

Abstract
Understanding pathogen infectivity and virulence requires combining insights from epidemiology, ecology, evolution and genetics. Although theoretical work in these fields has identified population structure as important for pathogen life-history evolution, experimental tests are scarce. Here, we explore the impact of population structure on life-history evolution in phage T4, a viral pathogen of Escherichia coli. The host–pathogen system is propagated as a metapopulation in which migration between subpopulations is either spatially restricted or unrestricted. Restricted migration favours pathogens with low infectivity and low virulence. Unrestricted migration favours pathogens that enter and exit their hosts quickly, although they are less productive owing to rapid extirpation of the host population. The rise of such ‘rapacious’ phage produces a ‘tragedy of the commons’, in which better competitors lower productivity. We have now identified a genetic basis for a rapacious life history. Mutations at a single locus (rI) cause increased virulence and are sufficient to account for a negative relationship between phage competitive ability and productivity. A higher frequency of rI mutants under unrestricted migration signifies the evolution of rapaciousness in this treatment. Conversely, spatially restricted migration favours a more ‘prudent’ pathogen strategy, in which the tragedy of the commons is averted. As our results illustrate, profound epidemiological and ecological consequences of life-history evolution in a pathogen can have a simple genetic cause.
Keywords: experimental evolution, life-history evolution, metapopulation, prudence, tragedy of the commons, virulence
1. Introduction
Ecosystems are filled with victim–exploiter interactions, including prey and predators, plants and herbivores and hosts and pathogens. There is a certain irony in these ecological interactions—exploiters routinely destroy the very thing that supports their existence. While the long-term persistence of exploiters is served by prudent use of victim ‘resources’, rapid or wasteful exploitation often ensures short-term competitive success. A ‘tragedy of the commons’ arises when selection favours inefficient or unproductive use of common resources (Hardin 1968; Rankin et al. 2007; MacLean 2008).
As exploiters of host resources, pathogens may face tradeoffs between short-term success and long-term persistence (Frank 1996). When a living host is required for transmission, high rates of disease-induced mortality (virulence) negatively affect long-term prospects, all else being equal. Consequently, we may ask: why is any pathogen virulent? The ‘conventional wisdom’ is that pathogens always evolve towards avirulence (see May & Anderson (1983), Bull (1994), Levin (1996) and Alizon et al. (2009) for discussion). In this view, virulence is a hallmark of recent associations between hosts and pathogens. An alternative view is that pathogen virulence positively covaries with other advantageous traits, such as the transmission rate between hosts (May & Anderson 1983) or the competitive ability within hosts (Nowak & May 1994). In this view, the very properties that make a pathogen more virulent (e.g. rapid proliferation within the host) also make it more transmissible or more competitive. Contrary to the conventional wisdom, increased pathogen virulence may evolve in such cases (May & Anderson 1983; Bull 1994; Nowak & May 1994; Levin 1996; Alizon et al. 2009).
Empirical support for a positive relationship between virulence and transmission (Fraser et al. 2007; de Roode et al. 2008) or within-host competitive ability (Bell et al. 2006) has been found in several, but not all, cases (Ebert & Bull 2003; Alizon et al. 2009). Several ecological factors are predicted to influence virulence evolution in the presence of such relationships, including: host density, the timing and mode of transmission, the frequency of superinfection, host heterogeneity and host population structure (Galvani 2003). Yet only in a few studies have these factors been manipulated experimentally to test theoretical predictions (Bull et al. 1991; Cooper et al. 2002; Abedon et al. 2003; Heineman & Bull 2007). Here, we focus on the evolution of pathogen virulence in experiments where ecological factors influencing population structure are manipulated.
Ecological conditions that promote highly structured host populations are predicted to favour low virulence (Claessen & deRoos 1995; Boots & Sasaki 1999; Haraguchi & Sasaki 2000; O'Keefe & Antonovics 2002; van Baalen 2002; Lion & van Baalen 2008; Wild et al. 2009). For instance, when host movement is restricted owing to poor dispersal (perhaps in a viscous milieu; see Boots & Mealor 2007), highly virulent pathogens risk extinction by decimating local hosts before successful invasion of a new patch of hosts. Less virulent variants persist longer with their local hosts, allowing time for transmission to a new patch. Consequently, the pathogen population as a whole is predicted to be more prudent under these circumstances. At this point, experimental research on the effects of population structure on virulence evolution is scarce (Kerr et al. 2006; Boots & Mealor 2007). Thus, it is valuable to explore host–pathogen systems in which factors affecting structure can be manipulated, constraints yielding tradeoffs can be elucidated and virulence evolution can be tracked.
To probe these issues, we previously manipulated the metapopulation structure of an evolving host–pathogen system (Kerr et al. 2006). The host was Escherichia coli B and the pathogen was the lytic bacteriophage T4. The bacteria–phage community was distributed heterogeneously into the wells of two microtitre plates, forming a metapopulation of 192 subpopulations. Each metapopulation was propagated by serial transfer, at which time inter-well migrations were permitted. In the restricted migration treatment, migrations occurred only between neighbouring wells (electronic supplementary material, figure S1a). In the unrestricted migration treatment, migrations potentially occurred between any two wells (electronic supplementary material, figure S1c). In both treatments, migrations occurred with the same probability.
After 20 transfers (approx. 65 phage generations), phage isolates from each treatment were assessed for productivity (the number of progeny per parent over an incubation cycle in a well with abundant hosts) and competitive ability (relative fitness when competing with a marked strain for common hosts). Phage from the restricted migration treatment were significantly more productive, whereas phage from the unrestricted migration treatment were significantly more competitive. We labelled these strategies ‘prudent’ and ‘rapacious’, respectively, and noted that the rapacious strategy allowed for a potential tragedy of the commons. Here, we address the phenotypic and genetic foundations of these strategies, their relation to virulence and the underlying constraints leading to a tradeoff between productivity and competitive ability. Not only do we find that population structure can affect life-history evolution, but also that ecologically important changes to the pathogen can have a simple genetic basis.
2. Life cycle of lytic phage
We focus on various stages of the life cycle of phage T4, which is illustrated in figure 1. A phage particle outside its host (step 1) starts the infection cycle when its tail fibres contact cell surface moieties (step 2). Sufficient contact leads to irreversible binding and injection of the phage genome into the host cell (step 3). The bacterium then becomes a phage factory: the phage genome is copied, structural components are synthesized and phage particles are assembled (step 4). The complete assembly of the first progeny particle marks the end of the eclipse period (steps 3–5); however, progeny production continues until host lysis liberates intracellular phage, which marks the end of the latent period (steps 3–6). The number of infective phage particles released via host lysis is termed the burst size. If the progeny phage particle remains intact long enough to contact another host, the cycle begins again. Phage life history is defined by the transition rates in this life cycle.
