

Abstract
The identification of loci influenced by positive selection is a major goal of evolutionary genetics. A popular approach is to perform scans of alignments on a genome-wide scale in order to find regions evolving at accelerated rates on a particular branch of a phylogenetic tree. However, positive selection is not the only process that can lead to accelerated evolution. Notably, GC-biased gene conversion (gBGC) is a recombination-associated process that results in the biased fixation of G and C nucleotides. This process can potentially generate bursts of nucleotide substitutions within hotspots of meiotic recombination. Here, we analyse the results of a scan for positive selection on genes on branches across the primate phylogeny. We show that genes identified as targets of positive selection have a significant tendency to exhibit the genomic signature of gBGC. Using a maximum-likelihood framework, we estimate that more than 20 per cent of cases of significantly elevated non-synonymous to synonymous substitution rates ratio (dN/dS), particularly in shorter branches, could be due to gBGC. We demonstrate that in some cases, gBGC can lead to very high dN/dS (more than 2). Our results indicate that gBGC significantly affects the evolution of coding sequences in primates, often leading to patterns of evolution that can be mistaken for positive selection.
Keywords: biased gene conversion, selection, recombination
1. Introduction
All evolutionary changes start out as polymorphisms segregating within populations and evolution results from the combined effect of the input of new mutations and subsequent changes in their allele frequencies. New mutations can be driven to fixation by natural selection, or by random genetic drift. In addition, several molecular mechanisms are known to affect fixation probability by causing non-Mendelian inheritance (Hurst 2009). These processes alter the probability with which a particular allele at a given locus is transmitted to the next generation, and hence its probability of fixation. For example, it has been shown in yeast that gene conversion events occurring during meiotic recombination result in the biased transmission of G and C (denoted by S, for strong) over A and T (W, for weak) alleles (Mancera et al. 2008). There is indirect evidence that this form of meiotic drive (termed gBGC, for GC-biased gene conversion) affects genome evolution in many other taxa (Duret & Galtier 2009a).
In primates, the evidence mainly comes from two observations. First, it has been demonstrated that the long-term average recombination rate strongly influences the rate of W → S nucleotide substitutions (Meunier & Duret 2004; Webster et al. 2005; Duret & Arndt 2008). Second, analyses of polymorphism frequency spectra showed that W → S mutations segregate at higher frequency than S → W mutations (Webster & Smith 2004), and that this frequency bias was maximal in regions of high recombination (Spencer 2006). These results indicate that there is fixation bias in favour of S alleles in regions of high recombination. Finally, it was observed that elevated W → S substitution rates show a much stronger association with male than female recombination rate (Webster et al. 2005; Dreszer et al. 2007; Duret & Arndt 2008), indicating that this bias does not result from selection, which would not predict a sex-specific pattern. Interestingly, analysis of mismatch repair in primate cell lines demonstrated a bias towards incorporation of S nucleotides (Brown & Jiricny 1989). Thus, the current working hypothesis is that, in primates, gBGC results from a bias in the repair of mismatches occurring in heteroduplex DNA during meiotic recombination (Duret & Galtier 2009a).
Human recombination occurs mainly in hotspots (typically less than 2 kb), where recombination rates can be hundreds of times higher than in surrounding DNA (Myers et al. 2005). Hotspots are not conserved between human and chimpanzee, which indicates that they have a short evolutionary lifespan (Winckler et al. 2005). It is, therefore, expected that gBGC could generate transient and local bursts of W → S nucleotide substitutions. Consistent with this prediction, genomic regions with high male recombination show a strong excess of lineage-specific hotspots of W → S nucleotide substitutions (Dreszer et al. 2007). In the long term, gBGC can affect large regions and there is strong evidence that this process can explain the origin of the large-scale variation in GC content (isochores) observed across mammalian genomes, owing to large scale variation in the average density of recombination hotspots (Duret & Arndt 2008).
Besides its effects on neutral regions of genomes, both empirical and theoretical results indicate that gBGC can affect the evolution of functional genomic elements (Galtier & Duret 2007; Berglund et al. 2009; Galtier et al. 2009). Specifically, gBGC is predicted to promote the fixation of slightly deleterious W → S substitutions, which would otherwise be discarded by natural selection (Duret & Galtier 2009a). Episodes of gBGC can, therefore, lead to bursts of neutral or deleterious substitutions at functional sites. This is problematic, because such episodes of accelerated evolution of non-neutral sequences are typically considered as evidence for positive selection in the literature—a very different interpretation.
Instances of human-specific acceleration of functional non-coding regions, for example, have been associated to adaptive evolution (Pollard et al. 2006; Prabhakar et al. 2006; Bird et al. 2007; Kim & Pritchard 2007), but a substantial fraction of these episodes were eventually found to be consistent with increased evolutionary rates owing to gBGC (Galtier & Duret 2007; Duret & Galtier 2009b). Most phylogenetic tests of positive selection on protein-coding genes use the ratio of non-synonymous (dN) to synonymous (dS) substitution rates. We have previously shown, however, that gBGC can lead to an increase of the dN/dS ratio, thus mimicking the effect of adaptive evolution (Berglund et al. 2009; Galtier et al. 2009). This effect of gBGC on the dN/dS ratio is due to the fact that non-synonymous codon positions generally have a lower GC content than synonymous codon positions (notably in GC-rich genes). Thus, there are more opportunities for W → S substitutions at non-synonymous sites compared with synonymous sites, and hence gBGC leads to increase dN relative to dS. An analysis of coding exons showing elevated dN/dS ratio in the human lineage revealed that these sequences had a significantly elevated proportion of W → S substitutions, consistent with an effect of gBGC (Berglund et al. 2009).
