

Abstract
The prevalence of recombination in eukaryotes poses one of the most puzzling questions in biology. The most compelling general explanation is that recombination facilitates selection by breaking down the negative associations generated by random drift (i.e. Hill–Robertson interference, HRI). I classify the effects of HRI owing to: deleterious mutation, balancing selection and selective sweeps on: neutral diversity, rates of adaptation and the mutation load. These effects are mediated primarily by the density of deleterious mutations and of selective sweeps. Sequence polymorphism and divergence suggest that these rates may be high enough to cause significant interference even in genomic regions of high recombination. However, neither seems able to generate enough variance in fitness to select strongly for high rates of recombination. It is plausible that spatial and temporal fluctuations in selection generate much more fitness variance, and hence selection for recombination, than can be explained by uniformly deleterious mutations or species-wide selective sweeps.
Keywords: Hill–Robertson interference, evolution of recombination, background selection, fixation probability, mutation load, drift load
1. Introduction
Evolutionary biology is a young field. We are celebrating the 150th anniversary of the publication of the Origin of Species; it is little more than a century since the establishment of classical genetics, which led to the development of population genetic theory; and it is only 50 years since the discovery of the structure of DNA and the genetic code, which gave us the tools for studying the genomes of all organisms, and which has culminated in large-scale genome sequencing. We now know far more than Darwin did about the relationships between all organisms, about the genetic system that they all share, about the genetic basis of phenotypic variation and about how selection works to build adaptations. Yet, despite this mass of detailed knowledge, several major questions that puzzled Darwin remain unanswered. I will focus on one of these: why are sex and recombination so widespread? I will argue that we have a good theoretical understanding of how this is likely to be explained, but that empirical confirmation depends on answering a larger and more fundamental question—what is the extent and genetic basis of fitness variation?
Virtually all organisms have evolved with the aid of recombination. Although bacteria, archaea and viruses do not have regular sex, their genomes can recombine through a variety of mechanisms, and recombination is essential for the spread of favourable alleles. In contrast, most eukaryotes have obligate meiotic sex—if not in every generation, then as part of a resting or dispersal stage (Bell 1982). Asexual eukaryote species are almost all young, and have a predominantly sexual ancestry. It is hardly conceivable that the elaborate machinery of sexual reproduction and meiosis is a by-product of selection for (say) DNA repair, though that may have been its origin (Maynard Smith 1988). I will take it that sex and recombination are adaptations whose purpose is to bring together new combinations of alleles.
Understanding sex and recombination has been perhaps the most puzzling issue in evolutionary biology—which suggests that its solution might give us a much better intuition about the evolutionary process in general. How can the random shuffling of genes be advantageous, when its immediate effect is to break up well-adapted sets of alleles? The strongest theoretical result in the field is the ‘reduction principle’ (Feldman et al. 1996): in a randomly mating population at equilibrium under selection, recombination can never increase. Moreover, there are all kinds of costs associated with recombination—the twofold cost of sex that arises with anisogamy, indirect costs of sexual selection, and so on (Maynard Smith 1978).
(a). Recombination facilitates selection
In fact, we have in principle known for a long time why sex and recombination are so widespread: they facilitate natural selection by generating useful variation (Weismann 1889). It is clear empirically that adaptation requires recombination as well as selection. Asexual species face a much higher rate of extinction (Maynard Smith 1978; Bell 1982; Engelstädter 2008), and non-recombining regions of genome degenerate (e.g. Y chromosomes; Charlesworth et al. 2009). Plant and animal breeders have long known the importance of outcrossing (Darwin 1876), and the extraordinary increases in crop yield that have been essential to feeding mankind have come from the efficient selection of large sexual populations. A less familiar example comes from evolutionary computation, where algorithms are competed against each other; almost all techniques include some kind of recombination (Mitchell 1998).
(b). Selection assembles adaptations step-by-step
The intimate relation between selection and recombination can be seen from an argument that was raised by the early geneticists (Provine 1971), and that is still found in the ‘intelligent design’ literature: that selection merely picks from pre-existing variation, and can create nothing new. In principle, an F2 cross between two true-breeding populations contains every possible genotype, and the fittest could be picked in a single step. However, if there is variation at more than a few loci, any particular genotype will be extremely improbable—with 20 differences, the chance of getting any particular homozygous genotype is 2−40 to 10−12. Indeed, if reproduction following the initial cross were entirely asexual, then only the fittest that actually arose could be selected, and these would be unlikely to be more than a few standard deviations away (electronic supplementary material, S1).
In any reasonably sized sexual population, selection works step-by-step. In the first generation, alleles that are individually favourable increase in frequency, until the fittest combination starts to appear at appreciable frequency, and can be picked up by selection. This argument requires a fairly smooth fitness landscape: alleles that have individually favourable effects when averaged over their current genetic background must give a still fitter genotype when brought together (Livnat et al. 2008). Allelic effects need not be strictly additive, but must generally keep the same sign across a variety of backgrounds. Recombination only aids selection if adaptation is largely a process of climbing the adaptive landscape—but fitness may nevertheless be far from additive. The key issue is whether the landscape is smooth enough that a population can always make progress, rather than being trapped at local optima.
(c). Recombination can help selection by increasing the additive genetic variance
In very large populations, selection can assemble successive mutations without any help from recombination: every possible single-step mutation arises in every generation, and so selection can work with mutation alone (see Maynard Smith 1978). However, in moderately large populations (Nμ < 1), recombination is more important than mutation in generating fitter combinations. To understand the importance of recombination in a more general way, we start from Fisher's (1930) ‘Fundamental Theorem’: the increase in mean fitness caused by selection on allele frequencies is equal to the additive genetic variance in relative fitness. Recombination can only help selection to increase mean fitness if it increases the additive genetic variance in fitness. Crucially, it can do this only if there are negative associations among favourable alleles. If alleles are already randomly shuffled, then recombination has no further effect: recombination can only alter the population if there is linkage disequilibrium between the alleles. If this is positive, so that there is an excess of ++ and − − combinations, then recombination reduces additive variance, and so slows down selection. It can only speed up selection if there is an excess of negative associations (+ with −, − with +) that tend to shield alleles from selection by reducing the additive variance in fitness (Charlesworth 1993; Barton 1995).
(d). Negative epistasis causes negative linkage disequilibria
Understanding how recombination can facilitate selection hinges on understanding why linkage disequilibria should be predominantly negative. One possibility is that selection favours negative associations—in other words, epistasis is systematically negative. This would be plausible if selection acts via quantitative traits that are under stabilizing selection, but with a moving optimum. Another argument is a population can only maintain itself, despite deleterious mutation at a total rate U > 1, if there is negative epistasis, combined with sex (Kimura & Maruyama 1966; Kondrashov 1988). However, there is no clear evidence that epistasis between spontaneous mutations is systematically negative (de Visser & Elena 2007; Halligan & Keightley 2009). Indeed, variance in epistasis tends to select against recombination: the recombination load depends on mean square epistasis, εij2, whereas the interaction between directional selection si,sj and epistasis εij selects for recombination via terms like siεijsj. Finally, simulations show that even in the most favourable cases, the negative associations caused by negative epistasis have less effect than do those caused by random drift, as long as there are very many selected loci and the population is not extremely large (Otto & Barton 2001; Iles et al. 2003; Keightley & Otto 2006).