Figure 1.

The life cycle of T4 phage (Mosig & Eiserling 2006). A phage particle outside its host (step 1) starts the infection cycle when its tail fibres contact the diglucosyl residues in the lipopolysaccharide (LPS) on the cell surface (step 2). These interactions are reversible and a phage particle may return to a free state. Eventually, the phage binds irreversibly (through interaction with heptose residues in the LPS core) and injects its genome into the bacterial cytoplasm (step 3). The bacterium now becomes a phage factory: the phage genome is copied, structural components are synthesized, the genome is packaged and phage are assembled (step 4). The complete assembly of the first infective particle marks the end of the eclipse period (steps 3–5). Following the production of more phage, the cell eventually lyses (step 6), which marks the end of the latent period (steps 3–6). Phage are still being constructed at the time of lysis (not shown) and any delay simply increases the number of mature progeny released (the burst size is larger). Upon release, the long tail fibres of phage T4 are stored near the base of the head before being deployed for the next infection. The cycle begins anew when a progeny phage contacts another host.
3. Materials and methods
Information about strains, media, acclimation procedures, genetic work (PCR, sequencing and engineering) and statistical analysis is provided in the electronic supplementary material.
(a). Experimental evolution
The full experimental set-up is explained in detail elsewhere (Kerr et al. 2006). Briefly, bacteria and phage were embedded in a metapopulation, consisting of two 96-well microtitre plates (each well was filled with 200 µl of supplemented MG growth medium). After 12 h of incubation, the ‘exhausted metapopulation’ was diluted 10-fold into a ‘fresh metapopulation’ with unused growth medium. This transfer preserved the metapopulation structure. Immediately following transfer, inter-well migrations occurred. Migration into any focal well happened with probability 0.45 and involved a 10-fold dilution from a single non-focal well in the exhausted metapopulation into the focal well in the fresh metapopulation. In the restricted migration treatment, migration into a focal well occurred from one of the north, south, east or west neighbouring wells (we used ‘wrap-around’ boundaries so that every well had four neighbours). In the unrestricted migration treatment, migration into a focal well occurred from one of any other well in the metapopulation. There were four replicates of the restricted and unrestricted migration treatments. We used a Biomek FX liquid-handling robot (Beckman-Coulter) to execute all dilutions and migrations. Plates were incubated and shaken between transfers (550 rpm on a microtitre shaker at 37°C). We ran the experiment for 20 transfers. One of the unrestricted replicates was terminated after 16 transfers owing to contamination.
(b). Adsorption assay
Acclimated phage were added to MG medium in a microtitre well to give a final volume of 180 µl (and an initial multiplicity of infection (MOI) ≈ 1 after bacterial addition). The initial titre of phage (p0) was estimated before 20 µl of fully grown acclimated bacteria was added to the well. The mixture was incubated at 37°C for exactly 10 min (the eclipse period for all tested phage isolates was greater than 10 min). At this time, the contents of the microtitre well were added to 30 µl of chloroform, mixed using a pipette, and spun for 2 min at 1300 rpm. Then the titre of phage in the supernatant (p10) was estimated. The adsorption rate, α, was calculated as α = ln[p0/p10]/10, which assumes that the concentration of free phage at minute T (pT) declines according to the exponential decay function pT = p0e−αT. Occasionally, our estimated α was slightly negative, which was probably owing to a very low adsorption rate combined with standard small plating errors; in such cases, α was reassigned to 0. The adsorption rate for each phage isolate was the average of three replicates.
(c). Spectrophotometric assay
The phage were added to a well with MG medium at a sufficient titre for an initial MOI ≈ 0.1 after bacterial addition. Acclimated bacteria were added by a 10-fold dilution from a stationary phase. At the same time, bacteria were added to four control wells without phage. The final volume in all wells was 200 µl. After bacterial addition, the optical density of each well was measured every 2 min for 8 h at 450 nm on a VersaMax spectrometer (Molecular Devices). The cultures were incubated at 37°C and shaken between each reading. Let B(t) be the average optical density of the wells with bacteria alone at time t. Let M(t) be the optical density of the well with phage and bacteria at time t. We measured virulence as the deviation in optical density between the well with bacteria and the well with both phage and bacteria: 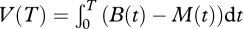 . Greater and earlier lysis of cells causes greater deviation. This index was found to give similar results to other measures of virulence (e.g. latent period from a one-step assay, see below) and the statistical results reported do not depend on the precise value of T (as long as it is sufficiently large). We approximated the above integral by using the trapezoidal Riemann sum at a resolution of 2 min. The virulence index for each phage isolate was the average of three or four replicates.
. Greater and earlier lysis of cells causes greater deviation. This index was found to give similar results to other measures of virulence (e.g. latent period from a one-step assay, see below) and the statistical results reported do not depend on the precise value of T (as long as it is sufficiently large). We approximated the above integral by using the trapezoidal Riemann sum at a resolution of 2 min. The virulence index for each phage isolate was the average of three or four replicates.
(d). Decay assay
Immediately after acclimation, the phage titre was estimated (p0). The phage were diluted 10-fold into a well with MG medium (final volume of 200 µl) without additional bacteria. After 12 h of incubation, the phage titre was estimated again (p12). This sequence of dilution and incubation was repeated twice more with the phage titre estimated at each time point (giving p24 and p36). The decay rate, δ, was calculated as the slope of the least-squares line between H (time in hours) and ln(pH) (the natural log of phage titre at hour H). This assumes that the titre of free phage at hour H decays (owing to disintegration and experimentally imposed dilution) exponentially: pH = p0e−δH. The decay rate for each phage isolate was the average of three replicates.