Three main features potentially enable us to distinguish gBGC from positive selection: (i) gBGC generates W → S biased patterns of substitution, whereas there is a priori no reason why selection should generally favour W → S substitutions (functional regions are not generally GC rich; Galtier & Duret 2007; Duret & Galtier 2009b); (ii) whereas selection operates only on functional sites, gBGC also affects flanking neutral sites; and (iii) gBGC is associated with regions of high male recombination (such as subtelomeric regions).
The aim of this paper is to apply these criteria to a set of genes previously identified as positively selected based on dN/dS genome scans, and to quantify the proportion of positively selected genes (PSGs) that could be false positives owing to gBGC. For this purpose, we have analysed patterns of substitution in codons and in flanking non-coding sites to examine the signature of gBGC in a dataset of PSG candidates detected across the primate phylogeny (Kosiol et al. 2008). To test the predictions that gBGC can in some cases lead to non-synonymous substitution rates that are higher than synonymous rates, we have performed theoretical modelling of ADCYAP1, a gene with a very high dN/dS ratio (more than 2) in the human lineage.
2. Material and Methods
(a). Dataset
We analysed alignments of orthologous genes from six mammalian genomes (human, chimpanzee, macaque, mouse, rat and dog) presented in Kosiol et al. (2008). Alignments and results from tests of positive selection across the tree were obtained from http://compgen.bscb.cornell.edu/projects/mammal-psg/. To clean the data further, we removed 43 alignments in which the observed rate of mismatches in four or more consecutive positions was greater than 50 000 times the value expected under the assumption of uniform rate across sites—such datasets were considered as potentially affected by hidden paralogy or alignment errors. We also obtained alignments of 100 bp of sequence flanking both sides of every exon (data provided by A. Siepel).
We obtained estimates of human recombination rates and the positions of human recombination hotspots determined from patterns of human linkage disequilibrium from the HapMap project at http://www.HapMap.org/downloads/. We downloaded male, female and sex-averaged human recombination rates based on pedigree analysis from the UCSC genome browser. We also noted whether each gene was located within a terminal chromosome band in the human genome.
(b). Analysis of nucleotide substitutions
We used the codeml program of PAML with F3 × 4 codon frequencies and the Goldman & Yang (1994) model of codon substitution to infer substitution patterns along branches of the known phylogenetic tree of the six mammals (figure 1). We inferred ancestral sequences at each node on the tree using the free ratios model of codeml, where the dN/dS ratio is allowed to vary along the different branches of the tree. The minimum number of species for which orthologues were required to accurately infer ancestral sequences at each node were specified in the supporting information of Kosiol et al. (2008). We analysed patterns of substitutions along each branch of the primate tree using the ancestral sequences constructed by codeml, classifying all substitutions as W → S, S → W, S → S or W → W, and either synonymous or non-synonymous. Substitutions in the non-coding regions flanking each exon were inferred using baseml using the HKY85 nucleotide substitution model.
Figure 1.

Phylogenetic tree of the species in the alignments. The branches in the lineage-specific tests for positive selection are marked: a, human; b, chimpanzee; c, hominid ancestral; d, macaque and e, primate ancestral.
The equilibrium GC content (GC*) can be inferred from the observed W → S and S → W substitution rates (denoted by u and v, respectively):
| 2.1 |
To identify genes with elevated rates of evolution in the human or chimpanzee lineage, we estimated the total number of substitutions on each lineage in each gene by comparison of the ML-inferred ancestral and extant sequences. A one-tailed Fisher's exact test was used to compare the ratio of the number of substitutions on the chimpanzee branch to the length of the inferred ancestral sequence with the corresponding ratio on the human branch. This test was performed separately on the human and chimpanzee branches to identify the set of genes with elevated substitution rates on one branch, compared with the other.
(c). Maximum likelihood analysis
Consider a sequence of W and S nucleotides evolving during a finite amount of time, the number x of W → S substitutions conditional on the total number x + y of W ↔ S mutations follows a binomial distribution with parameters n = x + y and p = uW/(uW + vS) = gW/( gW + (1 − g)S) so that
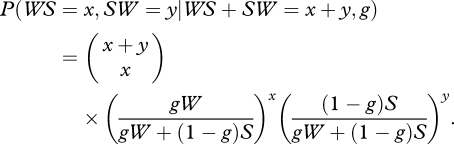 |
2.2 |
We used a model where genes belong to two classes with different GC* levels. In the first class, genes are not subject to gBGC, with a GC* of g1; in the second class genes are subject to gBGC, with a GC* of g2 > g1. The fraction of genes in the second class is called α.
A maximum-likelihood approach was used to infer g1, g2 and α from a sample of n genes, assuming that the numbers of W → S and S → W substitutions are known from each gene. From equation (2.2), we can infer the log-likelihood of the data given a set of parameters:
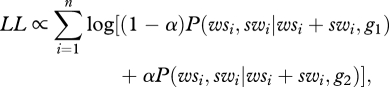 |
2.3 |
where wsi and swi are the observed numbers of W → S and S → W substitutions in the ith gene. P(wsi, swi|wsi + swig1) is obtained from equation (2.2).