(e). Random drift causes negative linkage disequilibria
How can random drift combine with directional selection to generate negative associations between alleles? Although drift is as likely to produce an excess of positive as negative associations, selection will sweep positive associations out of the population more rapidly, leaving behind negative associations that are selected more slowly. The extreme case is where there is no recombination at all. As Fisher (1930) and Muller (1932) argued, favourable mutations almost always arise on different genetic backgrounds and cannot be brought together. Instead, they compete with each other, so that only one can be fixed. (electronic supplementary material, S2 shows how this can be understood in terms of linkage disequilibria).
Hill & Robertson (1966) introduced another way to think about interference between selected loci. Any gene is embedded in a background that may affect its fitness. Thus, selection must disentangle the causal effect of the focal allele from the effect of the random genetic background in which it finds itself; because it stays with this background for approximately 1/r generations, where r is the recombination rate, the variance in allele frequency will be inflated by a factor 1/r2 (Robertson 1961). This random variance will, on average, reduce the response to selection, because under directional selection, random associations are converted into negative linkage disequilibria that reduce the response to selection.
What are the effects of Hill–Robertson interference (HRI) on neutral diversity, on the rate of adaptation from new mutations and on the mutation load? These are difficult theoretical questions, harder to answer than for the deterministic effects of epistasis. This is because the problem is stochastic, and also because we are interested in selection on very many loci. The theory is incomplete, but in the following section, I lay out the contribution of deleterious mutations, of balancing selection and of recurrent selective sweeps to HRI. I then consider whether, in the light of evidence from sequence variation, these sources of selection are sufficient to select for recombination.
2. Effects of HRI: theory
How do different kinds of selection affect linked loci through the inflation of random drift that is caused by HRI? Specifically, what are the effects on neutral diversity, on rates of adaptation and on mutation load? The basic measures of these various effects are explained in the electronic supplementary material, S3: HRI reduces the mean pairwise coalescence time; it reduces the fixation probability of beneficial mutations, and hence reduces the rate of adaptive substitution, Λ; and HRI increases the mutation load to the extent that drift impedes selection against deleterious alleles. Below, I summarize the existing theoretical results (and a few new ones)—aiming to find rough approximations that depend on measurable quantities.
(a). Deleterious mutations
First, consider the effect of deleterious mutations. This has been studied in a variety of ways: strongly deleterious mutations (Nes ≫ 1, where Ne is the effective population size, and s the selective disadvantage) in a sexual population cause ‘background selection’ (Nordborg et al. 1996); the cumulative effect of weakly deleterious mutations (Nes ∼ 1) cause ‘weak selection HRI’ (McVean & Charlesworth 2000); and the inevitable accumulation of deleterious alleles with one-way mutation in an asexual population is termed ‘Muller's ratchet’ (Muller 1964; Felsenstein 1974; Haigh 1978). Yet, all these are different aspects of the interaction between mutation, selection and drift, studied in different parameter ranges.
A sufficiently large asexual population will reach an equilibrium between mutation and selection. Assuming multiplicative effect s, then the fraction of genomes carrying no mutations has frequency exp(−U/s), which may be very low; U is the rate of deleterious mutation per genome. This fittest class is likely to be lost by chance if Nes exp(−U/s) is small (say, less than 10). The rate of Muller's ratchet has been studied intensely (reviewed by Rouzine et al. 2008). A modifier that causes recombination can gain a strong advantage, equal to the rate of the ratchet (Gordo & Campos 2008). However, once there is appreciable recombination, the advantage of a further increase becomes far smaller (figure 1).
Figure 1.

Tight linkage exacerbates the increase in mutation load owing to drift, via the Hill–Robertson effect. The horizontal line shows the baseline mutation load in an infinite population (L = U), and the lower curve shows the diffusion approximation assuming free recombination. The dots show simulated values, for r = 0.5 (grey dots), 0.01 (black dots) and 0 (unfilled dots) between adjacent loci. Simulations are of haploid individuals with n = 100 loci on a linear chromosome; U = nμ = 1, s = 0.05; runs were for 5000 generations with a burn-in of 1000 generations. Mutation was symmetric, so that an equilibrium is reached even with complete linkage.
In an asexual species that is maintaining itself despite an influx of deleterious mutation, all individuals descend from an ancestor that is in the fittest class; otherwise, the population would be fixing deleterious mutations (Charlesworth & Charlesworth 1997). Moreover, a random gene traces back into the fittest class in approximately 1/s generations, which is much shorter than the time of coalescence if Nes is large. Neutral diversity is approximately equal to that in the small fraction of very fit individuals, plus that owing to recent deleterious mutations.
The fixation probability of a favourable allele, and hence the rate of adaptation, is reduced in a similar way to the neutral diversity in an asexual population. A weakly favoured allele can only be established if it arises within the fittest class, and so the net rate of adaptation is reduced by exp(−U/s), just as for the neutral diversity. Alleles with a stronger advantage can fix within a wide range of backgrounds, and so allow a higher rate of adaptation. However, they will carry with them deleterious alleles, which may cause an irreversible loss of function that offsets the increase in fitness owing to the new mutations (Peck 1994; Johnson & Barton 2002; Hadany & Feldman 2005). Even though each substitution increases the overall fitness, the process may nevertheless lead to a long-term decline.
Unless an asexual population is extremely large, and all mutations are strongly selected (Nesexp(−U/s) > 1), one-way mutation leads to an indefinite increase in the mutation load via Muller's ratchet. This is a special case of HRI, with loss of the fittest class causing negative linkage disequilibrium, in which alleles with opposing effects are shielded from selection. However, low rates of recombination or back mutation will stop the ratchet (figure 1).
At the opposite extreme, with unlinked loci, genes find themselves on genetic backgrounds that change from generation to generation. With polygamy, the expected coalescence time is reduced by a factor of exp(−4v) under the infinitesimal model (Barton 2009). Thus, a relatively modest additive variance in fitness, v, can greatly reduce neutral diversity. (Note that the variance in fitness owing to deleterious mutation is Us, so that the effect of unlinked loci will tend to be due to strongly deleterious alleles.)
Because the effect of unlinked loci is mediated by short-term fluctuations, the reduction in the rate of adaptation and the increase in mutation load can both be described by the same change in effective population size, Ne/N = exp(−4v). Whether the mutational load is significantly increased depends on what fraction of that load is contributed by weakly selected alleles (Nes ∼ 1).
A linear genome lies somewhere between the extreme cases of asexuality and no linkage, and at first sight would seem harder to analyse. However, a remarkably simple approximation is available based on Robertson's (1961) argument. The effect of variance in fitness at map distance r is ∼v/r2, and so is dominated by tightly linked loci. However, the effect saturates at r ∼ s, and so the integral over a linear genome of length R is ∼v/Rs. Since the variance in fitness owing to deleterious mutations is v = Us, the net effect is ∼ exp(−U/R) (Hudson & Kaplan 1995; Nordborg et al. 1996). A more detailed calculation that takes account of variation in gene density along the map has been fitted to data on nucleotide diversity in Drosophila: a simple model of background selection accounts for the reduced diversity seen in regions of low recombination quite well, provided that the contribution from transposable elements is included (Charlesworth 1996).
