

Abstract
The P2Y6 receptor is a cytoprotective G protein-coupled receptor (GPCR) activated by UDP (EC50, 0.30 μM). We compared and combined modifications to enhance P2Y6 receptor agonist selectivity, including ribose ring constraint, 5-iodo and 4-alkyloxyimino modifications, and phosphate modifications such as α,β-methylene and extension of the terminal phosphate group into γ-esters of UTP analogues. The conformationally constrained (S)-methanocarba UDP is a full agonist (EC50 0.042 μM). 4-Methoxyimino modification of pyrimidine enhanced P2Y6, preserved P2Y2 and P2Y4, and abolished P2Y14 receptor potency, in the appropriate nucleotide. N4-Benzyloxy-CDP (15, MRS2964) and N4-methoxy-Cp3U (23, MRS2957) were potent, selective P2Y6 receptor agonists (EC50 0.026 μM and 0.012 μM, respectively). A hydrophobic binding region near the nucleobase was explored with receptor modeling and docking. UTP-γ-aryl and cycloalkyl phosphoesters displayed only intermediate P2Y6 receptor potency, but had enhanced stability in acid and cell membranes. UTP-glucose was inactive, but its (S)-methanocarba analogue and N4-methoxy-cytidine 5′-triphospho-γ-[1]glucose were active (EC50 of 2.47 μM and 0.18 μM, respectively). Thus, the potency, selectivity, and stability of pyrimidine nucleotides as P2Y6 receptor agonists may be enhanced by modest structural changes.
Keywords: G protein-coupled receptor, nucleotides, pyrimidines, phospholipase C, dinucleotide, uracil
Introduction
The P2Y receptors are G protein-coupled receptors (GPCRs) that respond to naturally-occurring extracellular nucleotides including ATP, ADP, UTP, UDP, and UDP-glucose.1 Five of the eight subtypes, P2Y1, P2Y2, P2Y4, P2Y6, and P2Y11 receptors, are preferentially coupled through Gq to activate phospholipase Cβ (PLC-β) and represent a distinct structural subfamily.2 The remaining P2Y12, P2Y13, and P2Y14 receptors are coupled to inhibition of adenylate cyclase. Activation of the P2Y6 receptor by its native ligand UDP 1 (Chart 1) is associated with vasoconstriction and cytoprotection.3-6 We reported the effects of P2Y6 receptor agonists to counteract apoptosis induced by tumor necrosis factor-α in astrocytoma cells stably expressing the human P2Y6 receptor and in mouse C2C12 skeletal muscle cells and MIN6 pancreatic cells that express an endogenous P2Y6 receptor.4-6 Furthermore, in vivo administration of 5-iodo-UDP (MRS2693) 3, which is a selective P2Y6 receptor agonist, induced protection in a model of ischemia in hindleg skeletal muscle.6 The dinucleoside triphosphate INS48823 4 activates the P2Y6 receptor in bone and increased survival of osteoclasts.7 A role for the P2Y6 receptor in inducing phagocytosis in microglial cells was recently discovered, suggesting the possible application of P2Y6 receptor ligands for the treatment of neurodegenerative conditions.8 The P2Y6 receptor is upregulated when neurons are damaged and in intestinal inflammation.8,9 UDP applied topically to the cornea reduced intraocular pressure, suggesting the use of P2Y6 receptor agonists for the treatment of ocular hypertension and glaucoma.10 Studies of mice lacking the P2Y6 receptor recently demonstrated a defective response to UDP in macrophages, endothelial cells, and vascular smooth muscle cells.11 Thus, pharmacological modulation of the P2Y6 receptor is of increasing interest for future therapeutics.12
Chart 1.

Structures of prototypical agonist ligands for studying P2Y6 receptors.
Potent P2Y6 receptor agonists, such as 3-phenacyl-UDP 5 with an EC50 value of 70 nM, have been reported.13-16 Recently, we reported that UDP also activates the P2Y14 receptor, and therefore, agonists that delineate between these two subtypes are needed.17,18 We have adopted a structure-based approach to developing P2Y6 receptor agonist ligands with improved potency, selectivity, and stability. In the absence of an X-ray structure, rhodopsin-based homology modeling has predicted a putative binding site for nucleotides in the human P2Y6 receptor.13 The receptor recognition of the 5′-diphosphate group is associated with three cationic residues in the transmembrane region, which serve as counterions and which are conserved in charge across the five Gq-coupled P2Y receptors.2 A Monte Carlo analysis of conformations of receptor-docked UDP molecules, followed by molecular dynamics of the receptor-ligand complex in a lipid bilayer model, has predicted that the South (S) conformation of the ribose ring is preferred at the P2Y6 receptor.13 This is the only Gq-coupled P2Y subtype at which the (S) conformation of the ribose or a ribose-like moiety of nucleotide ligands appears to be favored, and our comparison of the weak agonist 2′-deoxy-UDP 2 and the corresponding 2′-deoxy (S)-methanocarba analogue 613,14 supported this idea.
We recently reported the first synthetic route to the enantiomerically pure (S)-methanocarba-uridine,19 and in this study we examined the hypothesis that the (S) conformation is preferred at the P2Y6 receptor in the more potent 2′-hydroxynucleotide series. Modifications of the pyrimidine ring of UDP, including 5-iodo and 4-alkyloxyimino, and of the phosphate moiety, including α,β-methylene, and extension of the terminal phosphate group, were compared and combined in an effort to further enhance selectivity for the P2Y6 receptor.14,16 Chemical and metabolic instability of mononucleotides is well-documented.20 Mononucleotides are degraded enzymatically by ectonucleotidases into the lower nucleotides and eventually into the nucleoside. Chemical degradation of nucleotides is also evident at pH ranges less than 4 or greater than 10. The degradation products of nucleotides may interact with multiple P2Y receptors or with adenosine receptors and lead to side effects. Since various dinucleotides, such as diuridine triphosphates, show greater stability than analogues of mononucleotides,21,22 we hypothesized that blockade of the terminal phosphate with an alkyl or aryl group may also increase its stability.
Results
Chemical synthesis
A series of known (1-6, 8, 10, and 11) and newly synthesized derivatives of UDP (7, 9) and CDP (12-16) was assembled and compared (Tables 1 and 2). Novel analogues of UTP and Up3U (18, 20, and 22-33) also were included in this structure activity relationship (SAR) analysis (Table 3). The chemical synthesis of ribonucleoside 5′-di and triphosphate derivatives (Scheme 1) was performed by standard phosphorylation methods using the unprotected nucleoside precursors.23,24 Various 5′-diphosphate derivatives were then extended to form dinucleoside triphosphates or other 5′-triphosphate γ-esters (Scheme 2). (S)-Methanocarba nucleotides were synthesized as shown in Scheme 3. The synthesis of several of the pyrimidine nucleotides was reported in previous studies of the P2Y receptor (3, 5, 6, 8, 10, 19, 21, and 26).14-16
Table 1.