Maximum-likelihood estimates of g1, g2 and α were obtained by maximizing LL with the nlminb function in R (R Development Core Team 2009). In equation (2.3), the number of substitutions, wsi and swi, were counted either globally, or in specific branches or sets of branches of the tree. Model parameters were estimated using all genes (PSGs and neutraly evolving genes).
Once the model parameters are estimated, individual genes are assigned to class 1 or class 2 using Bayes' formula. The posterior probability that gene i belongs to the first GC* class is
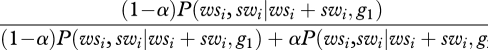 |
2.4 |
Gene i was assigned to class 1 if this probability was above 0.5 (and to class 2 otherwise). The proportion of genes assigned to the two classes of GC* were calculated for all genes, PSGs and non-PSGs.
3. Results
We analysed the dataset presented in Kosiol et al. (2008), consisting of 17 489 human genes with orthologues in at least two of the following mammalian genomes: chimpanzee, macaque, mouse, rat and dog. We estimated patterns of substitution along the human, chimpanzee, macaque, hominid ancestral and primate ancestral branches (figure 1). Non-primate branches were excluded from the analysis as the larger evolutionary distances make ancestral sequence reconstruction problematic, particularly in flanking non-coding regions.
(a). Does gBGC affect substitution rates measured on the whole gene scale?
Single exons with evidence for accelerated evolutionary rates have been shown to exhibit evolutionary features consistent with a strong effect of gBGC (Berglund et al. 2009; Galtier et al. 2009), suggesting that this process can promote fixation of both synonymous and non-synonymous mutations. However, phylogenetic scans for positive selection are typically performed on whole genes rather than individual exons and it is unclear whether the effects of gBGC are detectable at the whole gene level. We, therefore, scanned our dataset to identify genes with significantly elevated evolutionary rates on the human or chimpanzee lineages since their common ancestor.
Using a Fisher's exact test, we identified 450 out of 14 519 genes with evidence for elevated substitution rates on the human lineage and 360 on the chimpanzee lineage (table 1). Genes with faster evolutionary rates on the human or chimpanzee lineages show significantly elevated equilibrium GC content (GC*). We measured sex-specific and sex-averaged recombination rates from the DeCode map, the recombination rate and distance to nearest hotspot from HapMap data (which reflect historical recombination rates in human populations), and the proportion of genes in the final band of a human chromosome. HapMap data indicate that faster-evolving human genes have significantly elevated recombination rates and are significantly closer to recombination hotspots. Importantly, both in human and chimpanzee lineages, faster-evolving genes are significantly enriched in subtelomeric regions, and exhibit significantly elevated levels of male, but not female, recombination. These findings are consistent with the hypothesis that gBGC has generated accelerated evolutionary rates in a proportion of these genes.
Table 1.
Genes with significantly different rates in the human and chimpanzee lineages. The significance of departures of accelerated genes (acc.) from the mean (total) was determined by a randomization test (significant tests are indicated in bold).
| no. genes | GC content | W → S subs. | S → W subs. | GC* | male recomb. (cM/Mb) | female recomb. (cM/Mb) | HapMap recomb. (cM/Mb) | mean distance to hotspot (kb) | prop. terminal chromosome band | |
|---|---|---|---|---|---|---|---|---|---|---|
| human lineage | ||||||||||
| acc. | 450 | 0.54 | 1834 | 2260 | 0.48 (p = 10−4) | 1.20 (p = 4 × 10−4) | 1.74 | 1.56 (p = 0.006) | 44 (p = 0.005) | 0.18 (p < 10−5) |
| total | 14 519 | 0.52 | 17 301 | 25 589 | 0.41 | 0.98 | 1.66 | 1.18 | 54 | 0.11 |
| chimpanzee lineage | ||||||||||
| acc. | 360 | 0.54 | 1459 | 1756 | 0.48 (p = 10−3) | 1.32 (p < 10−5) | 1.65 | 1.30 | 55 | 0.26 (p < 10−5) |
| total | 14 519 | 0.52 | 16 643 | 23 371 | 0.43 | 0.98 | 1.66 | 1.18 | 56 | 0.11 |
(b). Does gBGC affect PSG scans based on branch tests of dN/dS in primates?
Generally, comparative genomic scans for protein-coding genes under positive selection are performed by estimating dN/dS, rather than substitution rate. The dN/dS test is expected to be more robust to gBGC than simple acceleration tests, because gBGC affects both synonymous and non-synonymous sites. However, theoretical modelling indicates that the dN/dS ratio can also be affected by gBGC, particularly in GC-rich genes (Berglund et al. 2009; Galtier et al. 2009). In practice, it is unclear how many of the genes identified as PSGs in genome scans for selection might be false positives owing to gBGC.