(e). One-step assay
This assay is described in detail elsewhere (Carlson 1994). Briefly, an overnight culture of bacteria was diluted one-eighth into MG medium and grown for 2 h at 37°C. Then, 500 µl of the growing bacterial culture was mixed in MG medium with the phage strain of interest to achieve an initial MOI ≈ 0.1 and a final volume of 1 ml. This mixture was incubated in a shaking water bath at 37°C. After 4 min, the mixture was diluted (1/5000 and 1/50 000) in MG and returned to the shaking water bath. At various time points over the next 75 min, samples from the incubating dilutions were plated directly with abundant bacteria in top agar to estimate the titre of free phage and infected cells. Also at various time points, samples from these incubating dilutions were mixed with chloroform, vortexed and placed on ice (later samples of the chloroform-free supernatant were plated with abundant bacteria to estimate the titre of free phage and mature intracellular phage). At time t, let X(t) be the titre of free phage and mature intracellular phage and let Y(t) be the titre of free phage and infected cells. Before intracellular maturation of phage, X(t) and Y(t) are fairly constant. At the end of the eclipse period, tE, X increases and Y remains unchanged. At the end of the latent period, tL (where tL > tE), Y increases. The Y data resemble a Hill function, y(t) = y0 + yf{ts/[(tL,e)s + ts]}. The inflection point parameter, tL,e, from the Hill least-squares fit to the data serves as our approximation to the latent period. The burst size is approximated by (yf − x0)/(y0 − x0), where x0 is the average of the first three X values (all before tE). Six replicate assays were run for each strain.
(f). Productivity assay
Acclimated bacteria were diluted 10-fold from the stationary phase into a well with MG medium. The phage strain of interest was added to achieve an initial MOI ≈ 1 and a final volume of 200 µl. The phage titre was estimated immediately after the bacteria and phage were mixed (p0) and then again 12 h later (p12). The productivity of the phage strain is defined to be p12/p0. Four replicate assays were run for each phage strain.
(g). Competition assay
Acclimated bacteria were diluted 10-fold from the stationary phase into a well with MG medium. A 1 : 10 mixture of the phage strain of interest and an rII mutant of T4 was added to achieve an initial MOI ≈ 1 and a final volume of 200 µl. The titres of both phage competitors were estimated immediately after the bacteria and phage were mixed (p0 and R0) and then again 12 h later (p12 and R12). These strains were distinguished by plating appropriate dilutions in top agar with either E. coli B or E. coli K-12 λ lysogen. Both strains will form plaques on E. coli B, but the rII mutant will not form plaques on λ lysogen hosts. Thus, the titre of the phage strain of interest was determined using the plaque count from the lysogen lawns and the titre of the rII mutant was determined by subtraction. Relative fitness is defined as the ‘per transfer’ fitness (Kerr et al. 2006), w(p, R) = p12R0/p0R12. Four replicate assays were run for each phage strain.
4. Results
Phage isolates from the last transfer of each metapopulation were characterized phenotypically and genetically (four isolates from each of the four replicate metapopulations in the two treatments for a total of 32 isolates). To avoid pseudoreplication, we averaged all isolates to give metapopulation replicate values for statistical analysis.
(a). Life-history changes
The first step in phage infection is adsorption, involving irreversible attachment of the phage particle to its host cell (figure 1, step 3). Adsorption rates were estimated by mixing phage with bacteria and quantifying extracellular phage over 10 min (§3). Adsorption rate serves as a measure of pathogen infectivity. Phage in the unrestricted migration treatment evolved to be significantly more infective than phage in the restricted migration treatment (figure 2a; Welch's two-sample t-test, 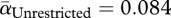 ,
, 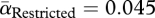 , t = 5.0738, d.f. = 5.606, p = 0.0028). The common ancestor for both treatments had an infectivity (αAncestor = 0.053; blue line in figure 2a) similar to its phage descendants in the restricted migration treatment (one-sample t-test, t = −1.345, d.f. = 3, p = 0.2713), whereas its descendants in the unrestricted migration treatment were more infective (one-sample t-test, t = 6.6056, d.f. = 3, p = 0.0071).
, t = 5.0738, d.f. = 5.606, p = 0.0028). The common ancestor for both treatments had an infectivity (αAncestor = 0.053; blue line in figure 2a) similar to its phage descendants in the restricted migration treatment (one-sample t-test, t = −1.345, d.f. = 3, p = 0.2713), whereas its descendants in the unrestricted migration treatment were more infective (one-sample t-test, t = 6.6056, d.f. = 3, p = 0.0071).
Figure 2.

Phage life-history evolution. When compared with phage evolved in the restricted migration treatment, phage in the unrestricted migration treatment have evolved (a) significantly faster adsorption (i.e. higher infectivity), (b) significantly faster lysis of the host population (i.e. higher virulence), and (c) non-significantly faster decay in the absence of host cells. The infectivity, virulence and decay of the ancestral phage are given by the dot-dashed blue lines. Virulence in (b) is measured by the integral difference in spectrophotometric absorbance between a well with bacteria alone and a well with both bacteria and phage. This ‘deviation integral’ measures how rapidly the phage clears its host population. Single examples are shown for (d) the ancestral phage, and phage isolates from (e) the restricted migration treatment and (f) the unrestricted migration treatment. In each case, the black curve is the absorbance of bacteria growing alone and the other coloured curve is the absorbance of the bacteria and phage incubating together. The area of the light shading is the virulence index in each case.
Phage virulence is inversely related to the length of the latent period, the time between phage adsorption and the release of phage progeny (figure 1, from step 3 to step 6). Time-intensive measures of the latent period (e.g. via one-step assays) were impractical given the large number of assays needed to characterize all evolved phage. Instead, we used a spectrophotometric assay in which the absorbance of a microtitre well with diluted phage and bacteria was compared with the absorbance of a well with diluted bacteria alone. The integral difference in absorbance between the two curves serves as an operational measure of virulence (§3). The more virulent the phage, the more they disrupt bacterial growth and the larger the difference in absorbance from the bacterial control (example absorbance profiles are shown in figure 2d–f, in the order of increasing virulence).
Phage in the unrestricted migration treatment evolved higher virulence than phage in the restricted migration treatment (figure 2b; Welch's two-sample t-test, 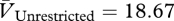 ,
, 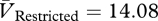 , t = 3.1893, d.f. = 5.502, p = 0.0213). The common ancestor was less virulent (VAncestor = 8.11; blue line in figure 2b) than its descendants in both treatments (one-sample t-tests: unrestricted: t = 12.4172, d.f. = 3, p = 0.0011; restricted: t = 5.148, d.f. = 3, p = 0.0142). By our measure, higher virulence can be caused by higher adsorption rates, shorter latent periods, greater burst sizes or some combination of these factors. However, one-step assays with genetically engineered mutants (see below) show that the latent period probably contributes to observed differences in virulence.