The probability of fixation of a favourable allele, embedded in a linear chromosome, can be approximated by the same formula—just as for asexual and for unlinked loci, the effects of deleterious mutations on fixation probability are the same as those on neutral diversity (Barton 1995; Santiago & Caballero 1998). Again, for mutation load, we expect an effect that is described by the change in Nes (figure 1).
This theory has largely been developed for the effects of strongly deleterious mutations (Nes > 1). If drift causes significant fluctuations in the frequency of deleterious mutations, then their effect on linked loci is reduced (Barton & Etheridge 2004). This helps explain why diversity is not reduced as much in regions of very low recombination as expected (by exp(−U/R) on a linear genome, or exp(−U/s) with no recombination): interference between alleles reduces their joint effect (Kaiser & Charlesworth 2008; Charlesworth et al. 2009). Nevertheless, a very large number of weakly selected sites can have a significant cumulative effect (McVean & Charlesworth 2000).
(b). Balancing selection
In an asexual population, balancing selection can maintain multiple coexisting clones if these exploit different limiting resources (e.g. Vrijenhoek 1994). If such clones were maintained for extremely long times, then diversity between them could become extremely high, with coalescence times between genes in different clones being at least as long as the ages of those clones. Indeed, Cohan (2002) has argued that such distinct asexual ecotypes should be regarded as species, since drift and selective sweeps within them will keep each relatively homogeneous.
Even though balancing selection may greatly increase coalescence times, the probability that a favourable mutation will fix within an ecotype is not affected. With strict asexuality, different adaptive alleles will fix within different ecotypes, causing functional divergence between them. However, even an extremely low rate of recombination will allow universally favoured alleles to spread across the whole set of clones, in which case the overall rate of adaptation will not be altered by balancing selection.
How will the mutation load be altered by balancing selection on an asexual population? Muller's ratchet will act within each ecotype, and so rarer types will collapse under their load of mutations. Thus, balancing selection can increase the mutation load even as it increases the net diversity across the whole set of ecotypes.
Balancing selection among unlinked loci will have little effect, though the additive variance in fitness may perhaps be slightly reduced. On a linear genetic map, neutral diversity is increased only within a very narrow region (r ∼ 1/Ne; Kreitman 1983, but see Begun et al. 1999). The fixation probability is not altered by balancing selection, since favourable mutations can establish within either genetic background in the usual way, without being affected appreciably while they are rare. Similarly, the mutation load will hardly be affected by balancing selection in a sexual population.
Navarro & Barton (2002) showed that balancing selection on multiple tightly linked sites could greatly increase diversity, because each of a very large number of selected genotypes could accumulate different neutral mutations. However, in a finite population, only a limited number of different selected genotypes can be maintained, and so the effect on neutral diversity reaches an upper limit as the number of sites under balancing selection increases.
It seems that long-established balanced polymorphisms that show a signature of increased diversity are rare (Bubb et al. 2006). However, in reality, balanced polymorphisms will fluctuate and so will show the signature of reduced diversity characteristic of directional selection. This seems to be the case for chromosomal inversions in Drosophila (e.g. Andolfatto et al. 2001).
(c). Recurrent selective sweeps
In an asexual population, favourable mutations compete with each other for fixation (Fisher 1930; Muller 1932). It takes about (1/s)log(4Nes) generations for an allele to fix, and other mutations that arise during this time can only themselves fix if they are on the successful background. When advantageous mutations are frequent, successful mutations arise within clones while they are still rare. Nevertheless, adaptation is greatly slowed because it must happen in series, rather than in parallel (Rouzine et al. 2008).
The effect on neutral diversity has not been studied explicitly, but is closely tied to the adaptive process. With strict asexuality, and in a very large population, lineages coalesce when a new favourable mutation appears. Thus, if two sampled genes are in the same clone, they will coalesce at the most recent mutation, while if they are in different clones, they coalesce just before the origin of the most recent mutation that they share. The mean coalescence time is implicit in the approximations for the rate of adaptation, Λ. For well-spaced events at rate Λ, it is ∼1/Λ, while for overlapping sweeps, driven by selection of strength S, it is ∼(1/S)log(2NeS).
Recurrent sweeps will carry with them any deleterious mutation that was on the original background. In a population that is maintaining itself despite deleterious mutations (i.e. when the ratchet is not clicking), a favoured mutation can only fix if the net effect, when combined with the background on which it arises, raises fitness above that of the currently fittest class. Since, assuming multiplicative effects, the typical individual has fitness a factor exp(−U) less than the fittest class; this means that a beneficial allele with an effect greater than exp(U) is likely to be fixed, whereas those with weaker effects can only fix if they are on an exceptionally fit background (Johnson & Barton 2002). Moreover, those strongly favoured alleles that do fix will typically carry with them approximately U/s deleterious alleles. Although in a narrow sense such strong selective sweeps increase overall fitness, they may cause an irreversible loss of function through the fixation of large numbers of weakly deleterious alleles (Hadany & Feldman 2005).
With no linkage, the Hill–Robertson effect is again mediated by the net heritable variance in log fitness, v, which reduces diversity and fixation probability by exp(−4v) (assuming polygamy), and which increases the mutation load by allowing the fixation of deleterious mutations with Nes < 1. Since the variance in fitness owing to sweeps at a rate Λ is just 2ΛS, this effect may usually be negligible: as discussed below, Λ is much less than the rate of deleterious mutation, U (at least, in the few species for which we have data). So, unless the selection on favourable substitutions is far stronger than against deleterious mutations, the latter are likely to contribute much more fitness variance. However, any directional selection will contribute to the heritable variance in fitness, and so, seen broadly, unlinked loci may have a substantial influence. Also, the Hill–Robertson effect sets an upper limit to the rate of adaptation, because if the fitness variance becomes large, most beneficial alleles will be lost by chance. This limit rises only logarithmically with the baseline rate of adaptation (see above).
Following Maynard Smith & Haigh's (1974) seminal paper, most theoretical work has focused on the effects of selective sweeps on a linear genome. To a good approximation, neutral diversity immediately after a sweep is reduced on average by (2NeS)−2r/S (see electronic supplementary material, S4). We have seen that background selection has a similar effect on neutral diversity and on the rate of adaptation via new favourable mutations. In contrast, selective sweeps have a much weaker effect on strongly selected alleles, but a much stronger effort on weakly favoured alleles, relative to their effects on neutral diversity; this gives a strong bias towards adaptation based on strongly selected alleles. A strongly favoured mutation is likely to either be lost, or to be established in large numbers, before the next nearby sweep, and so its chances are hardly affected (cf. Karasov et al. in press). In contrast, a weakly favoured mutation will tend to grow slowly, and will experience many sweeps, which will almost always occur on a different background and so will tend to knock it down to a lower frequency (figure 2). To a good approximation, the probability of fixation of an allele with a small advantage s is reduced by a factor of 1 − (s/S)r/S (Barton 1995). Thus, adaptation is impeded over a much wider region of genome than is neutral diversity if log(2NeS) ≫ 2 log(S/s). Averaging over the genome, we find that its fixation probability is given simply by the classical formula 2s*, where s* = s − sc is its net rate of increase, allowing for the average rate sc at which it is knocked back by linked sweeps. Thus, there is a critical threshold, sc, below which a weakly favoured allele is very unlikely to fix (Barton 1994). The critical selection coefficient is proportional to the heritable variance in fitness caused by sweeps per map unit: sc ∼ (2ΛS/R) (π2 /(3log(S/s))). This sensitivity of weakly favoured alleles is also seen in the effects of multiple bottlenecks, which have the same disproportionate effect on weakly favoured alleles (Barton 1987).