To quantify this effect, we analysed a set of genes that Kosiol et al. (2008) identified as PSGs by using a test searching for branch-specific elevated dN/dS (n = 88 for the five branches under investigation here). These genes are hereafter denoted branch-PSGs. There is a significant overlap between genes with evidence for lineage-specific acceleration on the human and chimpanzee branches and those identified as PSGs on the same branch with a false discovery rate (FDR) less than 5 per cent (four out of seven human PSGs, p = 0.0002; five out of 10 chimpanzee PSGs, p = 6 × 10−6). More branch-PSGs were identified on the longer branches, probably as a result of increased power. In four of the five branches, GC* in branch-PSGs is higher than in all genes (table 2). We estimated GC* across all branches by summing all W → S and S → W substitutions, weighting by the total number of these substitutions on each branch. The average GC* in all genes was 0.45, whereas in the 88 branch-PSGs it was 0.51. This difference is significant by a randomization test across all branches (p = 0.015). We performed a ‘combined’ randomization test by resampling genes on each branch independently and counting the number of randomized samples with four or more out of five branches with GC* in branch-PSGs greater than their observed values. This method indicated that the probability of observing the data by chance was p = 0.007. Hence, branch-PSGs have significantly elevated GC*, consistent with an effect of gBGC.
Table 2.
Substitution patterns in coding and non-coding regions of PSGs identified by branch-site tests.
| all genes |
PSGs (FDR <10%) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| region | branch | no. genes | GC content | S → W subs. | W → S subs. | GC* | no. genes | GC content | S → W subs. | W → S subs. | GC* |
| coding | human (a) | 14 519 | 0.52 | 25 589 | 17 301 | 0.41 | 7 | 0.61 | 40 | 43 | 0.62 |
| chimpanzee (b) | 14 519 | 0.52 | 23 371 | 16 643 | 0.43 | 10 | 0.56 | 76 | 64 | 0.49 | |
| macaque (d) | 12 476 | 0.52 | 96 626 | 83 494 | 0.48 | 17 | 0.52 | 245 | 176 | 0.41 | |
| hominid (c) | 10 961 | 0.52 | 53 055 | 48 874 | 0.49 | 6 | 0.53 | 59 | 86 | 0.56 | |
| primate (e) | 9553 | 0.52 | 154 795 | 121 669 | 0.46 | 48 | 0.52 | 1805 | 1585 | 0.49 | |
| non-coding | human (a) | 14 519 | 0.47 | 29 736 | 26 203 | 0.41 | 7 | 0.56 | 19 | 24 | 0.64 |
| chimpanzee (b) | 14 519 | 0.47 | 30 570 | 27 039 | 0.41 | 10 | 0.56 | 34 | 35 | 0.48 | |
| macaque (d) | 12 476 | 0.47 | 156 020 | 145 160 | 0.43 | 17 | 0.43 | 124 | 117 | 0.43 | |
| hominid (c) | 10 961 | 0.47 | 86 262 | 78 406 | 0.41 | 6 | 0.53 | 34 | 43 | 0.41 | |
| primate (e) | 9553 | 0.47 | 470 613 | 431 444 | 0.42 | 48 | 0.47 | 2960 | 3004 | 0.44 | |
We performed a similar analysis on non-coding regions flanking the exons of branch-PSGs, considering all regions within 100 bp of an exon of the gene (table 2). In this case five out of five branches tested had higher GC* in branch-PSGs compared with the entire dataset. Weighting the substitutions by branch length, average GC* in branch-PSGs is 0.47, significantly higher than 0.41 in all genes (randomization test across all branches p = 0.012). This was also significant using the ‘combined’ randomization test (p = 0.005). These results indicate that branch-PSGs tend to lie within regions of elevated GC*, consistent with a regional effect of gBGC.
In the human lineage the excess of W → S substitutions in branch-PSGs is mainly owing to the presence of two genes: (i) ADCYAP1 on chromosome 7, which has 20 substitutions, all of which are W → S and (ii) OR3A3 on chromosome 17, which has 12 W → S substitutions but only five S → W substitutions. Both of these genes are found in the terminal chromosome band within a region of high (more than 3.5 cM/Mb) male recombination rate.
(c). What proportion of PSG candidates in primates are due to gBGC?
We performed maximum likelihood modelling to determine the proportion of branch-PSGs that could potentially be due to gBGC. We considered a model assuming two classes of genes with distinct GC* (gBGC-free: GC* = g1; gBGC-affected: GC* = g2), and made use of inferred numbers of W → S and S → W substitutions to estimate the parameters. Model fitting was performed for each of the branches labelled in figure 1, for the hominid subtree (branches a, b and c), and for all branches. The analysis was performed separately for both coding substitutions, and non-coding flanking substitutions. In every case, the model with two GC* classes (g1 and g2) provided significantly better fit than a model with only one GC* class (p < 10−10).
Table 3 shows the model parameters estimated for each subset of the data. On average, 40 per cent of genes across the whole tree are assigned to model g2 based on patterns of substitution in the coding alignments, although this figure is lower for shorter branches. Similar estimates are obtained from the analysis of flanking non-coding substitutions. We then examined the proportion of genes assigned to each GC* class in branch-PSG candidates compared with other genes on each branch. When all branches are considered together, there is a significant excess of coding regions assigned to the model g2 class among PSG candidates compared with other genes. This excess corresponds to 14 per cent of the set of branch-PSGs. All branches show the same trend, but the excess of g2 genes is stronger among branch-PSGs identified within short phylogenetic branches (22% in all hominids). Owing to the limited amount of data, statistical tests on individual branches are significant only for the ones that show the strongest excess of g2 genes (human and chimpanzee). The analysis of substitutions in flanking non-coding regions shows the same trend as coding substitutions. These results indicate that more than 20 per cent of PSG candidates identified on the human lineage may be due to gBGC.