Potencies of (S)-methanocarba analogues 6, 7, and 9, in relation to known uridine 5′-diphosphate derivatives for activation of the human P2Y6 receptor. Unless noted: R = uridine; and X = O.
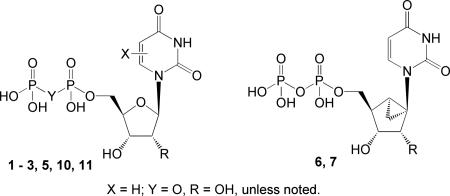 | |||
|---|---|---|---|
| Compound | Modification | Structure | EC50, μMa |
| hP2Y6 | |||
| 1 | UDP | - | 0.30±0.06 |
| 2 | dUDP | R = H | 1.72±0.76 |
| 3b | 5-I-UDP | X = 5-I | 0.015±0.002 |
| 5b | phenacylUDP | X = 3-Ph-COCH2 | 0.070±0.006 |
| 6b | (S)-methanocarba dUDP | R = H | 0.23±0.05 |
| 7 | (S)-methanocarba UDP | - | 0.042±0.008 |
| 8b,d | (N)-methanocarba UDP | NE | |
| 9 | (S)-methanocarba U-cyclic 3′,5′-diphosphate | NE | |
| 10b | α,β-CH2-UDP | Y = CH2 | 0.70±0.11 |
| 11 | α,β-CH2-5-I-UDP | X = 5-I, Y = CH2 | 0.13±0.02 |
Agonist potencies reflect stimulation of phospholipase C in 1321N1 human astrocytoma cells stably expressing the human P2Y6 receptor. Potencies are presented in the form of EC50values, which represent the concentration of agonist at which 50% of the maximal effect is achieved. These values were determined using a four-parameter logistic equation and the GraphPad software package (GraphPad, San Diego, CA). The results are presented as mean ± standard error and are the average of three to six different experiments with each molecule.
Values reported in literature: 3, Besada et al.14; 5, El-Tayeb et al.8; 6 and 8, Costanzi et al.13; 10, Ko et al.16
c6, MRS2633; 7, MRS2795.
Structure of 8 given in Chart 1.
eEC50 values33 (μM) reported in activation of the hP2Y14 receptor expressed in HEK-293 cells: 1, 0.16±0.04; 10, 0.011±0.005; 11, 0.014±0.07.
NE – no effect at 10 μM.
Table 2.
Potencies of N4-alkyloxy cytidine 5′-diphosphate derivatives for activation of the human P2Y2, P2Y4, and P2Y6 receptors.
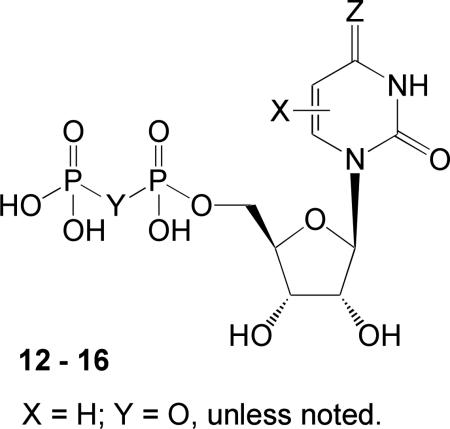 | |||||
|---|---|---|---|---|---|
| Compound | Modification | Structure | EC50, μMa | ||
| hP2Y2 | hP2Y4 | hP2Y6 | |||
| 12b | N4-OMe-CDP | Z = N-OCH3 | 3.60±1.08 | 6.45±1.55 | 0.070±0.007 |
| 13 | N4-OEt-CDP | Z = N-OCH2CH3 | 0.28±0.015 | 0.18±0.02 | 0.104±0.023 |
| 14 | N4-OtBu-CDP | Z = N-OC(CH3)3 | 3.51±1.52 | 6.23±1.88 | 2.55±0.70 |
| 15 | N4-OBn-CDP | Z = N-OCH2Ph | 2.13±0.64 | 1.15±0.15 | 0.026±0.002 |
| 16 | α,β-CH2-N4-OMe-CDP | Y = CH2, Z = N-OCH3 | >10c | NE | 0.678±0.018 |
Agonist potencies reflect stimulation of phospholipase C in 1321N1 human astrocytoma cells stably expressing the human P2Y2, P2Y4, or P2Y6 receptor. Potencies are presented in the form of EC50 values, which represent the concentration of agonist at which 50% of the maximal effect is achieved. These values were determined using a four-parameter logistic equation and the GraphPad software package (GraphPad, San Diego, CA). The results are presented as mean ± standard error and are the average of three to six different experiments with each molecule.
EC50 value33 reported in activation of the hP2Y14 receptor expressed in HEK-293 cells: 12, 3.32±1.62 μM.
≤50% effect at 10 μM.
ND – not determined. NE – no effect at 10 μM.
Table 3.
Potencies of nucleotide (5′-triphosphate) derivatives for activation of the human P2Y2, P2Y4, and P2Y6 receptors.
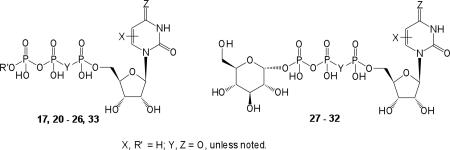 | |||||
|---|---|---|---|---|---|
| Compound | Modification | Structure | EC50, μMa | ||
| hP2Y2 | hP2Y4 | hP2Y6 | |||
| 17 | UTP | 0.060±0.00 | 0.090±0.01 | >10d | |
| 18 | (S)-methanocarba UTP | 0.08±0.02 | 0.30±0.10 | 1.37±0.05 | |
| 19b | (N)-methanocarba UTP | 0.016±0.007 | 0.085±0.005 | NE | |
| 20 | N4-OMe-CTP | Z = N-OCH3 | 0.05±0.00 | 0.05±0.02 | 0.08±0.02 |
| 21b | Up3U | R′ = [5′]uridine | 1.31±0.21 | 0.87±0.11 | 0.27±0.07 |
| 22 | α,β-CH2-Up3U | R′ = [5′]uridine, Y = CH2 | >10d | NE | 1.30±0.18 |
| 23 | N4-OMe-Cp3U | Z = N-OCH3 R′ = [5′]uridine | 0.17±0.04 | 0.79±0.12 | 0.012±0.0024 |
| 24 | N4-OMe-Cp3(N4-OMe)C | Z = N-OCH3 R′ = [5′](N4-methoxy)cytidine | 2.12±0.66 | 0.56±0.04 | 0.063±0.002 |
| 25 | Up3-O-cyclohexyl | R′ = cyclohexyl | 2.28±0.1 | NE | 6.99±0.18 |
| 26 | Up3-O-phenyl | R′ = phenyl | >10d | NE | 2.13±0.18 |
| 27 | Up3-gluc | NE | NE | >10d | |
| 28c | (S)-methanocarba-Up3-gluc | NE | NE | 2.47±0.39 | |
| 29 | 5-I-Up3-gluc | X = 5-I | NE | NE | 4.69±1.68 |
| 30 | 3-phenacyl -Up3 -gluc | X = 3-Ph-COCH2 | NE | NE | >10d |
| 31 | α,β-CH2-Up3-gluc | Y = CH2 | NE | NE | >10d |
| 32 | N4-OMe-Cp3-gluc | Z = N-OCH3 | >10d | NE | 0.18 ± 0.04 |
| 33 | N4-OMe-Cp3-O-Ph | R′ = phenyl, Z = N-OCH3 | >10d | NE | 3.49 ± 1.63 |
Agonist potencies reflect stimulation of phospholipase C in 1321N1 human astrocytoma cells stably expressing the human P2Y2, P2Y4, or P2Y6 receptor. Potencies are presented in the form of EC50 values, which represent the concentration of agonist at which 50% of the maximal effect is achieved. These values were determined using a four-parameter logistic equation and the GraphPad software package (GraphPad, San Diego, CA). The results are presented as mean ± standard error and are the average of three to six different experiments with each molecule.