Figure 2.

An allele with advantage s = 2 × 10−5 declines over time as a result of random selective sweeps at linked loci. In this example, sweeps with strength S = 0.01 occur at a density Λ/R = 0.01 per Morgan, in a population of 2N = 106. The expected rate of decline owing to linked sweeps is sc = (2ΛS/R)(π2/3log[S/s]) ∼ 0.0001. However, the actual decline is highly variable.
Because selective sweeps have a different effect from steady sampling drift, and from background selection, we may hope to be able to detect them from sequence data. Specifically, selective sweeps are expected to produce a characteristic pattern of linkage disequilibrium with common haplotypes, an excess of rare variants and heterogeneity along the genome. However, as just noted, their effects at any one location are similar to multiple bottlenecks, and though the latter have the same expected effects along the genome, they nevertheless cause strong heterogeneity. Thus, distinguishing recurrent selective sweeps from demography is difficult.
How do recurrent sweeps affect the mutation load? A strong selective sweep will raise bad alleles to high frequency, and may fix them if they are close to the favourable mutation. As discussed above, Johnson & Barton (2002) analysed this process for the asexual case, but the case of a linear genome seems not to have been studied. If the mutation load is U, on a map of length R, then the load that is swept up is roughly s ∼ (U/r)/(N∫pqPdt), where p is the fixation probability of the recombinant (see electronic supplementary material, S4). For example, if the density of the mutation load is U/R ∼ 0.2 per Morgan, 2N = 106, and the sweep is driven by selection S = 0.01, then s ∼ 4.1 × 10−5, due to fixation of a region r ∼ 0.01 cM on each side of the sweep.
A more detailed calculation is needed, that takes into account the probability of survival of recombinants of different lengths. Also, it is not clear what the equilibrium load would be when this increase is balanced by back-mutation. However, this rough argument suggests that selective sweeps will only completely fix an extremely small segment of genome in a large population, and so will not generate much additional load. The cumulative effect of random fluctuations caused by multiple sweeps may be much larger: the additional load is ∼U/(4Nes) (see electronic supplementary material, S3), and so substituting the rate of drift, 1/2Ne, owing to selective sweeps from §3d, we have ∼(U/(2s))(1/(2Ne)) ∼ ((U/R)(S/s)(Λ/log(2NS)). Nevertheless, this may be negligible relative to the mutation load, if the density of sweeps Λ/R ≪ 1.
3. Effects of HRI: facts
The overall effect of HRI on the rate of adaptation and the mutation load—and hence on the evolution of recombination—depends largely on the extent of selection, relative to recombination. For unlinked loci, what matters is the heritable variance in fitness, while for a linear genome, the key parameters are the variance of fitness owing to sweeps per map length (2ΛS/R) and the density of deleterious mutations (U/R). Sequence data provide us with good knowledge of the total rate of deleterious mutation, U, and the rate of species-wide selective sweeps, Λ, in Drosophila and a few other taxa. Such estimates of U and Λ are relatively straightforward and hence robust. However, it is difficult to go further and disentangle the strength, nature and spatial structure of selection.
(a). Recombination
We have known the total length of the genetic map since the early years of classical genetics, and we now know its detailed fine-scale structure. Both sperm typing and inference from linkage disequilibria show that in many taxa, recombination is concentrated at ‘hot spots’ (Singh et al. 2009); in primates at least, these move rapidly on evolutionary time scales (Coop & Przeworski 2007).
(b). Deleterious mutation
Sequencing of replicate inbred lines has given us the first direct estimates of the total mutation rate, which are consistent with the rate of the neutral molecular clock (3.5–8.4 × 10−9 point mutations per base per generation in Drosophila; e.g. Haag-Liautard et al. 2007; Keightley et al. 2009). When combined with the fraction of conserved (and presumably functional) sequence, this gives the total rate of deleterious mutation (U = 1.2 in D. melanogaster and U > 0.48 in Caenorhabditis elegans, respectively; Denver et al. 2004; Haag-Liautard et al. 2007; the latter counts only mutations that change amino acids). These estimates imply a high mutation load, which may be alleviated by sexual reproduction and negative epistasis (Kondrashov 1988; electronic supplementary material, S3). These estimates are far higher than indirect estimates from the mean and variance of fitness components across mutation accumulation lines, presumably because they include mutations of very small effect (Halligan & Keightley 2009; Keightley & Halligan 2009).
The distribution of effects of new non-synonymous mutations on fitness can be estimated from the distributions of rare synonymous versus non-synonymous alleles, and by comparing these distributions between species with different effective size (Kimura 1983; Ohta & Gillespie 1996; Loewe & Charlesworth 2006; Loewe et al. 2006; Eyre-Walker & Keightley 2007). Such estimates suggest a wide range of effects on a log-scale: for example, by comparing D. pseudoobscura and D. miranda, Loewe & Charlesworth (2006) found that a log-normal distribution could account for the existence of both dominant lethals and near-neutral alleles; assuming that all mutations had some non-zero effect, they estimated that similar proportions have negative selection coefficients below 2 × 10−5, between 2 × 10−5 and 2 × 10−3 and greater than 2 × 10−3; about 5 per cent were effectively lethal. This implies a mean deleterious effect of 2.8 per cent, and a root mean square effect of approximately 10 per cent—both these being dominated by the contribution from the small fraction of mutations of very large effect.
(c). Adaptive substitution
Both the rate of neutral divergence between species and neutral polymorphism within them is equal to the mutation rate. The observed excess of non-synonymous divergence over polymorphism implies that a large fraction of amino-acid differences between species have been positively selected (approx. 40% in Drosophila, say), and that at least as much divergence has been driven by positive selection in non-coding regions (McDonald & Kreitman 1991; Smith & Eyre-Walker 2002; Eyre-Walker & Keightley 2009). These estimates are sensitive to the presence of slightly deleterious mutations and to demography (Charlesworth & Eyre Walker 2007; Zeng & Charlesworth 2009), but different methods agree in giving an estimate of Λ which may be as high as 1/200 amino-acid substitutions per genome per generation in D. melanogaster, with still more from non-coding changes (Sella et al. 2009).
(d). Recombination correlates with sequence diversity
The strongest evidence for HRI in recombining genomes comes from the correlation between diversity and recombination, seen most clearly in D. melanogaster (Begun & Aquadro 1992; Sella et al. 2009). It is not easy to disentangle the effects of HRI from the mutagenic effects of recombination, but similar patterns are seen in other Drosophila species (Shapiro et al. 2007; Kulathinal et al. 2008; Sella et al. 2009). The positive relation between diversity and recombination could be accounted for by either background selection or recurrent selective sweeps acting alone or, more likely, by some combination of the two. It has proved difficult to distinguish between these different kinds of selection to estimate the effect of HRI on diversity in regions of high recombination, and to estimate the strength of the selection involved.