Table 3.
Maximum likelihood estimation of the number of genes evolving under model g1 or g2 in different branches of the tree, for the entire gene dataset and for PSG candidates identified in branch-specific tests. The proportion of genes following the model g2 is compared between the two datasets using Fisher's exact test (FET).
| number of genes following model g1 (Ng1) or g2 (Ng2) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| model parameters |
all genes |
PSG candidates |
||||||||
| region | branches | g1 | g2 | α | Ng1 | Ng2 | Ng1 | Ng2 | p FET | excess of Ng2 among PSG candidates, number (%) |
| coding regions | human | 0.397 | 0.742 | 0.069 | 14 382 | 130 | 5 | 2 | 0.002 | 1.9 (28) |
| chimpanzee | 0.399 | 0.647 | 0.139 | 14 316 | 193 | 8 | 2 | 0.008 | 1.9 (19) | |
| hominid ancestral | 0.447 | 0.648 | 0.249 | 9723 | 1232 | 4 | 2 | 0.14 | 1.3 (22) | |
| all hominids | 0.417 | 0.642 | 0.199 | 38 159 | 1817 | 17 | 6 | 0.0005 | 5.0 (22) | |
| macaque | 0.443 | 0.612 | 0.271 | 10 663 | 1796 | 13 | 4 | 0.29 | 1.5 (9) | |
| primate ancestral | 0.361 | 0.602 | 0.404 | 6293 | 3212 | 27 | 21 | 0.17 | 4.8 (10) | |
| all branches | 0.382 | 0.595 | 0.400 | 45 243 | 16 697 | 52 | 36 | 0.005 | 12.3 (14) | |
| flanking regions | human | 0.396 | 0.733 | 0.045 | 14 104 | 70 | 5 | 1 | 0.030 | 1.0 (16) |
| chimpanzee | 0.399 | 0.672 | 0.057 | 14 437 | 55 | 9 | 1 | 0.038 | 1.0 (10) | |
| hominid ancestral | 0.344 | 0.582 | 0.343 | 7988 | 2962 | 3 | 2 | 0.62 | 0.6 (13) | |
| all hominids | 0.354 | 0.579 | 0.286 | 34 674 | 4942 | 15 | 6 | 0.039 | 3.4 (16) | |
| macaque | 0.368 | 0.551 | 0.375 | 8657 | 3795 | 12 | 5 | 1.00 | −0.2 (−1) | |
| primate ancestral | 0.331 | 0.593 | 0.417 | 5592 | 3910 | 25 | 23 | 0.38 | 3.2 (7) | |
| all branches | 0.341 | 0.583 | 0.366 | 45 307 | 16 263 | 52 | 34 | 0.01 | 11.3 (13) | |
(d). The effect of recombination on PSGs
We examined human recombination rates of different subsets of PSGs compared with the average of all genes in the dataset (table 4). The branch-PSGs assigned to model g2 (high GC*) were found in regions of significantly higher male, but not female recombination rates. These genes also had significantly elevated HapMap recombination rates, were found significantly closer to recombination hotspots, and were found significantly more often in terminal chromosome bands. These observations are consistent with a strong influence of gBGC on the evolution of this subset of genes.
Table 4.
Genomic properties of genes identified as PSGs. The significance of departures from the mean was determined by a randomization test (significant tests are indicated in bold).
| class | subclass | no. genes | mean male recomb. rate (cM/Mb) | p | mean female recomb. rate (cM/Mb) | p | sex-averaged recomb. rate (cM/Mb) | p | HapMap recomb. rate (cM/Mb) | p | mean distance to hotspot (kb) | p | prop. final chrom. band | p |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| all genes | 16 030 | 1.00 | 1.65 | 1.35 | 1.21 | 55.9 | 0.14 | |||||||
| branch-PSGs | all | 88 | 1.16 | 0.137 | 1.54 | 0.758 | 1.41 | 0.274 | 2.90 | 0.002 | 50.2 | 0.593 | 0.15 | 0.361 |
| g1 | 57 | 0.90 | 0.703 | 1.52 | 0.792 | 1.26 | 0.732 | 2.07 | 0.048 | 64.3 | 0.814 | 0.07 | 0.96 | |
| g2 | 31 | 1.64 | 0.005 | 1.55 | 0.665 | 1.68 | 0.032 | 4.26 | < 0.001 | 23.2 | 0.006 | 0.30 | 0.025 | |
| site-PSGs | all | 349 | 1.14 | 0.025 | 1.78 | 0.025 | 1.47 | 0.012 | 1.69 | 0.002 | 43.2 | 0.012 | 0.12 | 0.158 |
(e). Is gBGC alone sufficient to produce the observed signature of positive selection in ADCYAP1?
ADCYAP1 is one of the seven PSG candidates identified in the human lineage. It is also in a region of highly elevated male recombination (4.17 cM/Mb) but not female recombination (0.16 cM/Mb), and is contained within a putative human recombination hotspot. The signal of human-specific accelerated evolution in ADCYAP1 is exhibited by exons 2 and 3 (respectively 132 and 99 bp, separated by an intron of 475 bp). Since the divergence from chimpanzee, these two exons have accumulated 20 substitutions, all W → S, among which 17 non-synonymous and three synonymous. Using an alignment of six mammalian species (Sorex araneus, Bos taurus, Canis lupus familiaris, Pongo pygmaeus, Pan troglodytes and Homo sapiens), we estimated dN/dS for these two exons to be 2.05 in the human lineage, and 0.21 in other branches (codeml program). The W → S pattern of substitution also extends into flanking non-coding regions (figure 2).