Structure of 28:
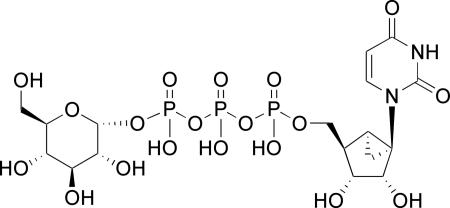
≤50% effect at 10 μM.
NE – no effect at 10 μM.
ND – not determined.
Scheme 1.

Synthesis of various pyrimidine ribonucleoside 5′-di and triphosphate derivatives. Reagents and conditions: (a) DCC, methylenebisphosphonate, rt, overnight, 11 % for 11, 48 % for 16; (b) RONH2, pyridine, 90°C, overnight, 55 – 100 %; (c) POCl3, Proton Sponge, PO(OMe)3, 0°C, 2 hr, followed by tributylammonium phosphate, rt, 0.5 hr (for 12 - 15), 14 – 47 %; (d) POCl3, Proton Sponge, PO(OMe)3, 0°C, 2hr, followed by tributylammonium pyrophosphate, rt, 0.5 hr, 31 % (for 20).
Scheme 2.

Synthesis of various pyrimidine ribonucleoside 5′-triphosphates and dinucleoside triphosphates. Reagents and conditions: (a) DIC, MgCl2, glucose 1-monophosphate tributylammonium salt, DMF, rt, overnight, 9 - 80 %; (b) DIC, MgCl2, respective monophosphate tributylammonium salt, e.g., uridine 5′-monophosphate, 35, cyclohexyl monophosphate16 or phenyl monophosphate, in DMF, rt, overnight, 9–80%. Unless noted, X = H and Y, Z = O.
Scheme 3.

Synthesis of uracil ribonucleoside phosphate derivatives constrained with a (S)-methanocarba ring. Reagents and conditions: (a) POCl3, Proton Sponge, PO(OMe)3, 0°C, 2 hr; then tributylammonium phosphate, rt, 0.5 hr, 45 %; (b) POCl3, Proton Sponge, PO(OMe)3, 0°C, 2 hr; then tributylammonium pyrophosphate, rt, 0.5 hr, 56 %; (c) CDI, DMF, rt, 1 hr, then glucose 1-monophosphate tributylammonium salt, overnight, 23 % for 28, 51 % for 9.
5-Iodouridine 34 was available from commercial sources, while the 4-methoxyimino derivative 36 and (S)-methanocarba-uridine 41 were prepared as described,19,25 and compounds 37-39 were prepared by the same procedure as 36. Compounds 34 and 36 were converted to the α,β-methylene diphosphonate analogues 11 and 16, respectively, by a carbodiimide condensation. N4-Alkoxy-cytidines 36-39 were converted to the corresponding 5′-diphosphates 12-15 and N4-methoxy-cytidine 36 to the 5′-triphosphate 20 using phosphorous oxychloride followed by either phosphate or pyrophosphate.24 Various 5′-diphosphates (1, 3, 5, 10, and 12) were converted to the corresponding 5′-triphosphate γ-esters by condensation with the appropriate monophosphate derivative in DMF.
Nucleotides 18 and 28 derived from (S)-methanocarba-uridine 40 were prepared by similar phosphorylation of the unprotected nucleoside (Scheme 3).24 The 3′,5′-cyclic diphosphate 9 was isolated as a side product in the synthesis of 28. A similar cyclic diphosphate was isolated following phosphorylation of other uridine analogues.14
Pharmacological evaluation
The nucleotide analogues (Tables 1 – 3) were assayed for capacity to promote activation of phospholipase C (PLC) in 1321N1 human astrocytoma cells stably expressing the human P2Y6 receptor.26 Receptor selectivity of the novel analogues of CDP and UTP in Tables 2 and 3 was assessed by quantification of PLC activation in 1321N1 cells stably expressing either the P2Y2 receptor or the P2Y4 receptor. Previously reported data for several analogues (2 – 6, 8, and 10) were included for comparison.
The enantiomerically pure (S)-methanocarba-UDP 7 was more potent at the P2Y6 receptor than either UDP 1 (7-fold) or the previously reported (S)-methanocarba-2′-deoxy-UDP 6 (5-fold). In contrast, the North (N)-methanocarba conformationally locked UDP analogue 8 was previously found to be inactive at the P2Y6 receptor, confirming a strong conformational preferrence in P2Y6 receptor recognition.13,14 The (S)-methanocarba-uridine-3′,5′-cyclic diphosphate 9 was also inactive at the P2Y6 receptor.
The carbon-bridged analogue α,β-methylene-UDP 10 was recently reported to be nearly as potent as UDP at the P2Y6 receptor.16 Compound 11 contained the 5′-α,β-methylenediphosphonate modification, which would increase the stability of a nucleotide in biological systems, and the 5-iodo modification (as in 3), both of which were previously noted to preserve potency at the P2Y6 receptor.14 This combination of modifications resulted in a molecule 11 that potently activated the P2Y6 receptor with an EC50 value of 127 nM.
Introduction of the 4-methoxyimino modification of UDP, i.e., N4-methoxy-CDP 12, resulted in high potency at the P2Y6 receptor (EC50 70 nM), which represents a 4-fold gain of potency over UDP (Table 2, Figure 1A). Compound 12 was 47-fold selective for activation of PLC via the P2Y6 receptor in comparison to inhibition of cyclic AMP accumulation in HEK-293 cells expressing the Gi-coupled human P2Y14 receptor.18 The native agonist for P2Y14, UDP-glucose, did not tolerate the 4-methoxyimino substitution.27 However, the 4-methoxyimino modification of the pyrimidine ring in the 5′-triphosphate derivative 20 produced a gain of potency (EC50 80 nM) of >100-fold over UTP 17 at the P2Y6 receptor (Table 3). The 4-methoxyimino modification of 20 was also well-tolerated at the P2Y2 and P2Y4 receptors, with EC50 values of 50 nM. Thus, compound 20 was a nonselective agonist at these three P2Y receptor subtypes.
Figure 1.