, t = 3.1893, d.f. = 5.502, p = 0.0213). The common ancestor was less virulent (VAncestor = 8.11; blue line in figure 2b) than its descendants in both treatments (one-sample t-tests: unrestricted: t = 12.4172, d.f. = 3, p = 0.0011; restricted: t = 5.148, d.f. = 3, p = 0.0142). By our measure, higher virulence can be caused by higher adsorption rates, shorter latent periods, greater burst sizes or some combination of these factors. However, one-step assays with genetically engineered mutants (see below) show that the latent period probably contributes to observed differences in virulence.
After host lysis (figure 1, step 6), liberated phage are free to infect new hosts—if they remain intact (De Paepe & Taddei 2006). Loss of phage before infection occurs through two routes in our evolution experiment: periodic dilution and particle disintegration. We refer to these processes collectively as ‘decay’. Decay rates were measured by titrating phage over successive dilutions and incubations in the absence of bacteria (§3). No significant differences in decay rates were found, although phage from the unrestricted migration treatment tend to decay faster than phage from the restricted migration treatment (figure 2c; Welch's two-sample t-test, 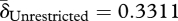 ,
, 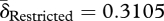 , t = 1.8366, d.f. = 5.421, p = 0.1212). Phage from both treatments had lower decay rates than the ancestor (δAncestor = 0.3586, one-sample t-tests: unrestricted: t = −4.2419, d.f. = 3, p = 0.024; restricted: t = −5.2765, d.f. = 3, p = 0.0133).
, t = 1.8366, d.f. = 5.421, p = 0.1212). Phage from both treatments had lower decay rates than the ancestor (δAncestor = 0.3586, one-sample t-tests: unrestricted: t = −4.2419, d.f. = 3, p = 0.024; restricted: t = −5.2765, d.f. = 3, p = 0.0133).
Unrestricted migration favoured the evolution of a ‘live-fast, die-young’ strategy. The phage from this treatment were more infective, more virulent and tended to be shorter lived than their counterparts evolving under restricted migration. We now turn to the genetic causes and population-level consequences of this evolved strategy.
(b). Genetics of virulence
The timing of host lysis after T4 infection is determined by the phage's holin, a protein that forms oligomeric complexes in the host's inner membrane. Membrane disruption by holin complexes allows a phage-encoded endolysin to access the host cell wall, causing degradation and host lysis (Ramanculov & Young 2001a; Young 2002; Tran et al. 2005; Young & Wang 2006). Mutations in holin genes in different species of phage have been shown to alter the latent period (Grundling et al. 2000; Ramanculov & Young 2001b; Abedon et al. 2003; Wang 2006). However, sequencing all of our isolates revealed no mutations at gene t, which codes for the holin in T4 (electronic supplementary material, Methods).
Phage T4 also encodes an antiholin, the product of gene rI, which binds to the holin and antagonizes holin-mediated inner-membrane disruption (Ramanculov & Young 2001a; Young 2002; Tran et al. 2005, 2007; Young & Wang 2006). This antiholin has been studied mostly for its role in the ‘lysis inhibition’ (LIN) phenotype in T4 (Paddison et al. 1998; Ramanculov & Young 2001a). LIN involves a delay in lysis when a T4-infected cell is secondarily infected by another T4. As the ratio of phage to bacteria increases, such secondary infections become more likely. Mutations in rI can lead to a LIN-defective phenotype: rapid lysis under multi-infection conditions (Paddison et al. 1998). In glucose-limited conditions, rI mutants display earlier lysis of their host even when the ratio of phage to bacteria is low and, thus, multiple infection is unlikely (Los et al. 2003). Therefore, under glucose-limited conditions (such as those present in our experiment), mutations in rI may decrease the latent period over a spectrum of ratios of phage to bacteria.
Sequencing revealed two frameshift mutations in rI (electronic supplementary material, Methods). The first, rIA, is a deletion of an adenine in a trimeric A repeat region on the coding strand (positions 59 400–59 402). The second, rIB, is a deletion of an adenine in a pentameric A repeat region (positions 59 299–59 303). Both frameshifts cause premature termination in the C-terminal domain of the RI antiholin (electronic supplementary material, figure S3).
In the unrestricted migration treatment, mutations at the rI locus were greater in prevalence (three rIA mutants and one rIB mutant out of 16 isolates) than in the restricted migration treatment (one rIA mutant out of 16 isolates). This trend, though intriguing, was based on too small a sample size for confidence. Thus, we sequenced an additional 96 randomly chosen isolates at the rI locus (12 isolates from each of the four replicates of the two migration treatments from the last transfer of the experiment). New rI mutations, including insertions, deletions and non-synonymous substitutions, were discovered in this collection of isolates (see the electronic supplementary material for details). Using all of the genetic data, we found that mutations at the rI locus were significantly more prevalent in the unrestricted migration treatment (14 mutants out of 64 isolates) than in the restricted migration treatment (two mutants out of 64 isolates) (likelihood ratio test, 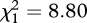 , p = 0.003, see the electronic supplementary material for details). Other statistical approaches, such as a generalized linear mixed-effects model, yielded similar conclusions (see the electronic supplementary material for details).
, p = 0.003, see the electronic supplementary material for details). Other statistical approaches, such as a generalized linear mixed-effects model, yielded similar conclusions (see the electronic supplementary material for details).
In our original collection of evolved phage isolates, strains with mutations at rI were the most virulent by our spectrophotometric assay. To assign a causal role for these mutations on virulence required controlling for genetic background. We therefore introduced the rIA and rIB alleles into the T4 wild-type background (electronic supplementary material, Methods). The rIA- and rIB-engineered strains were more virulent than the wild-type by the spectrophotometric assay (figure 3a; ANOVA, F2,6 = 80.25, p = 0.00005), but did not differ from wild-type in adsorption rate (data not shown).
Figure 3.

Virulence and the relation between productivity and competitive ability revealed by the rI mutants. (a) Using the spectrophotometric assay (§3), both rI mutants are significantly more virulent than the wild-type phage. Also, the rIA mutant is significantly more virulent than the rIB mutant. (b) Both rI mutants are significantly more competitive than the wild-type, and (c) significantly less productive than the wild-type. Also, the rIA mutant is significantly less productive than the rIB mutant. (d) The relationship between productivity and competitive ability is negative, driven largely by the poorly competing, highly productive wild-type and the highly competitive, less productive rI mutants.