(e). Variation in diversity along the genome
Additional information comes from variation in diversity along the genome: a single sweep eliminates variation at the selected site and reduces diversity in a region of width of approximately s/log(4Nes). Thus, a low rate of strongly selected sweeps causes extreme variation in diversity, whereas a higher rate of weaker sweeps gives a smoother pattern. In addition, if rates of adaptive divergence differ consistently from gene to gene, a negative correlation between amino-acid divergence and neutral diversity will be seen (Macpherson et al. 2007). Yet more information comes from distortions in the allele frequency spectrum (e.g. Tajima's D) and characteristic linkage disequilibria, with particular haplotypes being raised to high frequency by partial or ‘soft’ sweeps.
The Drosophila data have been used to estimate the rate and strength of sweeps (Kim & Stephan 2000; Nurminsky 2001; Macpherson et al. 2007; Jensen et al. 2008; Sella et al. 2009). However, such estimates are confounded by background selection (which reduces diversity while generating little variation along the genome) and by bottlenecks or other forms of population structure (which can generate strong variation along the genome). Current estimates of selection strength span a wide range (from s = 10−5 (Jensen et al. 2008) to s = 10−2 (Macpherson et al. 2007)), although this range may be compatible with a mixture of weakly and strongly selected sweeps (Sella et al. 2009), as is estimated to be the case for deleterious mutations. However, such a wide range of selection on adaptive substitutions would be hard to reconcile with strong HRI, which should suppress adaptation via weakly favoured alleles.
(f). Selection on recombination
The combined effects of HRI owing to background selection and to selective sweeps could be substantial, at least in multicellular eukaryotes—as can be seen directly in the reduced diversity in regions of low recombination. Indeed, as Maynard Smith & Haigh (1974) first argued, selective sweeps must be the dominant source of random drift in very large populations (Gillespie 2001). The rate of adaptation owing to weakly favoured alleles (i.e. those with 1/Ne < s ≪ 2ΛS/R) sets a lower limit to the contribution of weakly selected adaptations.
However, it is not clear that these sources of HRI are enough to maintain recombination despite its obvious costs. Simulation and analytical theory for two selected loci, plus a recombination modifier, show selection for recombination that is significant and stronger than the effect owing to epistasis even in moderately large populations (Otto & Barton 2001; Keightley & Otto 2006), but nevertheless is weak in absolute terms. Typically, the rate of fixation of the modifier is reported relative to the neutral expectation, which depends on (roughly speaking) Nes. In a large population, a substantial increase in the relative rate of fixation can be produced by very weak selection, approximately 2/Ne. HRI itself reduces Ne, implying somewhat stronger selection, s, for a given Nes. Nevertheless, it remains hard to see from this theoretical work that recombination could be maintained unless it has very little cost.
Although good approximations are available for models with two selected loci, it has not yet been possible to integrate over the effect of mutations that are scattered randomly in time and genomic location. Barton & Otto (2005) show how random drift generates negative linkage disequilibria between loci under directional selection, which leads to selection for recombination modifiers proportional to the product of the additive variance in fitness at the selected loci. Summing over loci implies that the total selection for recombination will be proportional to the square of the variance in fitness, divided by population size, which is likely to be small. But sadly, selection on recombination in this framework is dominated by alleles that increase from one or a few copies, and by very tightly linked loci—and in both cases, the approximations break down.
Otto & Barton (1997) and Roze & Barton (2006) develop a complementary approach which uses a branching process to model the increase in a recombination modifier through an entire selective sweep. Recombination is favoured for two distinct reasons: first, it increases the probability of fixation of a favourable allele, and second, it speeds up its fixation once it has become common. The net effect of recurrent sweeps of strength S was estimated as CS(Λ/R)2, with C ∼ 2–3 for N = 105–107 and S = 0.05. Even given the highest estimates for the rates of sweeps in Drosophila, this implies very weak selection.
Most seriously, it is difficult to extend analytical theory to large numbers of selected sites, and simulations suggest that the theory for pairs of selected loci just described fails to describe interactions across the whole genome. Selection for recombination can be fairly strong if very large numbers of sites are selected (Iles et al. 2003; Keightley & Otto 2006; Hartfield et al. 2010). Understanding the joint effect on recombination of a large number of loci presents the most important outstanding theoretical issue in this area.
4. Local adaptation
Even though both the rates of deleterious mutation and of species-wide sweeps are high enough to cause substantial HRI, they are unlikely to sustain much variance in fitness: this is equal to US, 2ΛS, respectively, and so will be small unless there is a large contribution from very strongly selected mutations (Keightley & Halligan 2009). Indeed, direct estimates of the increase in variance in fitness components across mutation accumulation lines are low: Halligan & Keightley (2009) give an average 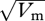 , relative to the mean, of 1.7 per cent across all studies (about half being of viability in D. melanogaster). Yet, the high additive genetic variance of fitness components in both laboratory and nature makes it plausible that the heritable variance in fitness itself could be high (Burt 2000; Merila & Sheldon 2000). For example, if the breeding value of log fitness were normally distributed with v = 0.1, then 95 per cent of individuals would have a breeding value for fitness between 0.43 and 1.65; with monogamy and unlinked loci, this would reduce neutral diversity and the rate of adaptation by a factor of e−9v = 0.57. Thus, it could be that most heritable variance in fitness is due to selection that fluctuates in space and time.
, relative to the mean, of 1.7 per cent across all studies (about half being of viability in D. melanogaster). Yet, the high additive genetic variance of fitness components in both laboratory and nature makes it plausible that the heritable variance in fitness itself could be high (Burt 2000; Merila & Sheldon 2000). For example, if the breeding value of log fitness were normally distributed with v = 0.1, then 95 per cent of individuals would have a breeding value for fitness between 0.43 and 1.65; with monogamy and unlinked loci, this would reduce neutral diversity and the rate of adaptation by a factor of e−9v = 0.57. Thus, it could be that most heritable variance in fitness is due to selection that fluctuates in space and time.
Lenormand & Otto (2000) showed that spatial subdivision can favour recombination, even with no epistasis or drift, provided that there is a negative covariance between selection coefficients at different loci. Moreover, this deterministic effect of gene flow can maintain high rates of recombination. Martin et al. (2006) analysed the case where recombination is favoured because it facilitates the spread of a new mutation that is favoured everywhere. In this model, random drift within local demes favours recombination through the Hill–Robertson effect. This can generate substantial selection for a modifier, even with deme sizes in the thousands. Martin et al.'s (2006) model involves species-wide sweeps and so is still limited by the slow rate of adaptive substitution, Λ. There may be a much higher rate of local sweeps, which would also select for recombination. However, if the various alleles are maintained polymorphic in the species as a whole, then linkage disequilibria, and hence selection for recombination, would presumably be weaker.
Can local adaptation be a source of the substantial fitness variance required to select strongly for recombination? While this is an attractive idea, there are some problems. First, consistent local selection for different sets of alleles in different places will select for tighter linkage (Lenormand & Otto 2000; Kirkpatrick & Barton 2006). Second, strong local adaptation leads to reproductive isolation that impedes further adaptation—suggesting that there may be an upper limit to the degree of local adaptation (see electronic supplementary material, S5). Thus, the rate of local sweeps might not be high enough to drive significant selection for recombination. However, if there is indeed a substantial barrier to gene flow into locally adapted populations, recombination will be favoured because it allows locally favoured alleles to escape from other locally unfavourable alleles; yet, this will be offset by the selection against introgression of multiple maladapted alleles. Moreover, in diploids, heterosis will aid introgression (Ingvarsson & Whitlock 2000) and may also select for sex and recombination. This is a complex question which needs more theoretical analysis, but which could also be studied empirically, through measurements of the extent of local adaptation and of barriers to introgression between local demes.