A) Activity of agonists 7, 12, 15, and 23 at the human P2Y6 receptor as indicated by activation of PLC in stably transfected 1321N1 human astrocytoma cells. The effect of UDP corresponds to 100%. B) Mobilization of intracellular calcium in astrocytoma cells expressing the human P2Y6 receptor (EC50 values shown in nM) by nucleotide agonists 3 (50), 21 (370), 23 (83), and 24 (630). The EC50 value of 1 was 200 nM.
In the series of 4-alkyloxyimino 5′-diphosphate analogues, the progression from MeO to larger O-alkyl substituents 12 – 15, first decreased then increased the P2Y6 receptor potency. The sterically bulky O-t-butyl group of 14 was not well tolerated. However, the O-benzyl analogue 15 was the most potent P2Y6 receptor agonist in the series of 4-imino 5′-diphosphate analogues with 82- and 44-fold selectivity vs. P2Y2 and P2Y4 receptors, respectively. Curiously, the O-ethyl analogue 13 was potent at P2Y2 and P2Y4 receptors and was thus a non-selective hP2Y2/4/6 agonist. The potency-enhancing 4-methoxyimino modification was combined with α,β-methylene substitutions in compound 16. However, there was no enhancement by the 4-methoxyimino modification, as 10 and 16 were equipotent at the P2Y6 receptor.
Up3U 21 slightly favored activation of the P2Y6 receptor, in comparison to the P2Y2 and P2Y4 receptors, with an EC50 value of 270 nM.16 Introduction of a carbon bridge in the corresponding α,β-methylene analogue 22 reduced P2Y6 receptor potency 5-fold, but with increased selectivity. Given the P2Y6 receptor potency of extended phosphate derivatives, such as dinucleoside triphosphates related to Up3U 21 (including 4), we tested other γ-phosphoesters of UTP for P2Y6 receptor activity, including alkyl and aryl esters and Up3-sugar derivatives. The analogue of Up3U containing a single 4-methoxyimino modification 23 displayed 22-fold enhanced potency at the P2Y6 receptor with an EC50 value of 12 nM and selectivity versus the P2Y2 and P2Y4 receptors of 14- and 66-fold, respectively. The γ-cyclohexyl 25 and γ-phenyl 26 esters of UTP displayed moderate potency at the P2Y6 receptor, with EC50 values of 6.99 and 2.13 μM, respectively. However, Up3-[1]glucose 27 only weakly activated the P2Y6 receptor, and an EC50 value was not determined. The introduction of the potency enhancing (S)-methanocarba modification in the corresponding ring-constrained analogue 28 increased potency, as predicted. Nevertheless, only a moderate potency of 2.47 μM was attained.
Combinations of Up3-glucose with other known modifications that are tolerated or favored at the P2Y6 receptor were evaluated. Introduction of the 5-iodo modification in 29 only slightly enhanced potency in comparison to 27. The corresponding 3-phenacyl 30 and α,β-methylene 31 analogues were only weakly active at the P2Y6 receptor. However, the P2Y6 receptor potency of the corresponding N4-methoxy-cytidine analogue 32 was greatly increased with an EC50 value of 180 nM, which also displayed >100-fold selectivity versus P2Y2 and P2Y4 receptors. This finding suggested another analogue, N4-methoxy-cytidine-5′-triphosphate γ-phenyl ester 33, but an additive effect on potency was absent, as it weakly activated the P2Y6 receptor (EC50 3.49 nM).
Selected nucleotide analogues were also examined for the ability to raise levels of intracellular calcium ions in 1321N1 human astrocytoma cells expressing the human P2Y6 receptor (Figure 1B). The rank order of potency was 23, 3 > 21, 24, which was similar to the order of potency in PLC activation.
Stability of Nucleotide Derivatives
The chemical and enzymatic instability of nucleotide analogues in biological systems is a major limitation in their use as pharmacological probes. Therefore, to assess their potential for broad use, selected nucleotide derivatives were evaluated for stability during prolonged exposure to two different conditions. The derivatives were incubated at 37°C in aqueous medium at low pH or in the presence of 1321N1 astrocytoma membranes, which are known to contain ectonucleotidases, as a representative mammalian cell of the central nervous system. At regular intervals aliquots were taken for HPLC analysis (Table 4 and Supporting Information). The injection sample was pre-diluted with 5 mM tetrabutylammonium dihydrogenphosphate (TBAP) to allow complete equilibration with the mobile phase, in order to avoid the early HPLC elution at around one minute.
Table 4.
Half-lives of nucleotide (5′-triphosphate) derivatives when incubated at 37°C in acidic aqueous solution or in the presence of mammalian (1321N1 human astrocytoma) cell membranes, determined by HPLC.
| Compound | t1/2, h (pH 1.2) | t1/2, h (membranes) | Pharmacological properties at hP2Y6 receptor |
|---|---|---|---|
| 12, N4-MeO-CDP | 116 | 55 | selective, potent agonist |
| 15, N4-BnO-CDP | 126 | 26 | selective, potent agonist |
| 17, UTP | 23 | 31 | ~inactive |
| 21, Up3U | 218 | 42 | moderately potent agonist, only 4-5 fold selective |
| 22, α,β-me-Up3U | 34 | 62 | weak agonist, selective |
| 23, N4-MeO-Cp3U | 120 | 23 | selective, potent agonist |
| 25, Up3-O-cyclohexyl | 193 | 62 | weak agonist, nonselective vs. P2Y2 receptor |
| 26, Up3-O-Ph | 164 | 43 | weak agonist, selective |
| 27, Up3-gluc | 5.1 | 29 | ~inactive |
| 31, α,β-me-Up3-gluc | 2.8 | 19 | ~inactive |
The stability in the presence of cell membranes was 25, 22 > 12 >21, 26 > 17, 27 > 15, 23 > 31. The potent P2Y6 receptor agonist 12 was more stable than the related N4-benzyloxy cytidine 5′-diphosphate 15. The γ-cyclohexyl ester 25 was more stable than either Up3U 21, which has a uridyl group at this terminal position, or the γ-phenyl ester 26. The α,β-methylene analogue of Up3U 22 was more stable than the parent Up3U 21 in the presence of cell membranes, which may indicate that a α,β-methylene bridges blocks enzymatic breakdown, as has been observed for other P2 receptor agonists containing phosphonates.16,28,29 However, the introduction of a methylene bridge into Up3U unfortunately also reduced potency at the P2Y6 receptor 5-fold.
In acidic solution, the CDP derivatives 12 and 15 and the dinucleoside triphosphates were of enhanced chemical stability. Also, the γ-phenyl phosphoester 26 was relatively stable. However, the Up3U analogue 22 containing a α,β-methylene bridge was significantly less stable than the parent Up3U 21. Likewise, the Up3-[1]glucose analogue 31 containing a α,β-methylene bridge was less stable than the parent Up3-[1]glucose 27, which itself was very labile. Since both of the glucose derivatives 27 and 31 were rapidly degraded, the introduction of glucose was not a favored modification.
