI] [PubMed] [Google Scholar]
- Sabri A, Guo J, Elouardighi H, Darrow AL, Andrade-Gordon P, Steinberg SF. Mechanisms of protease-activated receptor-4 actions in cardiomyocytes: role of Src tyrosine kinase. J Biol Chem. 2003;278:11714–11720. doi: 10.1074/jbc.M213091200. [DOI] [PubMed] [Google Scholar]
- Shryock JC, Belardinelli L. Adenosine and adenosine receptors in the cardiovascular system: biochemistry, physiology, and pharmacology. Am J Cardiol. 1997;79:2–10. doi: 10.1016/s0002-9149(97)00256-7. [DOI] [PubMed] [Google Scholar]
- Snabaitis AK, Muntendorf A, Wieland T, Avkiran M. Regulation of the extracellular signal-regulated kinase pathway in adult myocardium: differential roles of Gq/11, Gi and G12/13 proteins in signalling by [alpha]1-adrenergic, endothelin-1 and thrombin-sensitive protease-activated receptors. Cell Signal. 2005;17:655–664. doi: 10.1016/j.cellsig.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Sommerschild HT, Kirkeboen KA. Adenosine and cardioprotection during ischaemia and reperfusion: an overview. Acta Anaesthesiol Scand. 2000;44:1038–1055. doi: 10.1034/j.1399-6576.2000.440903.x. [DOI] [PubMed] [Google Scholar]
- Strande JL, Hsu A, Su J, Fu X, Gross GJ, Baker JE. SCH 79797, a selective PAR1 antagonist, limits myocardial ischemia/reperfusion injury in rat hearts. Basic Res Cardiol. 2007;102:350–358. doi: 10.1007/s00395-007-0653-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touyz RM. Regulation of endothelial nitric oxide synthase by thrombin. Hypertension. 2007;49:429–431. doi: 10.1161/01.HYP.0000255955.75119.1a. [DOI] [PubMed] [Google Scholar]
- Yang X-M, Krieg T, Cui L, Downey JM, Cohen MV. NECA and bradykinin at reperfusion reduce infarction in rabbit hearts by signaling through PI3K, ERK, and NO. J Mol Cell Cardiol. 2004;36:411–421. doi: 10.1016/j.yjmcc.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Yellon DM, Alkhulaifi AM, Browne EE, Pugsley WB. Ischaemic preconditioning limits infarct size in the rat heart. Cardiovasc Res. 1992;26:983–987. doi: 10.1093/cvr/26.10.983. [DOI] [PubMed] [Google Scholar]
- Yoshioka K, Hosoda R, Kuroda Y, Nakata H. Hetero-oligomerization of adenosine A1 receptors with P2Y1 receptors in rat brains. FEBS Lett. 2002;531:299–303. doi: 10.1016/s0014-5793(02)03540-8. [DOI] [PubMed] [Google Scholar]