We performed one-step assays to explore the components of the enhanced virulence in the engineered mutants (Carlson (1994) and §3). Briefly, phage were mixed with bacteria at low multiplicity of infection, diluted and then samples were titrated at various time points. Initial plaque counts estimated the concentration of free phage and infected cells in the original mixture. The latent period was determined from the time point at which the plaque numbers experienced a dramatic increase owing to host lysis and progeny release in the mixture (the ‘one-step’; figure 4a). The average number of phage released per infected cell (burst size) can also be computed from the one-step assay (§3).
Figure 4.

One-step analysis of the rI mutants. Tracking the abundance of plaque-forming units (PFUs) following infection of host cells under low multiplicity of infection generates a step-like increase in PFUs when the host cells are lysed. The timing of the step marks the end of the latent period. (a) Here we show single-example one-step curves for the rIA mutant (red), the rIB mutant (orange) and the wild-type (blue). For readability, we have normalized PFU abundance on the ordinate and focused on the period of time between 35 and 75 min (where most lysis occurs). (b) The number of phage progeny released per infection (the burst size) was significantly smaller for the rIA mutant than for the wild-type. The rIB mutant did not differ significantly from either the wild-type or the other mutant. (c) The latent periods of the rIA- and rIB-engineered mutants are significantly shorter than the latent period of the wild-type. (d) The relationship between latent period and burst size is positive: the longer the infection, the larger the number of progeny. The colours in (b) to (d) match the colours of the different genotypes given for (a).
The latent periods of rIA- and rIB-engineered mutants are significantly shorter than that of wild-type T4 (figure 4c; ANOVA, F2,15 = 8.1801, p = 0.004; multiple comparisons using Tukey's HSD). The burst size of the rIA and rIB mutants are smaller than the burst size of wild-type (figure 4b; ANOVA, F2,15 = 3.8215, p = 0.0456), the rIA mutant significantly so (Tukey's HSD). The positive relationship between latent period and burst size (figure 4d) is consistent with previous studies (Hutchison & Sinsheimer 1966; Josslin 1970; Wang 2006). The increased virulence attributable to rI mutations comes with a concomitant decrease in progeny production. Thus, we find that a tradeoff between generation time and fecundity is mediated through mutations in a single gene.
(c). The tragedy of the commons
The rI mutations have population-level consequences. A defining feature of r-mutants in T4 (including rI mutants) is an altered plaque morphology (Paddison et al. 1998; Ramanculov & Young 2001a; Young 2002). We find that plaques of rIA and rIB mutants are large and clear with crisp boundaries, whereas wild-type plaques are smaller and cloudy with indistinct boundaries (figure 5a). Also, when incubated in a tube at the same initial titre, rIA and rIB phage completely clear a bacterial culture, whereas the wild-type T4 leaves the tube cloudy over the same time period (figure 5b). Owing to their enhanced virulence, the engineered rI mutants rapaciously exploit their host (figure 3a). Such rapaciousness could equate with good competitive ability when common host resources are at stake. However, rapid host use may not translate to efficient host use. When competitive displacement leads to lower group productivity, the ingredients for a tragedy of the commons are present (Hardin 1968; Rankin et al. 2007; MacLean 2008).
Figure 5.

Population-scale phenotypes of the rI mutants. (a) On a lawn of sensitive bacteria, the rI mutants form large clear plaques with crisp boundaries. The wild-type forms smaller turbid plaques with fuzzy boundaries. (b) After 2 h of incubation with a population of host cells in a test tube, the rI mutants completely clear the bacterial culture while the wild-type leaves the tube turbid. (All cultures in this photograph were initiated with the same concentrations of phage and bacteria).
In our original experiment, we measured the productivity and competitive ability of evolved phage isolates (Kerr et al. 2006). The productivity of an isolate is given by the number of progeny phage per parent phage over a 12 h incubation period in a well, while the competitive ability of an isolate is its relative fitness in pair-wise competition with a marked phage strain (§3). Phage isolates from the unrestricted migration treatment were more competitive, whereas phage isolates from the restricted migration treatment were more productive (Kerr et al. 2006). Here we find that the wild-type and engineered rI mutants echo this basic pattern. The rI mutants are significantly more competitive than the wild-type (figure 3b; ANOVA, F2,9 = 28.376, p = 0.0001; comparisons by Tukey's HSD), but the wild-type is significantly more productive (figure 3c; F2,9 = 909.54, p ≪ 0.0001; comparisons by Tukey's HSD). Pooling the data on these three strains, we see a negative relationship between productivity and competitive ability similar to the relationship in our original study (figure 3d). Thus, the higher frequency of rI mutants in the unrestricted migration treatment is consistent with a tragedy of the commons, whereas this tragedy is largely averted in the restricted migration treatment.
5. Discussion
Given their relatively simple life cycles and the ease of measuring various life stages, lytic bacteriophages make ideal subjects for the study of life-history evolution (Chao et al. 1977; Lenski & Levin 1985; Abedon 1989, 2008; Wang et al. 1996; Abedon et al. 2001, 2003; Bull et al. 2004; Bull 2006; Wang 2006; Heineman & Bull 2007; Shao & Wang 2008). One life-history component of particular interest is the length of the latent period. Because phage progeny accumulate over time within a host cell (Hutchison & Sinsheimer 1966; Josslin 1970; Wang 2006), longer latent periods translate to more progeny. However, a shorter latent period can decrease the time to the next infection. Thus, lytic phage face a basic tradeoff between fecundity and generation time, which is mediated by their latent period (Frank 1996; Abedon et al. 2003; Bull et al. 2004; Abedon 2008).
Some insight into latent-period evolution is gained from optimal foraging theory. Phage can be likened to foragers in a patchy environment, where host cells constitute resource ‘patches’ (see the electronic supplementary material for details). Theory predicts that if rates of resource acquisition diminish with patch use, then shorter travel times between patches promote shorter patch residence times (Charnov 1976; Stephens & Krebs 1986). Similarly, shorter dispersal periods (figure 1, from step 6 to step 2) should favour shorter latent periods (Abedon 1989, 2008; Wang et al. 1996; Abedon et al. 2001; Bull et al. 2004; electronic supplementary material). For a phage ‘forager,’ the ‘inter-patch travel time’ (dispersal period) is influenced by host density and adsorption rate. Experimental manipulation of host density (Abedon et al. 2003; Wang 2006; Heineman & Bull 2007) and adsorption rate (Shao & Wang 2008) in competing or evolving phage populations shows qualitative agreement with theoretical predictions.