Molecular Modeling
A docking of the semi-rigid (S)-methanocarba analogue 7 in complex with our rhodopsin-based P2Y6 receptor model13 (Figure 2A) suggested that 7 establishes with the receptor the same putative interactions characteristic of UDP. In particular, three cationic residues located in transmembrane domain 3 (TM3), TM6, and TM7 coordinate the diphosphate of 7, namely R103(3.29), K259(6.55), and R287(7.39), while the base is coordinated by Y33(1.39) and S291(7.43), located in TM1 and TM7, respectively – see Experimental Section for residue indexing. The higher potency of 7 compared to UDP is induced by the rigid methanocarba scaffold, which constrains the ribose in the bioactive conformation, likely with a consequent gain in the entropic component of the binding affinity. A hydrogen bond is possibly formed between the ribose 2′-hydroxy group and the uracil 2-oxo group of 7 (Figure 2A). Furthermore, as shown in Figure 2A, the molecular dynamics simulation predicted the access of several water molecules to the receptor binding pocket from the extracellular milieu. These molecules surround the portion of the ligand exposed to the extracellular opening of the binding site, including the 5 position of the uracil ring. For entropic reasons, substituents capable of displacing these water molecules from the binding cavity to the bulk of the solvent are expected to enhance the affinity of the compounds. The increase in affinity noted upon the introduction of the iodo-substituent at the 5-position of the uracil ring (see compounds 1 and 3) is consistent with this reasoning and supports the orientation of this moiety of the ligand towards the extracellular opening of the helical bundle of the receptor as suggested by our models.
Figure 2.


Molecular modeling of the human P2Y6 receptor. A) Details of the nucleotide binding site of the receptor complexed with the agonist 7 as obtained after a fully flexible Monte Carlo conformational search. The ligand is represented as balls and sticks, colored by element. The binding pocket is represented as a van der Waals surface colored according to the electrostatic potential, with positive charges in blue and negative charges in red. For clarity, the residues located in front of the ligand are represented as sticks, colored by element. Labels indicate all of the residues represented as sticks (adjacent) and the key residues represented as van der Waals surfaces (trough arrows). A schematic representation of the receptor-ligand complex is given in the lower left inset. In the tube representations the receptor is colored according to residue positions, with a spectrum of colors that ranges from red (N-terminus) to purple (C-terminus): TM1 is in orange, TM2 in ochre, TM3 in yellow, TM4 in green, TM5 in cyan, TM6 in blue, TM7 in purple. B) Docking of the potent agonist N4-methoxy-cytidine-5′-diphosphate 12. A hydrophobic binding pocket for the N4-alkyloxy substituent is defined in this docking model. Distances in Å from the methoxy carbon atom to neighboring amino acid residues are shown.
A molecular model of N4-methoxy-CDP 12 docked in the P2Y6 receptor showed similar coordination to that of UDP, i.e., S291(7.43) coordinates the base, and four cationic residues coordinate the diphosphate moiety (Figure 2B). The methyl group at the 4 position seems to fit well in a sterically restricted hydrophobic pocket, which consisted of V32(1.38), Y33(1.39), I83(2.61) and P288(7.40). These hydrophobic interactions may be the reason for the increased activity of 12 (Table 2).
Discussion
The present set of analogues provides new insights into the structural requirements for activation of the P2Y6 receptor at each of the three regions modified: ribose, nucleobase, and terminal phosphate. In addition to studying the SAR of pyrimidine nucleotides in receptor activation, we have also confirmed that blockade of the terminal phosphate with an alkyl or aryl group or other groups may increase its stability.
Using sterically constrained carbocyclics, we have extended to the ribo (2′-hydroxy) series the prediction that the (S) conformation of a ribose-like moiety is preferred for recognition at the human P2Y6 receptor, consistent with previous activity of the 2′-deoxy analogues. The first synthesis of enantiomerically pure (S)-methanocarba-uridine enabled the preparation of nucleotide derivatives in the 2′-hydroxy series (7, 18, and 28). These (S)-methanocarba nucleotides exhibited enhanced potency over the corresponding natural ribose analogue in each case.
In homology-based modeling of the P2Y6 receptor and ligand docking, an intramolecular hydrogen bond was detected between the 2′-hydroxy group and the uracil 2-oxo group of the conformationally constrained, potent analogue of UDP 7. We suggest that this interaction might further restrain 7 in its bioactive conformation, and thus, contribute to the greater potency of 7 over the corresponding 2′-deoxy analogue 6.
One of the striking findings of this study was that the 4-methoxyimino and 4-benzyloxyimino modifications of the pyrimidine ring in the appropriate nucleotide derivative served to enhance potency at the P2Y6 receptor. This is one of the few modifications of the uracil ring that is tolerated for activation of the P2Y6 receptor. CDP itself is nearly inactive at the human P2Y6 receptor, and its EC50 at the P2Y14 receptor is >10 μM.17,30 The 4-methoxyimino 5′-diphosphate analogue 12 displayed a selectivity of 51- and 47-fold versus the P2Y2 and P2Y14 receptors, respectively, and a substituent as large as O-benzyl 15 was tolerated. In the 5′-triphosphate analogue 20, the 4-methoxyimino modification also preserved potency at P2Y2 and P2Y4 receptors and abolished potency at the P2Y14 receptor.27 In Schemes 1 and 2, the imino group of N4-methoxycytidine is depicted as the imino-oxo tautomer, based on an early study of the poly C derivative after treatment with methoxyamine.31 This predominant form, which resembles the major lactam (keto) tautomer of UDP, was used in the molecular modeling. However, the amino tautomeric form, which is found in native cytosine in DNA, might also be present. The 4-methoxyimino group in a γ-[1]glucose triphosphate ester 32 also enhancedpotency. However, the γ-[1]glucose triphosphate in another analogue 27 suffered from low stability, and therefore, 32 is not a likely candidate for generation of a potent and stable agonist.
The introduction of a phenyl or cyclohexyl ring at the terminal phosphate of UTP in compounds 25 and 26, respectively, enhanced stability in both acidic solution and in the presence of membranes to at least the same extent as Up3U. Both 25 and 26 containing terminal hydrophobic groups showed higher potency than UTP 17 at the P2Y6 receptor, and the more hydrophilic Up3-[1]glucose 27 was nearly inactive. Thus, the nature of the terminal group can greatly affect agonist activity. Since Up4-[1]glucose is a potent agonist of the P2Y2 receptor, and Up2-[1]glucose is a native agonist of the P2Y14 receptor, we speculated that the intermediate triphosphate homologue 27 might be an effective agonist of the P2Y6 receptor. However, this receptor is not as tolerant of a terminal glucose moiety as are the P2Y2 and P2Y14 receptors. Nevertheless, the introduction of a rigid (S)-methanocarba ring system in 18 and 28 resulted in agonists that exceeded the P2Y6 potency of both UTP and Up3-[1]glucose.