5. Conclusions
The population genetics of molecular evolution currently focuses almost exclusively on two kinds of selection: deleterious mutation and beneficial mutation. This is understandable since the effects of both can be summarized by their total rates (U,Λ), and these rates can be measured quite reliably. The evidence reviewed above shows that in Drosophila and a few other model eukaryotes, both rates are high, with a genomic rate of deleterious mutation around 1, and species-wide selective sweeps occurring at rates as high as around 0.005 per generation. These rates approach the upper limits set by load arguments, though those limits may be surpassed if there is negative epistasis (Haldane 1957; Kimura & Maruyama 1966). Though detailed studies concentrate on D. melanogaster, these estimates are likely to be typical: mutation rates and the size of the functional genome are broadly similar across multicellular eukaryotes.
Deleterious mutation and adaptive substitution may be common enough to account for the correlation between neutral diversity and recombination, and may significantly reduce diversity in regions of high recombination in Drosophila (Sella et al. 2009). Indeed, selective sweeps are likely to be the main cause of genetic drift in very abundant species (Maynard Smith & Haigh 1974; Gillespie 2001). However, because both involve mutations with modest effects on fitness, they are unlikely to generate enough variance in fitness to select strongly for high rates of recombination. Although it is satisfying that sequence polymorphism and divergence can give us good estimates of U, Λ, neither provides a compelling explanation for the prevalence of sex and recombination.
Can we really believe that natural populations are subject to the homogeneous selection envisaged by the current view of molecular evolution? Almost all traits show high genetic variance, and their heritable variance is just as high for components of fitness (Roff & Mousseau 1987). Moreover, quantitative traits typically are under strong selection in nature (directional, stabilizing and disruptive; Kingsolver et al. 2001). It is possible in principle that abundant heritability is maintained in a mutation–selection balance, but if so, the alleles involved would have to be under quite weak selection (s ∼ Vm/Vg ∼ 10−3; Johnson & Barton 2005). It seems to me more plausible that changes in environment in space and time cause corresponding fluctuations in selection on the underlying alleles. This may maintain balanced polymorphism, or may more broadly maintain a flux of alleles that leads to much higher diversity in selected alleles and traits than would arise from a static mutation–selection balance. This view is still constrained by the puzzling constancy of the molecular clock and by the modest rate of species-wide sweeps. Nevertheless, the extraordinary diversity of natural populations—the crux of Darwin's argument in The Origin and the most important discovery since the evolutionary synthesis—support this view, and suggests strongly that local populations experience much more selected change than does the species as a whole.
If the additive genetic variance in fitness were as high as approximately 0.1, then there would be substantial HRI between even unlinked loci. A modifier of sex or recombination would experience a more or less immediate advantage through experiencing an effectively lower rate of drift. (Modifiers that altered the variance in offspring number—‘bet hedging'—would have similar population genetics.)
Of course, Hamilton (1996) strongly argued for the importance of interactions between pathogen and host as a cause of strongly fluctuating selection that can drive the evolution of sex. However, though simulations show how such coevolution can select for recombination, it remains unclear what population genetic mechanisms are involved (though see Peters & Lively 1998; Otto & Nuismer 2004; Salathé et al. 2009). Host–pathogen coevolution can be seen as one source of fluctuating selection that may select for recombination via HRI.
It is a major theoretical challenge to understand the consequences of spatially and temporally fluctuating selection, and to find how to estimate its strength and nature from sequence data. Indirect inferences will need to be reconciled with direct observations of gene flow, variance in fitness and local adaptation. While it will be difficult to analyse the real complexity of the ‘entangled bank’, models of fluctuating and heterogeneous selection will give us a richer and more realistic understanding of natural populations.
Acknowledgements
I would like to thank the Royal Society and Wolfson Foundation for their support, and Brian Charlesworth and Sally Otto for their helpful comments.
Footnotes
One contribution of 18 to a Discussion Meeting Issue ‘Genetics and the causes of evolution: 150 years of progress since Darwin’.
References
- Andolfatto P., Depaulis F., Navarro A.2001Inversion polymorphisms and nucleotide polymorphism: lessons from Drosophila. Genet. Res. 77, 1–8 [DOI] [PubMed] [Google Scholar]
- Barton N. H.1987The probability of establishment of an advantageous mutation in a subdivided population. Genet. Res. 50, 35–40 (doi:10.1017/S0016672300023314) [DOI] [PubMed] [Google Scholar]
- Barton N. H.1994The reduction in fixation probability caused by substitutions at linked loci. Genet. Res. 64, 199–208 (doi:10.1017/S0016672300032857) [Google Scholar]
- Barton N. H.1995Linkage and the limits to natural selection. Genetics 140, 821–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton N. H.2009Why sex and recombination? Cold Spring Harb. Symp. Quant. Biol. 74 (doi:10.1101/sqb.2009.74.030) [DOI] [PubMed] [Google Scholar]
- Barton N. H., Etheridge A. M.2004The effect of selection on genealogies. Genetics 166, 1115–1131 (doi:10.1534/genetics.166.2.1115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton N. H., Otto S. P.2005Evolution of recombination due to random drift. Genetics 169, 2353–2370 (doi:10.1534/genetics.104.032821) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun D. J., Aquadro C. F.1992Levels of naturally occurring DNA polymorphism correlate with recombination rate in Drosophila melanogaster. Nature 356, 519–520 (doi:10.1038/356519a0) [DOI] [PubMed] [Google Scholar]
- Begun D. J., Betancourt A. J., Langley C. H., Stephan W.1999Is the fast/slow allozyme variation at the Adh locus of Drosophila melanogaster an ancient balanced polymorphism? Mol. Biol. Evol. 16, 1816–1819 [DOI] [PubMed] [Google Scholar]
- Bell G.1982The masterpiece of nature: the evolution and genetics of sexuality. Berkeley, CA: University of California Press [Google Scholar]
- Bubb K. L., Bovee D., Buckley D., Haugen E., Kibukawa M., Paddock M., Palmieri A., Subramanian S., Zhou Y., Kaul R., Green P., Olson M. V.2006Scan of human genome reveals no new loci under ancient balancing selection. Genetics 173, 2165–2177 (doi:10.1534/genetics.106.055715) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt A.2000Sex, recombination and the efficacy of selection—was Weissman right? Evolution 54, 337–351 [DOI] [PubMed] [Google Scholar]
- Charlesworth B.1993Directional selection and the evolution of sex and recombination. Genet. Res. 61, 205–224 [DOI] [PubMed] [Google Scholar]