Figure 2.

Location of human-specific substitutions (W → S in red, S → W in blue, others in black) and W → S bias (proportion of all substitutions that are W → S, computed in sliding windows of 20 substitutions), along the ADCYAP1 gene. Substitutions were inferred by parsimony from UCSC blastz human–chimpanzee–macaque genomic alignments. There is no orthologous macaque sequence present in part of the alignment, and these positions are marked. W → S substitutions are notably very dense in and around exons 2 and 3.
Responding to Duret & Galtier (2009a,b), Prabhakar et al. (2009) analysed the evolution of ADCYAP1 on the human lineage. They argued that the observation of a dN/dS ratio greater than 1 in exons 2 and 3 of this gene, coupled with their biased substitution pattern, reflected the joint action of gBGC and positive selection. However, theoretical modelling indicates that gBGC can generate dN/dS ratios greater than 1 in the absence of positive selection. We made use of the gBGC model introduced by Galtier et al. (2009, Box 2) to investigate this question. Specifically, we ask is the pattern observed in ADCYAP1 plausible invoking gBGC alone, or is positive selection required?
We assumed that population-scaled selection coefficients (S = 4Nes) in ADCYAP1 exons 2 and 3 are gamma distributed across sites, and we varied the shape parameter, a, from 0.1 to 5 (Piganeau & Eyre-Walker 2003). The mutational bias was set to 2, i.e. we assume twice as many S → W than W → S mutations. Several values of the population-scaled gBGC coefficient (B = 4Neb) were used for the human branch, including the estimated average for human hotspots (8.7; Galtier et al. 2009) and higher values (very hot spots). In the absence of any specific information, we assumed that the percentage of non-synonymous sites for which G or C is the optimal state is 50 per cent. Given the AT-biased mutation process, this would imply a lower than 50 per cent GC12 at selection/mutation/drift equilibrium. To account for the observed GC12 (GC content at the first and second codon positions) of 54 per cent, we assumed that moderate gBGC has been acting in mammals prior to a strong human-specific gBGC episode. We adjusted the background gBGC level and mean value of S to the observed GC12 and dN/dS ratio in non-human branches (0.21). Under each combination of parameters, we computed the expected dN/dS ratio during the episode, and the probability of observing no S → W substitutions, and at least 17 W → S non-synonymous substitutions out of 20.
As shown in table 5, the gBGC model predicts a strong increase in dN/dS in the human branch. This is because GC3 (GC content at third codon positions) is much greater than GC12, and hence the impact of gBGC is stronger on non-synonymous sites than on synonymous sites. The increase in dN/dS depends on the shape parameter of the gamma distribution: when the fitness effect of mutations tends to be uniformly distributed over all sites (high a) dN/dS can be higher than 1 (up to 2 for very strong recombination hotspots); but for lower values of a then the expected dN/dS ratio under this gBGC model is lower than the observed one. However, the probability of observing a pattern as extreme as in the data is always higher than 5 per cent, except for one parameter combination (table 5). For example, with a = 0.5 and B = 8.7 (i.e. a moderate recombination hotspot), the expected dN/dS is 0.6, but the probability of observing dN/dS =2.05 is 10 per cent. We also tested scenarios assuming no gBGC before the episode, and higher than 50 per cent optimal GC12 (from 58% to 61%, depending on a). Again, the expected dN/dS ratio was generally lower than the observed one, but the probability of observing the data was most frequently higher than 0.1, and always higher than 0.04 (not shown). In other words, the evolution of ADCYAP1 can be explained by a wide range of scenarios that only invoke gBGC, in the absence of adaptive evolution.
Table 5.
Expected dN/dS in the human branch and probability of observing the dataset under the gBGC model with various parameters. (S, population-scaled selection coefficient; a, shape parameter of the gamma distribution of S; B, population-scaled gBGC coefficient.)
| model parameters |
|||||
|---|---|---|---|---|---|
| a | B | background B | S | human dN/dS | p (nonsyn ≥ 17/20) |
| 0.1 | 8.7 | 1.5 | 950 000 | 0.437 | 0.039 |
| 15 | 0.468 | 0.053 | |||
| 20 | 0.486 | 0.062 | |||
| 30 | 0.514 | 0.077 | |||
| 0.2 | 8.7 | 1.5 | 1200 | 0.479 | 0.052 |
| 15 | 0.545 | 0.096 | |||
| 20 | 0.586 | 0.123 | |||
| 30 | 0.651 | 0.169 | |||
| 0.5 | 8.7 | 1.6 | 30 | 0.596 | 0.109 |
| 15 | 0.789 | 0.277 | |||
| 20 | 0.916 | 0.376 | |||
| 30 | 1.121 | 0.517 | |||
| 1 | 8.7 | 1.6 | 10 | 0.800 | 0.240 |
| 15 | 1.164 | 0.543 | |||
| 20 | 1.387 | 0.657 | |||
| 30 | 1.695 | 0.767 | |||
| 5 | 8.7 | 2 | 5 | 1.139 | 0.448 |
| 15 | 1.691 | 0.766 | |||
| 20 | 1.910 | 0.820 | |||
| 30 | 2.130 | 0.861 | |||
4. Discussion
We analysed patterns of nucleotide substitutions across the primate phylogeny in a genome-wide set of coding alignments. We first demonstrated that genes with evidence for elevated rates of evolution on either the human or chimpanzee branch show clear signatures of gBGC, characterized by W → S biased substitution patterns, and elevated male recombination rates. We then examined a set of 88 branch-PSG candidates identified on five separate branches of the primate tree. These genes also exhibit an excess of W → S substitutions in both coding and flanking non-coding regions, consistent with an effect of gBGC in generating elevated dN/dS, thus mimicking positive selection.