Evolution in our experiment is consistent with these predictions as well. Average ‘travel time’ between hosts is shorter in our unrestricted migration treatment. Phage isolates from this treatment possess a higher frequency of mutations at the rI locus that shortens latent period (figure 4a,c). The majority of rI mutations were frameshift mutations, which disrupt the antiholin and cause rapid lysis (under low multiplicities of infection) and a breakdown of LIN (under high multiplicities of infection). Interestingly, we did discover significant differences in virulence between two of these frameshift mutations (rIA and rIB), suggesting that there could be some fine-tuning of virulence at this genetic locus.
In addition to latent-period evolution, phage in the unrestricted migration treatment evolved significantly faster adsorption than phage from the restricted migration treatment. In optimal foraging models, the forager should always decrease travel time between patches if there are no costs involved. One way phage can reduce dispersal time is to increase their adsorption rate. Increased infectivity in the unrestricted migration treatment accomplishes this end. So why does infectivity not increase in the restricted migration treatment (figure 2a)? One answer is that while infectivity always evolves to high levels, phage in the restricted migration treatment have experienced fewer generations than phage in the unrestricted migration treatment. However, phage in both treatments have experienced about the same number of generations (Kerr et al. 2006). The answer we favour focuses on a violation of an assumption of the optimality model.
This is the assumption that travel time is constant. In our case, this would mean host density is constant (but see Abedon et al. (2001) for a caveat). We note that there would be no advantage to maintain low adsorption rate if every subpopulation within a metapopulation were continuously supplied with a constant density of bacterial cells. However, given that migration immediately followed serial dilution, the phage in our experiment always encountered bacterial populations growing with limited resources. The speed at which hosts are infected directly influences the number of future hosts, as only uninfected hosts reproduce. Slower adsorption (and a longer latent period) could favour higher productivity in the long run as phage use an expanded host population. Regardless of the group-level benefits of restraint, phage that adsorb faster enjoy a local competitive advantage, because they access shared hosts first. Thus, myopic gain works against long-term welfare.
Evidently, patterns of migration affect how the conflict between short-term and long-term payoffs is resolved. Phage possessing rapid adsorption and lysis have short-term competitive advantages (figure 3b and Kerr et al. 2006). However, these rapacious phage are less productive (figure 3c and Kerr et al. 2006). Given the periodic dilution that characterizes our experiment, less productive phage are less persistent when host cells are absent. All else being equal, hosts are encountered at a lower rate in the restricted migration treatment, which places a premium on persistence. With restricted access to hosts, subpopulations of rapacious phage are frequently diluted out before reaching hosts, whereas subpopulations of phage with slower adsorption, longer latent period and LIN persist long enough to contact new hosts. In the unrestricted migration treatment, global access to host resources allows the tragedy of the commons generated by rapacity within a subpopulation to be transferred to a tragedy at the metapopulation scale.
Our findings echo a number of theoretical predictions suggesting spatial structure favours prudent use of resources when within-group competitive ability conflicts with between-group productivity or persistence (Frank 1996; Keeling 2000; Sella & Lachmann 2000). For instance, in cyclical communities, such as molecular hypercycles or non-transitive triplets of competitors, competitive restraint is predicted given spatial structure (Boerlijst & Hogeweg 1991; Johnson & Seinen 2002; Prado & Kerr 2008). Boots & Mealor (2007) addressed these issues in an elegant experiment with a granulosis virus that infects phycitiid moth larvae. They controlled the population structure of the host by manipulating the viscosity of the food medium in which the larvae moved. The virus evolved lower infectivity in the most viscous treatment, a result consistent with the idea that spatial structure favours restraint.
Several theoretical studies predict that spatial structure also favours lower levels of pathogen virulence (Claessen & deRoos 1995; Boots & Sasaki 1999; Haraguchi & Sasaki 2000; O'Keefe & Antonovics 2002; van Baalen 2002; Lion & van Baalen 2008; Wild et al. 2009). Our results provide some of the first experimental support of these predictions (figures 2b, 3a, 4b and 5). We also found that other life-history components of the pathogen evolve in response to spatial structure. Similar to the study by Boots & Mealor (2007), we found that phage in our restricted migration treatment were significantly less infective than phage from our unrestricted migration treatment (figure 2a). In our study, both adsorption rate and latent period influence how quickly a growing bacterial population is used. The prudence found under spatially restricted migration is achieved through relatively lower infectivity and virulence (figure 2a,b). This is consistent with the prediction that spatial structure generally favours prudent parasite exploitation of the local host supply (van Baalen 2002; Lion & van Baalen 2008).
Most discussions about the evolution and potential management of virulence have hinged on the existence and form of virulence tradeoffs (Bull 1994; Levin 1996; Ebert & Bull 2003; Alizon et al. 2009). Recently, a positive relationship between virulence and transmission was described for HIV-1 (Fraser et al. 2007) and a protozoan parasite of monarch butterflies (de Roode et al. 2008). More empirical data are needed to gauge the existence, type and form of tradeoffs in different pathogens (for a recent survey of tradeoffs within coliphage, see De Paepe & Taddei (2006)). Some of the basic tradeoffs faced by phage T4 are well-understood. Because phage progeny accumulate within an infected cell, the phage face a tradeoff between generation time and fecundity (Abedon et al. 2003; Bull et al. 2004; Abedon 2008). In a growing population of hosts, this basic tradeoff underlies a productivity tradeoff between rate and yield (figure 3). Thus, virulence can be seen as a strategy for rapid increase (but, ultimately, low yield) within a host subpopulation. Infectivity can be seen in a similar light. In our host–pathogen system, the degree of spatial structure influences the success of these rapacious tendencies and thus affects the evolution of pathogen virulence and infectivity.
Acknowledgements
We thank J. Cooper, B. Miner, J. Nahum, K. Walag, J. Wald and J. West for helpful discussion, S. Decew, T. Edwards, K. Laegreid, J. Nahum, K. Reagan and B. Rogers for laboratory help and J. Hille Ris Lambers for statistical advice. M. Bonsall, B. Charlesworth, S. Frank and one anonymous reviewer provided very helpful comments on previous versions of this manuscript. B.K. would like to thank M. Bonsall and B. Charlesworth for the invitation to participate in the Royal Society meeting: ‘Genetics and the causes of evolution: 150 years of progress since Darwin’. This research was supported by NSF grant 071746 awarded to B.K. and A.M.D.
Footnotes
One contribution of 18 to a Discussion Meeting Issue ‘Genetics and the causes of evolution: 150 years of progress since Darwin’.