- Charlesworth B.1996Background selection and patterns of genetic diversity in Drosophila melanogaster. Genet. Res. 68, 131–150 (doi:10.1017/S0016672300034029) [DOI] [PubMed] [Google Scholar]
- Charlesworth B., Charlesworth D.1997Rapid fixation of deleterious alleles can be caused by Muller's ratchet. Genet. Res. 70, 63–74 (doi:10.1017/S0016672397002899) [DOI] [PubMed] [Google Scholar]
- Charlesworth J., Eyre-Walker A.2007The other side of the nearly neutral theory, evidence of slightly advantageous back-mutations. Proc. Natl Acad. Sci. USA 104, 16992–16997 (doi:10.1073/pnas.0705456104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B., Betancourt A. J., Kaiser V. B., Gordo I.2009Genetic recombination and molecular evolution. Cold Spring Harbor Symp. Quant. Biol. 74 (doi:10.1101/sqb.2009.74.015) [DOI] [PubMed] [Google Scholar]
- Cohan F. M.2002What are bacterial species? Annu. Rev. Microbiol. 56, 457–487 (doi:10.1146/annurev.micro.56.012302.160634) [DOI] [PubMed] [Google Scholar]
- Coop G., Przeworski M.2007An evolutionary view of human recombination. Nat. Rev. Genet. 8, 23–34 (doi:10.1038/nrg1947) [DOI] [PubMed] [Google Scholar]
- Darwin C. R.1876The effects of cross and self fertilisation in the vegetable kingdom. London, UK: John Murray [Google Scholar]
- de Visser J. A. G. M., Elena S. F.2007The evolution of sex: empirical insights into the roles of epistasis and drift. Nat. Rev. Genet. 8, 139–149 (doi:10.1038/nrg1985) [DOI] [PubMed] [Google Scholar]
- Denver D. R., Morris K. H., Lynch M., Thomas W. K.2004High mutation rate and predominance of insertions in the C. elegans nuclear genome. Nature 430, 679–682 (doi:10.1038/nature02697) [DOI] [PubMed] [Google Scholar]
- Engelstadter J.2008Constraints on the evolution of asexual reproduction. Bioessays 30, 1138–1150 (doi:10.1002/bies.20833) [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A., Keightley P. D.2007The distribution of fitness effects of new mutations. Nat. Rev. Genet. 8, 610–618 (doi:10.1038/nrg2146) [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A., Keightley P. D.2009Estimating the rate of adaptive molecular evolution in the presence of slightly deleterious mutations and population size change. Mol. Biol. Evol. 26, 2097–2108 (doi:10.1093/molbev/msp119) [DOI] [PubMed] [Google Scholar]
- Feldman M. W., Otto S. P., Christiansen F. B.1996Population genetic perspectives on the evolution of recombination. Ann. Rev. Genet. 30, 261–295 (doi:10.1146/annurev.genet.30.1.261) [DOI] [PubMed] [Google Scholar]
- Felsenstein J.1974The evolutionary advantage of recombination. Genetics 78, 737–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher R. A.1930The genetical theory of natural selection. Oxford, UK: Oxford University Press [Google Scholar]
- Gillespie J. H.2001Is the population size of a species relevant to its evolution? Evolution 55, 2161–2169 [DOI] [PubMed] [Google Scholar]
- Gordo I., Campos P. R. A.2008Sex and deleterious mutations. Genetics 179, 621–626 (doi:10.1534/genetics.108.086637) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag-Liautard C., Dorris M., Maside X., Macaskill S., Halligan D. L., Houle D., Charlesworth B., Keightley P. D.2007Direct estimation of per nucleotide and genomic deleterious mutation rates in Drosophila. Nature 445, 82–85 (doi:10.1038/nature05388) [DOI] [PubMed] [Google Scholar]
- Hadany L., Feldman J. F.2005Evolutionary traction: the cost of adaptation and the evolution of sex. J. Evol. Biol. 18, 309–314 (doi:10.1111/j.1420-9101.2004.00858.x) [DOI] [PubMed] [Google Scholar]
- Haigh J.1978The accumulation of deleterious genes in a population–Muller' s ratchet. Theoret. Popul. Biol. 14, 251–267 (doi:10.1016/0040-5809(78)90027-8) [DOI] [PubMed] [Google Scholar]
- Haldane J. B. S.1957The cost of natural selection. J. Genet. 55, 511–524 (doi:10.1007/BF02984069) [Google Scholar]
- Halligan D., Keightley P. D.2009Spontaneous mutation accumulation studies in evolutionary genetics. Ann. Rev. Ecol. Evol. Syst. 40, 151–172 (doi:10.1146/annurev.ecolsys.39.110707.173437) [Google Scholar]
- Hamilton W. D.1996Narrow roads of gene land 2 Evolution of sex and sexual selection. Oxford, UK: W.H. Freeman [Google Scholar]
- Hartfield M., Otto S. P., Keightley P. D.2010The role of advantageous mutations in enhancing the evolution of a recombination modifier. Genetics 88, 1153–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill W. G., Robertson A.1966The effect of linkage on limits to artificial selection. Genet. Res. 8, 269–294 (doi:10.1017/S0016672300010156) [PubMed] [Google Scholar]
- Hudson R. R., Kaplan N. L.1995Deleterious background selection with recombination. Genetics 141, 1605–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iles M. M., Walters J. R., Cannings C.2003Selection for recombination in large genomes. Genetics 165, 2249–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingvarsson P. K., Whitlock M. C.2000Heterosis increases the effective migration rate. Proc. R. Soc. Lond. B. 267, 1321–1326 (doi:10.1098/rspb.2000.1145) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen J. D., Thornton K. R., Andolfatto P.2008An approximate Bayesian estimator suggests strong recurrent selective sweeps in Drosophila. PLoS Genet. 4, e1000198 (doi:10.1371/journal.pgen.1000198) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T., Barton N. H.2002The effect of deleterious alleles on adaptation in asexual populations. Genetics 162, 395–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T., Barton N. H.2005Theoretical models of selection and mutation on quantitative traits. Phil. Trans. R. Soc. B 360, 1411–1425 (doi:10.1098/rstb.2005.1667) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser V., Charlesworth B.2008The effects of deleterious mutations on evolution in non-recombining genomes. Trends Genet. 25, 9–12 [DOI] [PubMed] [Google Scholar]
- Karasov T., Messer S. W., Petrov D. A.In press Evidence that adaptation in Drosophila is not limited by mutation at single sites. PLoS Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley P. D., Halligan D. L.2009Analysis and implications of mutational variation. Genetica 136, 359–369 (doi:10.1007/s10709-008-9304-4) [DOI] [PubMed] [Google Scholar]
- Keightley P. D., Otto S. P.2006Interference among deleterious mutations favours sex and recombination in finite populations. Nature 443, 89–92 (doi:10.1038/nature05049) [DOI] [PubMed] [Google Scholar]
- Kim Y., Stephan W.2000Joint effects of hitch–hiking and background selection on neutral variation. Genetics 155, 1415–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M.1983The neutral theory of molecular evolution. Cambridge, UK: Cambridge University Press [Google Scholar]
- Kimura M., Maruyama T.1966Mutational load with epistatic gene interactions in fitness. Genetics 54, 1337–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick M., Barton N. H.2006Chromosome inversions, local adaptation, and speciation. Genetics 173, 419–434 (doi:10.1534/genetics.105.047985) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov A. S.1988Deleterious mutations and the evolution of sexual reproduction. Nature 336, 435–441 (doi:10.1038/336435a0) [DOI] [PubMed] [Google Scholar]