The combination of two modifications in the present set of compounds either produced an additive effect on P2Y6 receptor potency, as in γ-[1]glucose triphosphate ester 32, or failed to increase potency, as in the γ-phenyl ester 33 and the α,β-methylene analogue 16. The inconsistent effects of combination of various structural modifications on receptor recognition suggested multiple modes of binding of these nucleotide series.
The homology modeling and docking, interpreted in light of the biological activity, indicate conformational cross-talk between the nucleobase and the terminal phosphate substituent. Although the molecular model of N4-methoxy-CDP 12 docked in the P2Y6 receptor showed similar coordination to that of UDP, Up3-O-phenyl 26 docked a different mode (not shown). The base of 26 did not coordinate with Y33(1.39) and S291(7.43), because the position of the base had shifted within the binding site. Also, the ribose ring showed a planar rather than a (S) conformation. The terminal phenyl group of 26 seemed to fit in a hydrophobic region in the model, which might be the reason for the dislocation of the nucleobase, i.e. by withdrawal of the entire molecule. On the other hand, the docked model of N4-methoxy-Cp3-O-phenyl 33 in the receptor showed a position and coordination similar to N4-methoxy-CDP 12, but the phenyl ring did not interact with the distal hydrophobic region occupied by 26. The strong interaction of the methyl group of 33 in a hydrophobic pocket near the nucleobase may cause the phenyl group to exit its large hydrophobic binding region. This interpretation is consistent with the lack of enhanced activity of compound 33 in comparison to Up3-O-phenyl 26, despite the presence of methoxyimino moiety at the 4 position.
The modeling and receptor docking also predicted that various water molecules are located in proximity of the 5 position of the ligand. The enhanced affinity of the 5-iodo-substituted analogues could be a result of the fact that the halogen displaced some of the structured water molecules present in the binding site, with a consequent entropic gain.
Conclusions
We have identified novel nucleotides, such as N4-alkoxy-cytidine derivatives, that are more potent, selective, and potentially more stable in vivo than both the native agonist and previously reported synthetic agonists of the P2Y6 receptor. Thus, N4-methoxy-cytidine nucleotides 12, 15, and 23 were relatively potent and selective agonists, and moreover, compound 23 displayed favorable stability. Thus, in future SAR studies (S)-methanocarba-(α,β-CH2)N4-OMe-CDP could be a suitable combination to maximize the potency, while retaining selectivity and stability.
The P2Y6 receptor is implicated in neurodegeneration, osteoporosis, ischemic effects in skeletal muscle, and possibly diabetes.4,32,33 Therefore, new potent and selective ligands are of interest in the search for new treatments for these conditions. Agonists of the P2Y6 receptor are predicted to produce beneficial responses in for example muscle wasting and possibly neurodegeneration, and several compounds such as 23 from the present SAR analysis could provide useful pharmacological tools.
Experimental Section
Synthesis
1H NMR spectra were obtained with a Varian Gemini 300 or Varian Mercury 400 spectrometer using D2O, CDCl3 or DMSO-d6 as a solvent. The chemical shifts are expressed as relative ppm from HOD (4.80 ppm). 31P NMR spectra were recorded at room temperature (rt) by use of Varian XL 300 (121.42 MHz) or Varian Mercury 400 (162.10 MHz) spectrometers; orthophosphoric acid (85%) was used as an external standard. In several cases the signal of the terminal phosphate moiety was not visible due to high dilution.
Purity of compounds was checked using a Hewlett–Packard 1100 HPLC equipped with a Zorbax Eclipse 5 μm XDB-C18 analytical column (250 × 4.6 mm; Agilent Technologies Inc, Palo Alto, CA). Mobile phase: linear gradient solvent system: 5 mM TBAP-CH3CN from 80:20 to 40:60 in 20 min; the flow rate was 1 mL/min (System A) or Zorbax SB-Aq 5 μm analytical column (50 × 4.6 mm; Agilent Technologies Inc, Palo Alto, CA). Mobile phase: linear gradient solvent system: 5 mM TBAP-CH3CN from 80:20 to 40:60 in 13 min; the flow rate was 0.5 mL/min (System B). Peaks were detected by UV absorption with a diode array detector at 254, 275, and 280 nm. All derivatives tested for biological activity showed >99% purity by HPLC analysis (detection at 254 nm).
High-resolution mass measurements were performed on Micromass/Waters LCT Premier Electrospray Time of Flight (TOF) mass spectrometer coupled with a Waters HPLC system, unless noted. Purification of the nucleotide analogues for biological testing was carried out on (diethylamino)ethyl (DEAE)-A25 Sephadex columns with a linear gradient (0.01–0.5 M) of 0.5 M ammonium bicarbonate as the mobile phase. Compounds 7 were purified by Sephadex alone (and isolated in the ammonium salt form), and all other compounds were purified by HPLC with a Luna 5 μ RP-C18(2) semipreparative column (250 × 10.0 mm; Phenomenex, Torrance, CA) and using the following conditions: flow rate of 2 mL/min; 10 mM triethylammonium acetate (TEAA)-CH3CN from 100:0 to 95:5 (or up to 100:0 to 75:25) in 20-40 min (and isolated in the triethylammonium salt form).
All reagents were of analytical grade. 3-Phenacyl-UDP 5 was purchased from Tocris. 5-Iodouridine 34 was purchased from Berry Associates (Ann Arbor, MI). Compound 36 were synthesized as reported.19,25 Other reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO).
(S)-Methanocarba-uridine-5′-diphosphate triethylammonium salt (7)
A solution of the (S)-methanocarba-uridine 40 (1-[(1S,2S,5S,3R,4R)-2,3-dihydroxy-4-(hydroxymethyl) bicyclo[3.1.0]hexyl]-1,3-dihydropyrimidine-2,4-dione, 10 mg, 0.04 mmol, ref. 13) and Proton Sponge (17 mg, 0.08 mmol) in trimethyl phosphate (1 mL) was stirred for 10 min at 0 °C. Then phosphorous oxychloride (0.008 mL, 0.08 mmol) was added dropwise, and the reaction mixture was stirred for 2 h at 0 °C. A mixture of tributylamine (0.8 mL, 0.35 mmol) and a solution 0.35 M of the bis(tributylammonium) salt of phosphoric acid in DMF (0.7 mL) was added at once. After 10 min, 0.2 M triethylammonium bicarbonate solution (1.5 mL) was added, and the clear solution was stirred at rt for 1 h. After removal of solvents, the residue was purified using the same method as the general procedure using Sephadex-DEAE A-25 resin and HPLC to get compound 7 (1.3 mg, 45%) as a white solid. 1H NMR (D2O) δ 7.79 (d, J = 7.8 Hz, 1H), 5.86 (d, J = 8.1 Hz, 1H), 4.69 (m, 1H), 4.15 (m, 3H), 2.39 (t, J = 6.3 Hz, 1H), 1.87 (m, 1H), 1.71 (t, J = 6.0 Hz, 1H), 1.30 (ddd, J = 2.1, 6.3, 8.4 Hz, 1H); 31P NMR (D2O) δ -5.87 (d, J = 22.0 Hz), -10.10 (d, J = 23.2 Hz); HRMS-EI found 413.0170 (M – H+)-. C11H15N2O11P2 requires 413.01511; purity > 99% by HPLC (System A: 11.1 min).