We divided genes on each lineage into a high GC* and low GC* class using maximum likelihood. Across the primate phylogeny, there is a significant excess of 14 per cent of branch-PSG candidates in the high GC* category. The analysis suggests that 14 per cent of the branch-PSG candidates might in fact have been subject to gBGC and not positive selection. This fraction is higher in short branches (22% in human, chimpanzee and the hominid ancestor), which is consistent with the short lifespan of recombination hotspots: episodes of gBGC are expected to be limited in time, and hence to leave a signature only over short phylogenetic branches. As predicted by the gBGC model, the high GC* class of branch-PSGs are significantly enriched in regions of high recombination, specifically in males.
Kosiol et al. (2008) also reported a second set of PSG candidates, identified by searching for elevated dN/dS across the whole tree (hereafter denoted site-PSGs). Thus, whereas branch-PSGs were inferred by searching the signature of selection in specific branches of the tree, site-PSGs were identified by analysing substitution patterns over the entire tree. These site-PSGs show a less pronounced increase in GC* than branch-PSGs, with no noticeable effect in the flanking non-coding substitutions (data not shown). Site-PSGs have a significant tendency to occur in regions of elevated recombination, but male recombination is not specifically elevated (table 4). These two observations indicate that patterns of evolution in site-PSGs have not been influenced strongly by gBGC. This is again consistent with the short-lived nature of hotspots: genome scans for PSGs based on the analysis of substitution patterns over long evolutionary times are expected to be robust to transient episodes of gBGC. Thus, the effects of gBGC appear to be more problematic for genome scans aiming at identifying PSGs over short branches (typically, to search for PSGs responsible for human-specific adaptations). The reason why site-PSGs tend to occur in regions of high recombination is not clear. It should be noted that, because of Hill-Robertson effects (Hill & Robertson 1966), selection is expected to be more efficient in regions of high recombination. It is, therefore, possible that a greater number of true PSGs exist in regions of high recombination for this reason. However, a recent study found no evidence that recombination affects the efficacy of selection in the human genome (Bullaughey et al. 2008).
The potential effect of gBGC on the evolution of coding sequences is clearly illustrated by the striking example of ADCYAP1: 20 substitutions that have occurred in exons 2 and 3 of this gene, of which 17 are non-synonymous, three are synonymous and all are W → S. These extremely W → S biased patterns of substitution extend into non-coding flanking regions and the gene is located in a region of high male recombination. Altogether, these observations strongly suggest that this gene has been subject to gBGC. We, therefore, tested whether the peculiar substitution pattern in ADCYAP1 could be due to gBGC alone, or whether positive selection was required to explain the high dN/dS ratio (more than 2). Across a wide range of realistic scenarios, we were unable to reject the hypothesis that gBGC alone is responsible for the elevated non-synonymous substitution rate in this gene. Thus, the high dN/dS ratio in ADCYAP1 might simply reflect the accumulation of neutral or weakly deleterious mutations driven to fixation by gBGC alone. Note that selection and gBGC are not exclusive hypotheses: we cannot exclude that gBGC could have favoured the fixation of beneficial W → S mutations, or that ADCYAP1 was also influenced by positive selection on the human lineage. It also seems plausible that an episode of gBGC driving the fixation of deleterious mutations might be followed by an accumulation of compensatory substitutions—favoured by positive selection. However, this corresponds more to selection without adaptation (sensu Hartl & Taubes 1996) than to ‘true’ adaptation to new environmental conditions. Overall, gBGC alone remains the most parsimonious hypothesis to explain the peculiar substitution pattern in ADCYAP1.
Acknowledgements
This work was supported by the Swedish Research Council, the Centre National de la Recherche Scientifique, and by the Agence Nationale de la Recherche (ANR-08-GENM-036-01).
Footnotes
One contribution of 18 to a Discussion Meeting Issue ‘Genetics and the causes of evolution: 150 years of progress since Darwin’.