- Kreitman M.1983Nucleotide polymorphisms at the alcohol dehydrogenase locus of Drosophila melanogaster. Nature 304, 412–417 (doi:10.1038/304412a0) [DOI] [PubMed] [Google Scholar]
- Kulathinal R. J., Bennett† S. M., Fitzpatrick C. L., Noor M. A. F.2008Fine-scale mapping of recombination rate in Drosophila refines its correlation to diversity and divergence. Proc. Natl Acad. Sci. USA 105, 10 051–10 056 (doi:10.1073/pnas.0801848105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenormand T., Otto S. P.2000The evolution of recombination in a heterogeneous environment. Genetics 156, 423–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livnat A., Papadimitriou C., Dushoff J., Feldman M. W.2008A mixability theory for the role of sex in evolution. Proc. Natl Acad. Sci. USA 105, 19 803–19 808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewe L., Charlesworth B.2006Inferring the distribution of mutational effects on fitness in Drosophila. Biol. Lett. 2, 426–430 (doi:10.1098/rsbl.2006.0481) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewe L., Charlesworth B., Bartolome C., Noel V.2006Estimating selection on nonsynonymous mutations. Genetics 172, 1079–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson J. M., Sella G., Davis J. C., Petrov D. A.2007Genome-wide spatial correspondence between nonsynonymous divergence and neutral polymorphism reveals extensive adaptation in Drosophila. Genetics 177, 2083–2099 (doi:10.1534/genetics.107.080226) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G., Otto S.P., Lenormand T.2006Selection for recombination in structured populations. Genetics 172, 593–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith J.1978The evolution of sex. Cambridge, UK: Cambridge University Press [Google Scholar]
- Maynard Smith J.1988The evolution of recombination. In The evolution of sex (eds Michod R. E., Levin B. R.). Sunderland, MA: Sinauer Associates [Google Scholar]
- Maynard Smith J., Haigh J.1974The hitch-hiking effect of a favourable gene. Genet. Res. 23, 23–35 [PubMed] [Google Scholar]
- McDonald J. H., Kreitman M.1991Adaptive protein evolution at the Adh locus in Drosophila. Nature 351, 652–654 (doi:10.1038/351652a0) [DOI] [PubMed] [Google Scholar]
- McVean G. A. T., Charlesworth B.2000The effects of Hill - Robertson interference between weakly selected sites on patterns of molecular evolution and variation. Genetics 155: 929–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merila J., Sheldon B.2000Lifetime reproductive success and heritability in nature. Am. Nat. 155, 301–310 (doi:10.1086/303330) [DOI] [PubMed] [Google Scholar]
- Mitchell M.1998An introduction to genetic algorithms. Cambridge, MA: MIT Press [Google Scholar]
- Muller H. J.1932Some genetic aspects of sex. Am. Nat. 66, 118–138 (doi:10.1086/280418) [Google Scholar]
- Navarro A., Barton N. H.2002The effects of multilocus balancing selection on neutral variability. Genetics 161, 849–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg M., Charlesworth B., Charlesworth D.1996The effect of recombination on background selection. Genet. Res. 67, 159–174 (doi:10.1017/S0016672300033619) [DOI] [PubMed] [Google Scholar]
- Nurminsky D. I.2001Genes in sweeping competition. Cell. Mol. Life Sci 58, 125–134 (doi:10.1007/PL00000772) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T., Gillespie J. H.1996Development of neutral and nearly—neutral theories. Theoret. Popul. Biol. 49, 128–142 (doi:10.1006/tpbi.1996.0007) [DOI] [PubMed] [Google Scholar]
- Otto S. P., Barton N. H.1997The evolution of recombination: removing the limits to natural selection. Genetics 147, 879–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto S. P., Barton N. H.2001Selection for recombination in small populations. Evolution 55, 1921–1931 [DOI] [PubMed] [Google Scholar]
- Otto S. P., Nuismer S. L.2004Species interactions and the evolution of sex. Science 304, 1018–1020 (doi:10.1126/science.1094072) [DOI] [PubMed] [Google Scholar]
- Peck J. R.1994A ruby in the rubbish: beneficial mutations and the evolution of sex. Genetics 137, 597–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A. D., Lively C. M.1998The Red Queen and fluctuating epistasis: a population genetic analysis of antagonistic coevolution. Am. Nat. 154, 393–405 [DOI] [PubMed] [Google Scholar]
- Provine W.1971The origins of theoretical population genetics. Chicago, IL: University of Chicago Press [Google Scholar]
- Robertson A.1961Inbreeding in artificial selection programmes. Genet. Res. 2, 189–194 (doi:10.1017/S0016672300000690) [DOI] [PubMed] [Google Scholar]
- Roff D. A., Mousseau T. A.1987Quantitative genetics and fitness: lessons from Drosophila. Heredity 58, 103–118 (doi:10.1038/hdy.1987.15) [DOI] [PubMed] [Google Scholar]
- Rouzine I. M., Brunet E., Wilke C. O.2008The traveling—wave approach to asexual evolution: Muller's ratchet and speed of adaptation. Theoret. Popul. Biol. 73, 24–46 (doi:10.1016/j.tpb.2007.10.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roze D., Barton N. H.2006The Hill–Robertson effect and the evolution of recombination. Genetics 173, 1793–1811 (doi:10.1534/genetics.106.058586) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salathé M., Kouyos R. D., Bonhoeffer S.2009On the causes of selection for recombination underlying the Red Queen hypothesis. Am. Nat. 174, S31–S42 (doi:10.1086/599085) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago E., Caballero A.1998Effective size and polymorphism of linked neutral loci in populations under directional selection. Genetics 149, 2105–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sella G., Petrov D. A., Przeworski M., Andolfatto P.2009Pervasive natural selection in the Drosophila genome? PLoS Genet. 5, e1000495 (doi:10.1371/journal.pgen.1000495) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro J. A., Huang W., Zhang C. H., Hubisz M. J., Lu J., Turissini D. A., Fang S., Wang H. Y., Hudson R. R., Nielsen R., Chen Z., Wu C. I.2007Adaptive genic evolution in the Drosophila genomes. Proc. Natl Acad. Sci. USA 104, 2271–2276 (doi:10.1073/pnas.0610385104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N. D., Aquadro C. F., Clark A. G.2009Estimation of fine -scale recombination intensity variation in the white–echinus interval of D. melanogaster. J. Mol. Evol. 69, 42–53 (doi:10.1007/s00239-009-9250-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith N. G. C., Eyre–Walker A.2002Adaptive protein evolution in Drosophila. Nature 415, 1022–1024 (doi:10.1038/4151022a) [DOI] [PubMed] [Google Scholar]
- Vrijenhoek R. C.1994Unisexual fish: model systems for studying ecology and evolution. Ann. Rev. Ecol. Syst. 25, 71–96 (doi:10.1146/annurev.es.25.110194.000443) [Google Scholar]
- Weismann A.1889The significance of sexual reproduction in the theory of natural selection. In Essays upon heredity and kindred biological problems (eds Poulton E. B., Schönland S., Shipley A. E.). Oxford, UK: Clarendon Press [Google Scholar]
- Zeng K., Charlesworth B.2009Estimating selection intensity on synonymous codon usage in a nonequilibrium population. Genetics 183, 651–662 (doi:10.1534/genetics.109.101782) [DOI] [PMC free article] [PubMed] [Google Scholar]