5-Iodo-uridine-5′-α,β-methylenediphosphate triethylammonium salt (11)
Methylene diphosphonic acid (14 mg, 0.08 mmol) and DCC (25 mg, 0.121 mmol) were added to a solution of 5-iodo-uridine 34 (15 mg, 0.040 mmol) in DMF (1.0 mL). The mixture was stirred for 2 d at rt under a nitrogen atmosphere. Solvent was evaporated and the residue was purified on Sephadex-DEAE A-25 resin and HPLC using the same method as the general procedure, to give the compound 11 (2 mg, 11%) as a white powder. 1H NMR (D2O) δ 8.26 (s, 1H), 5.98 (d, J = 6.1 Hz, 1H), 4.39-4.46 (m, 2H), 4.29-4.34 (m, 1H), 4.19-4.24 (m, 2H), 2.23 (t, J = 19.1 Hz, 2H); 31P NMR (D2O) δ 19.02 (d, J = 8.34 Hz), 15.04 (d, J = 8.34 Hz); HRMS-EI found 526.9108 (M-H), C10H15IN2O11P2 requires 527.9109.
N4-Methoxycytidine-5′-diphosphate ammonium salt (12)
A solution of the N4-methoxycytidine 36 (10 mg, 0.037 mmol) and Proton Sponge (17 mg, 0.08 mmol) in trimethyl phosphate (1 mL) was stirred for 10 min at 0 °C. Then, phosphorous oxychloride (0.008 mL, 0.08 mmol) was added dropwise, and the reaction mixture was stirred for 2 h at 0 °C. A mixture of tributylamine (0.8 mL, 0.35 mmol) and a solution 0.35 M of the bis(tributylammonium) salt of phosphoric acid in DMF (0.7 mL) was added at once. After 10 min 0.2 M triethylammonium bicarbonate solution (1.5 mL) was added, and the clear solution was stirred at rt for 1 h. After removal of solvents, the residue was purified using the same method as the general procedure using Sephadex-DEAE A-25 resin and HPLC to get compound 12 (4.4 mg, 25%) as a white solid. 1H NMR (D2O) δ 7.23 (d, J = 8.1 Hz, 1H), 5.92 (d, J = 5.7 Hz, 1H), 5.79 (d, J = 7.8 Hz, 1H), 4.35 (m, 2H), 4.21 (m, 1H), 4.15 (m, 2H), 3.78 (s, 3H); 31P NMR (D2O) δ -7.74 (d, J = 21.4 Hz), -10.6 (d, J = 21.4 Hz); HRMS-EI found 432.0207 (M – H+)-. C10H16N3O12P2 requires 432.0209; purity > 98% by HPLC (System A: 13.6 min).
N4-Ethoxycytidine-5′-diphosphate ammonium salt (13)
A solution of the cytidine 35 (200 mg, 0.82 mmol) and O-ethylhydroxylamine hydrochloride (160 mg, 1.64 mmol) in pyridine (2 mL) was stirred overnight at 100 °C. The reaction mixture was concentrated, and the residue was purified on a silica gel column (CHCl3/MeOH = 10/1) to obtain compound 37 (230 mg, 97%) as a yellow amorphous solid. 1H NMR (acetone-d6) δ 7.32 (d, J = 8.2 Hz, 1H), 5.86 (d, J = 5.1 Hz, 1H), 5.62 (d, J = 8.2 Hz, 1H), 4.27-4.21 (m, 2H), 4.07-3.95 (m, 3H), 3.79 (dd, J = 12.0, 2.8 Hz, 1H), 3.73 (dd, J = 12.0, 2.8 Hz, 1H), 1.22 (t, J = 7.0 Hz, 3H); HRMS-EI found 288.1196 (M + H+)-. C11H18N3O6 requires 288.1185
A solution of the N4-ethoxycytidine 37 (85 mg, 0.30 mmol) and Proton Sponge (95 mg, 0.44 mmol) in trimethyl phosphate (2.5 mL) was stirred for 10 min at 0 °C. Then, phosphorous oxychloride (0.054 mL, 0.59 mmol) was added dropwise, and the reaction mixture was stirred for 2 h at 0 °C. A mixture of tributylamine (0.56 mL, 2.4 mmol) and a 0.35 M solution of the bis(tributylammonium) salt of phosphoric acid in DMF (6.6 mL) was added at once. After 10 min 0.2 M triethylammonium bicarbonate solution (6.9 mL) was added, and the clear solution was stirred at rt for 1 h. After removal of solvents, the residue was purified using the same method as the general procedure using Sephadex-DEAE A-25 resin and HPLC to obtain compound 13 (21.3 mg, 14%) as a white amorphous solid. 1H NMR (D2O) δ 7.24 (d, J = 8.2 Hz, 1H), 5.96 (d, J = 5.6 Hz, 1H), 5.82 (d, J = 8.2 Hz, 1H), 4.42-4.33 (m, 2H), 4.26-4.14 (m, 3H), 4.05 (dd, J = 14.1, 7.0 Hz, 2H); HRMS-EI found 446.0366 (M – H+)-. C10H16N3O12P2 requires 446.0373; purity > 98% by HPLC (System B: 7.54 min).
N4-t-Butyloxycytidine-5′-diphosphate ammonium salt (14)
A solution of the cytidine 35 (200 mg, 0.82 mmol) and O-t-butylhydroxylamine hydrochloride (207 mg, 1.64 mmol) in pyridine (2 mL) was stirred overnight at 100 °C. The reaction mixture was concentrated, and the residue was purified on a silica gel column (CHCl3/MeOH = 10/1) to obtain compound 38 (143 mg, 55%) as a white solid. 1H NMR (acetone-d6) δ 7.18 (d, J = 8.2 Hz, 1H), 5.85 (d, J = 5.7 Hz, 1H), 5.56 (d, J = 8.2 Hz, 1H), 4.27-4.17 (m, 2H), 3.95 (dd, 1H, J = 6.1, 3.0 Hz), 3.80-3.68 (m, 2H), 1.26 (s, 9H); HRMS-EI found 316.1509 (M + H+)-. C13H22N3O6 requires 316.1516.