References
- Berglund J., Pollard K. S., Webster M. T.2009Hotspots of biased nucleotide substitutions in human genes. PLoS Biol. 7, e26 (doi:10.1371/journal.pbio.1000026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird C. P., Stranger B. E., Liu M., Thomas D. J., Ingle C. E., Beazley C., Miller W., Hurles M. E., Dermitzakis E. T.2007Fast-evolving noncoding sequences in the human genome. Genome Biol. 8, R118 (doi:10.1186/gb-2007-8-6-r118) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown T. C., Jiricny J.1989Repair of base-base mismatches in simian and human cells. Genome 31, 578–583 [DOI] [PubMed] [Google Scholar]
- Bullaughey K., Przeworski M., Coop G.2008No effect of recombination on the efficacy of natural selection in primates. Genome Res. 18, 544–554 (doi:10.1101/gr.071548.107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreszer T. R., Wall G. D., Haussler D., Pollard K. S.2007Biased clustered substitutions in the human genome: the footprints of male-driven biased gene conversion. Genome Res. 17, 1420–1430 (doi:10.1101/gr.6395807) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L., Arndt P. F.2008The impact of recombination on nucleotide substitutions in the human genome. PLoS Genet. 4, e1000071 (doi:10.1371/journal.pgen.1000071) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L., Galtier N.2009aBiased gene conversion and the evolution of mammalian genomic landscapes. Annu. Rev. Genom. Hum. Genet. 10, 285–311 (doi:10.1146/annurev-genom-082908-150001) [DOI] [PubMed] [Google Scholar]
- Duret L., Galtier N.2009bComment on ‘Human-specific gain of function in a developmental enhancer’. Science 323, 714. [DOI] [PubMed] [Google Scholar]
- Galtier N., Duret L.2007Adaptation or biased gene conversion? Extending the null hypothesis of molecular evolution. Trends Genet. 23, 273–277 (doi:10.1016/j.tig.2007.03.011) [DOI] [PubMed] [Google Scholar]
- Galtier N., Duret L., Glemin S., Ranwez V.2009GC-biased gene conversion promotes the fixation of deleterious amino acid changes in primates. Trends Genet. 25, 1–5 (doi:10.1016/j.tig.2008.10.011) [DOI] [PubMed] [Google Scholar]
- Goldman N., Yang Z.1994A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 11, 725–736 [DOI] [PubMed] [Google Scholar]
- Hartl D. L., Taubes C. H.1996Compensatory nearly neutral mutations: selection without adaptation. J. Theor. Biol. 182, 303–309 (doi:10.1006/jtbi.1996.0168) [DOI] [PubMed] [Google Scholar]
- Hill W. G., Robertson A.1966The effect of linkage on limits to artificial selection. Genet. Res. 8, 269–294 (doi:10.1017/S0016672300010156) [PubMed] [Google Scholar]
- Hurst L. D.2009Fundamental concepts in genetics: genetics and the understanding of selection. Nat. Rev. Genet. 10, 83–93 (doi:10.1038/nrg2506) [DOI] [PubMed] [Google Scholar]
- Kim S. Y., Pritchard J. K.2007Adaptive evolution of conserved noncoding elements in mammals. PLoS Genet. 3, 1572–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosiol C., Vinar T., da Fonseca R. R., Hubisz M. J., Bustamante C. D., Nielsen R., Siepel A.2008Patterns of positive selection in six mammalian genomes. PLoS Genet. 4, e1000144 (doi:10.1371/journal.pgen.1000144) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancera E., Bourgon R., Brozzi A., Huber W., Steinmetz L. M.2008High-resolution mapping of meiotic crossovers and non-crossovers in yeast. Nature 454, 479–485 (doi:10.1038/nature07135) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier J., Duret L.2004Recombination drives the evolution of GC-content in the human genome. Mol. Biol. Evol. 21, 984–990 (doi:10.1093/molbev/msh070) [DOI] [PubMed] [Google Scholar]
- Myers S., Bottolo L., Freeman C., McVean G., Donnelly P.2005A fine-scale map of recombination rates and hotspots across the human genome. Science 310, 321–324 (doi:10.1126/science.1117196) [DOI] [PubMed] [Google Scholar]
- Piganeau G., Eyre-Walker A.2003Estimating the distribution of fitness effects from DNA sequence data: implications for the molecular clock. Proc. Natl Acad. Sci. USA 100, 10 335–10 340 (doi:10.1073/pnas.1833064100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard K. S., et al. 2006An RNA gene expressed during cortical development evolved rapidly in humans. Nature 443, 167–172 (doi:10.1038/nature05113) [DOI] [PubMed] [Google Scholar]
- Prabhakar S., Noonan J. P., Paabo S., Rubin E. M.2006Accelerated evolution of conserved noncoding sequences in humans. Science 314, 786 (doi:10.1126/science.1130738) [DOI] [PubMed] [Google Scholar]
- Prabhakar S., et al. 2009Response to comment on ‘Human-specific gain of function in a developmental enhancer’. Science 323, 714 (doi:10.1126/science.1166571) [DOI] [PubMed] [Google Scholar]
- R Development Core Team 2009R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing [Google Scholar]
- Spencer C. C.2006Human polymorphism around recombination hotspots. Biochem. Soc. Trans. 34, 535–536 [DOI] [PubMed] [Google Scholar]
- Webster M. T., Smith N. G.2004Fixation biases affecting human SNPs. Trends Genet. 20, 122–126 (doi:10.1016/j.tig.2004.01.005) [DOI] [PubMed] [Google Scholar]
- Webster M. T., Smith N. G., Hultin-Rosenberg L., Arndt P. F., Ellegren H.2005Male-driven biased gene conversion governs the evolution of base composition in human alu repeats. Mol. Biol. Evol. 22, 1468–1474 (doi:10.1093/molbev/msi136) [DOI] [PubMed] [Google Scholar]
- Winckler W., et al. 2005Comparison of fine-scale recombination rates in humans and chimpanzees. Science 308, 107–111 (doi:10.1126/science.1105322) [DOI] [PubMed] [Google Scholar]