References
- Abedon S. T.1989Selection for bacteriophage latent period length by bacterial density—a theoretical-examination. Microb. Ecol. 18, 79–88 (doi:10.1007/BF02030117) [DOI] [PubMed] [Google Scholar]
- Abedon S. T.2008Phage population growth: constraints, games, adaptation. In Bacteriophage ecology: population growth, evolution, and impact of bacterial viruses (ed. Abedon S. T.), pp. 64–93 Cambridge, UK: Cambridge University Press [Google Scholar]
- Abedon S. T., Herschler T. D., Stopar D.2001Bacteriophage latent-period evolution as a response to resource availability. Appl. Environ. Microbiol. 67, 4233–4241 (doi:10.1128/AEM.67.9.4233-4241.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abedon S. T., Hyman P., Thomas C.2003Experimental examination of bacteriophage latent-period evolution as a response to bacterial availability. Appl. Environ. Microbiol. 69, 7499–7506 (doi:10.1128/AEM.69.12.7499-7506.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizon S., Hurford A., Mideo N., Van Baalen M.2009Virulence evolution and the trade-off hypothesis: history, current state of affairs and the future. J. Evol. Biol. 22, 245–259 (doi:10.1111/j.1420-9101.2008.01658.x) [DOI] [PubMed] [Google Scholar]
- Bell A. S., De Roode J. C., Sim D., Read A. F.2006Within-host competition in genetically diverse malaria infections: parasite virulence and competitive success. Evolution 60, 1358–1371 [PubMed] [Google Scholar]
- Boerlijst M. C., Hogeweg P.1991Spiral wave structure in pre-biotic evolution: hypercycles stable against parasites. Physica D 48, 17–28 (doi:10.1016/0167-2789(91)90049-F) [Google Scholar]
- Boots M., Mealor M.2007Local interactions select for lower pathogen infectivity. Science 315, 1284–1286 (doi:10.1126/science.1137126) [DOI] [PubMed] [Google Scholar]
- Boots M., Sasaki A.1999‘Small worlds’ and the evolution of virulence: infection occurs locally and at a distance. Proc. R. Soc. Lond. B 266, 1933–1938 (doi:10.1098/rspb.1999.0869) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull J. J.1994Virulence. Evolution 48, 1423–1437 (doi:10.2307/2410237) [DOI] [PubMed] [Google Scholar]
- Bull J. J.2006Optimality models of phage life history and parallels in disease evolution. J. Theor. Biol. 241, 928–938 (doi:10.1016/j.jtbi.2006.01.027) [DOI] [PubMed] [Google Scholar]
- Bull J. J., Molineux I. J., Rice W. R.1991Selection of benevolence in a host–parasite system. Evolution 45, 875–882 (doi:10.2307/2409695) [DOI] [PubMed] [Google Scholar]
- Bull J. J., Pfennig D. W., Wang I. N.2004Genetic details, optimization and phage life histories. Trends Ecol. Evol. 19, 76–82 (doi:10.1016/j.tree.2003.10.008) [DOI] [PubMed] [Google Scholar]
- Carlson K.1994Single-step growth. In Molecular biology of bacteriophage T4 (ed. Karam J. D.), pp. 434–437 Washington, DC: ASM Press [Google Scholar]
- Chao L., Levin B. R., Stewart F. M.1977Complex community in a simple habitat—experimental study with bacteria and phage. Ecology 58, 369–378 (doi:10.2307/1935611) [Google Scholar]
- Charnov E. L.1976Optimal foraging, marginal value theorem. Theor. Popul. Biol. 9, 129–136 (doi:10.1016/0040-5809(76)90040-X) [DOI] [PubMed] [Google Scholar]
- Claessen D., deRoos A. M.1995Evolution of virulence in a host–pathogen system with local pathogen transmission. Oikos 74, 401–413 (doi:10.2307/3545985) [Google Scholar]
- Cooper V. S., Reiskind M. H., Miller J. A., Shelton K. A., Walther B. A., Elkinton J. S., Ewald P. W.2002Timing of transmission and the evolution of virulence of an insect virus. Proc. R. Soc. Lond. B 269, 1161–1165 (doi:10.1098/rspb.2002.1976) [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paepe M., Taddei F.2006Viruses' life history: towards a mechanistic basis of a trade-off between survival and reproduction among phages. PLoS Biol. 4, e193 (doi:10.1371/journal.pbio.0040193) [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Roode J. C., Yates A. J., Altizer S.2008Virulence-transmission trade-offs and population divergence in virulence in a naturally occurring butterfly parasite. Proc. Natl Acad. Sci. USA 105, 7489–7494 (doi:10.1073/pnas.0710909105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert D., Bull J. J.2003Challenging the trade-off model for the evolution of virulence: is virulence management feasible? Trends Microbiol. 11, 15–20 (doi:10.1016/S0966-842X(02)00003-3) [DOI] [PubMed] [Google Scholar]
- Frank S. A.1996Models of parasite virulence. Q. Rev. Biol. 71, 37–78 (doi:10.1086/419267) [DOI] [PubMed] [Google Scholar]
- Fraser C., Hollingsworth T. D., Chapman R., de Wolf F., Hanage W. P.2007Variation in HIV-1 set-point viral load: epidemiological analysis and an evolutionary hypothesis. Proc. Natl Acad. Sci. USA 104, 17 441–17 446 (doi:10.1073/pnas.0708559104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvani A. P.2003Epidemiology meets evolutionary ecology. Trends Ecol. Evol. 18, 132–139 (doi:10.1016/S0169-5347(02)00050-2) [Google Scholar]
- Grundling A., Blasi U., Young R.2000Genetic and biochemical analysis of dimer and oligomer interactions of the lambda S holin. J. Bacteriol. 182, 6082–6090 (doi:10.1128/JB.182.21.6082-6090.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraguchi Y., Sasaki A.2000The evolution of parasite virulence and transmission rate in a spatially structured population. J. Theor. Biol. 203, 85–96 (doi:10.1006/jtbi.1999.1065) [DOI] [PubMed] [Google Scholar]
- Hardin G.1968The tragedy of the commons. Science 162, 1243–1248 [PubMed] [Google Scholar]
- Heineman R. H., Bull J. J.2007Testing optimality with experimental evolution: lysis time in a bacteriophage. Evolution 61, 1695–1709 (doi:10.1111/j.1558-5646.2007.00132.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison C. A., Sinsheimer R. L.1966Process of infection with bacteriophage fX174. X. Mutations in a fX lysis gene. J. Mol. Biol. 18, 429–447 (doi:10.1016/S0022-2836(66)80035-9) [DOI] [PubMed] [Google Scholar]
- Johnson C. R., Seinen I.2002Selection for restraint in competitive ability in spatial competition systems. Proc. R. Soc. Lond. B 269, 655–663 (doi:10.1098/rspb.2001.1948) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josslin R.1970Lysis mechanism of phage T4: mutants affecting lysis. Virology 40, 719–726 (doi:10.1016/0042-6822(70)90216-3) [DOI] [PubMed] [Google Scholar]
- Keeling M.2000Evolutionary trade-offs at two time-scales: competition versus persistence. Proc. R. Soc. Lond. B 267, 385–391 (doi:10.1098/rspb.2000.1013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr B., Neuhauser C., Bohannan B. J. M., Dean A. M.2006Local migration promotes competitive restraint in a host–pathogen ‘tragedy of the commons'. Nature 442, 75–78 (doi:10.1038/nature04864) [DOI] [PubMed] [Google Scholar]
- Lenski R. E., Levin B. R.1985Constraints on the coevolution of bacteria and virulent phage—a model, some experiments, and predictions for natural communities. Am. Nat. 125, 585–602 [Google Scholar]
- Levin B. R.1996The evolution and maintenance of virulence in microparasites. Emerg. Infect. Dis. 2, 93–102 (doi:10.3201/eid0202.960203) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lion S., van Baalen M.2008Self-structuring in spatial evolutionary ecology. Ecol. Lett. 11, 277–295 (doi:10.1111/j.1461-0248.2007.01132.x) [DOI] [PubMed] [Google Scholar]
- Los M., Wegrzyn G., Neubauer P.2003A role for bacteriophage T4 rI gene function in the control of phage development during pseudolysogeny and in slowly growing host cells. Res. Microbiol. 154, 547–552 (doi:10.1016/S0923-2508(03)00151-7) [DOI] [PubMed] [Google Scholar]
- MacLean R. C.2008The tragedy of the commons in microbial populations: insights from theoretical, comparative and experimental studies. Heredity 100, 233–239 (doi:10.1038/sj.hdy.6801073) [DOI] [PubMed] [Google Scholar]
- May R. M., Anderson R. M.1983Parasite host coevolution. In Coevolution (eds Futuyama D. J., Slatkin M.), pp. 186–206 Sunderland, UK: Sinauer [Google Scholar]
- Mosig G., Eiserling F.2006T4 and related phages: structure and development. In The bacteriophages (eds Calendar R., Abedon S. T.), Washington, DC: ASM Press [Google Scholar]
- Nowak M. A., May R. M.1994Superinfection and the evolution of parasite virulence. Proc. R. Soc. Lond. B 255, 81–89 (doi:10.1098/rspb.1994.0012) [DOI] [PubMed] [Google Scholar]
- O'Keefe K. J., Antonovics J.2002Playing by different rules: the evolution of virulence in sterilizing pathogens. Am. Nat. 159, 597–605 (doi:10.1086/339990) [DOI] [PubMed] [Google Scholar]
- Paddison P., Abedon S. T., Dressman H. K., Gailbreath K., Tracy J., Mosser E., Neitzel J., Guttman B., Kutter E.1998The roles of the bacteriophage T4 r genes in lysis inhibition and fine-structure genetics: a new perspective. Genetics 148, 1539–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado F., Kerr B.2008The evolution of restraint in bacterial biofilms under nontransitive competition. Evolution 62, 538–548 (doi:10.1111/j.1558-5646.2007.00266.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanculov E., Young R.2001aAn ancient player unmasked: T4 rI encodes a t-specific antiholin. Mol. Microbiol. 41, 575–583 (doi:10.1046/j.1365-2958.2001.02491.x) [DOI] [PubMed] [Google Scholar]
- Ramanculov E., Young R.2001bGenetic analysis of the T4 holin: timing and topology. Gene 265, 25–36 (doi:10.1016/S0378-1119(01)00365-1) [DOI] [PubMed] [Google Scholar]
- Rankin D. J., Bargum K., Kokko H.2007The tragedy of the commons in evolutionary biology. Trends Ecol. Evol. 22, 643–651 (doi:10.1016/j.tree.2007.07.009) [DOI] [PubMed] [Google Scholar]
- Sella G., Lachmann M.2000On the dynamic persistence of cooperation: how lower individual fitness induces higher survivability. J. Theor. Biol. 206, 465–485 (doi:10.1006/jtbi.2000.2130) [DOI] [PubMed] [Google Scholar]
- Shao Y. P., Wang I. N.2008Bacteriophage adsorption rate and optimal lysis time. Genetics 180, 471–482 (doi:10.1534/genetics.108.090100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens D. W., Krebs J. R.1986Foraging theory. Princeton, NJ: Princeton University Press [Google Scholar]
- Tran T. A. T., Struck D. K., Young R.2005Periplasmic domains define holin–antiholin interactions in T4 lysis inhibition. J. Bacteriol. 187, 6631–6640 (doi:10.1128/JB.187.19.6631-6640.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran T. A. T., Struck D. K., Young R.2007The T4 RI antiholin has an N-terminal signal anchor release domain that targets it for degradation by DegP. J. Bacteriol. 189, 7618–7625 (doi:10.1128/JB.00854-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Baalen M.2002Contact networks and the evolution of virulence. In Adaptive dynamics of infectious diseases: in pursuit of virulence management (eds Dieckmann U., Metz J. A. J., Sabelis M. W., Sigmund K.), pp. 85–103 Cambridge, UK: Cambridge University Press [Google Scholar]
- Wang I. N.2006Lysis timing and bacteriophage fitness. Genetics 172, 17–26 (doi:10.1534/genetics.105.045922) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang I. N., Dykhuizen D. E., Slobodkin L. B.1996The evolution of phage lysis timing. Evol. Ecol. 10, 545–558 (doi:10.1007/BF01237884) [Google Scholar]
- Wild G., Gardner A., West S. A.2009Adaptation and the evolution of parasite virulence in a connected world. Nature 459, 983–986 (doi:10.1038/nature08071) [DOI] [PubMed] [Google Scholar]
- Young R.2002Bacteriophage holins: deadly diversity. J. Mol. Microbiol. Biotechnol. 4, 21–36 [PubMed] [Google Scholar]
- Young R., Wang I. N.2006Phage lysis. In The bacteriophages (eds Calendar R., Abedon S. T.), pp. 104–125 Oxford, UK: Oxford University Press [Google Scholar]