A solution of the N4-t-butyloxycytidine 38 (39 mg, 0.12 mmol) and Proton Sponge (40 mg, 0.19 mmol) in trimethyl phosphate (1.2 mL) was stirred for 10 min at 0 °C. Then, phosphorous oxychloride (0.023 mL, 0.25 mmol) was added dropwise, and the reaction mixture was stirred for 2 h at 0 °C. A mixture of tributylamine (0.24 mL, 1.0 mmol) and a 0.35 M solution of the bis(tributylammonium) salt of phosphoric acid in DMF (2.8 mL) was added at once. After 10 min 0.2 M triethylammonium bicarbonate solution (3 mL) was added, and the clear solution was stirred at rt for 1 h. After removal of solvents, the residue was purified using the same method as the general procedure using Sephadex-DEAE A-25 resin and HPLC to obtain compound 14 (12.5mg, 21%) as a white amorphous solid. 1H NMR (D2O) δ 7.12 (d, J = 8.3 Hz, 1H), 5.84 (d, J = 6.0 Hz, 1H), 5.74 (d, J = 8.3 Hz, 1H), 4.32-4.23 (m, 2H), 4.13-4.03 (m, 2H), 3.08 (dd, J = 14.7, 7.4 Hz, 12H), 1.18 (s, 9H), 1.16 (t, J = 7.3 Hz, 18H); HRMS-EI found 474.0679 (M – H+)-. C13H22N3O12P2 requires 474.0669; purity > 98% by HPLC (System B: 8.06 min).
N4-Benzyloxycytidine-5′-diphosphate ammonium salt (15)
A solution of the cytidine 35 (200 mg, 0.82 mmol) and O-benzylhydroxylamine hydrochloride (263 mg, 1.64 mmol) in pyridine (2 mL) was stirred overnight at 100 °C. The reaction mixture was concentrated, and the residue was purified on a silica gel column (CHCl3/MeOH = 10/1) to obtain compound 39 (288 mg, 100%) as pale yellow amorphous. 1H NMR (acetone-d6) δ 7.40-7.26 (m, 5H), 7.23 (d, J = 8.2 Hz, 1H), 5.85 (d, J = 5.6 Hz, 1H), 5.52 (d, J = 8.2 Hz, 1H), 5.00 (s, 2H), 4.27-4.18 (m, 2H), 3.96 (dd, J = 6.1, 3.0 Hz, 1H), 3.82-3.69 (m, 2H); HRMS-EI found 350.1352 (M + H+)-. C16H19N3O6 requires 350.1356.
A solution of the N4-benzyloxycytidine 39 (80 mg, 0.23 mmol) and Proton Sponge (74 mg, 0.34 mmol) in trimethyl phosphate (2.4 mL) was stirred for 10 min at 0 °C. Then, phosphorous oxychloride (0.042 mL, 0.46 mmol) was added dropwise, and the reaction mixture was stirred for 2 h at 0 °C. A mixture of tributylamine (0.43 mL, 1.83 mmol) and a 0.35 M solution of the bis(tributylammonium) salt of phosphoric acid in DMF (5.1 mL) was added at once. After 10 min 0.2 M triethylammonium bicarbonate solution (1.5 mL) was added, and the clear solution was stirred at rt for 1 h. After removal of solvents, the residue was purified using the same method as the general procedure using Sephadex-DEAE A-25 resin and HPLC to obtain compound 15 (54 mg, 47%) as a white amorphous solid. 1H NMR (D2O) δ 7.50-7.39 (m, 5H), 7.26 (d, J = 8.4 Hz, 1H), 5.95 (d, J = 5.9 Hz, 1H), 5.77 (d, J = 7.8 Hz, 1H), 5.07 (s, 2H), 4.46-4.43 (m, 1H), 4.37 (t, J = 5.7 Hz, 1H), 4.27-4.11 (m, 3H); 31P NMR (D2O) δ -7.33 (s), -11.04 (s), -11.18(s); HRMS-EI found 508.0522 (M – H+)-. C16H20N3O12P2 requires 508.0507; purity > 98% by HPLC (System B: 9.45 min).
N4-Methoxycytidine -5′-α,β-methylenediphosphate triethylammonium salt (16)
Methylene diphosphonic acid (13 mg, 0.07 mmol) and DCC (23 mg, 0.11 mmol) were added to a solution of N4-Methoxycytidine 36 (10 mg, 0.037 mmol) in DMF (1.0 mL). The mixture was stirred for 2 d at rt under a nitrogen atmosphere. Solvent was evaporated and the residue was purified on Sephadex-DEAE A-25 resin and HPLC using the same method as the general procedure, to give the compound 16 (7.5 mg, 48%) as a white powder. 1H NMR (D2O) δ 7.30 (d, J = 8.1 Hz, 1H), 5.97 (br, 1H), 5.84 (d, J = 8.1 Hz, 1H), 4.42-4.39 (m, 2H), 4.28-4.23 (m, 1H), 4.17-4.12 (m, 2H), 3.83 (s, 3H), 2.20 (t, J = 19.5 Hz, 2H); 31P NMR (D2O) δ 18.88 (br), 14.75 (br); HRMS-EI found 430.0425 (M-H+)-, C11H18N3O11P2 requires 430.0417; purity > 98% by HPLC (System B: 6.93 min).
(S)-Methanocarba-uridine-5′-triphosphate triethylammonium salt (18)
A solution of (S)-methanocarba-uridine 41 (10 mg, 0.04 mmol) and Proton Sponge (17 mg, 0.08 mmol) in trimethyl phosphate (1 mL) was stirred for 10 min at 0 °C. Then phosphorous oxychloride (0.008 mL, 0.08 mmol) was added dropwise, and the reaction mixture was stirred for 2 h at 0 °C. Tributylammonium pyrophosphate (1.6 moles C12H27N per mole H4P2O7, 110 mg, 0.23 mmol) in DMF (0.3 mL) was added at once to the reaction mixture. After 10 min, 0.2 M triethylammonium bicarbonate solution (1.5 mL) was added, and the clear solution was stirred at rt for 1 h. After removal of solvents, the residue was purified using the same method as the general procedure using Sephadex-DEAE A-25 resin and HPLC to get compound 18 (2.0 mg, 56%) as a white solid. 1H NMR (D2O) δ 7.78 (d, J = 7.8 Hz, 1H), 5.85 (d, J = 8.1 Hz, 1H), 4.67 (m, 1H), 4.16 (m, 3H), 2.38 (t, J = 6.3 Hz, 1H), 1.86 (m, 1H), 1.71 (t, J = 6.0 Hz, 1H), 1.30 (ddd, J = 2.1, 6.3, 8.4 Hz, 1H); 31P NMR (D2O) δ -6.76 (m), -10.7 (d, J = 20.2 Hz), -22.63 (app t); HRMS-EI found 492.9847 (M – H+)-. C11H16N2O14P3 requires 492.9814; purity > 98% by HPLC (System A: 17.5 min).
N4-Methoxycytidine-5′-triphosphate ammonium salt (20)
A solution of N4-methoxycytidine 36 (10 mg, 0.037 mmol) and Proton Sponge (17 mg, 0.08 mmol) in trimethyl phosphate (1 mL) was stirred for 10 min at 0 °C. Then phosphorous oxychloride (0.008 mL, 0.08 mmol) was added dropwise, and the reaction mixture was stirred for 2 h at 0 °C. Tributylammonium pyrophosphate (1.6 moles C12H27N per mole H4P2O7, 110 mg, 0.23 mmol) in DMF (0.3 mL) was added at once to the reaction mixture. After 10 min, 0.2 M triethylammonium bicarbonate solution (1.5 mL) was added, and the clear solution was stirred at rt for 1 h. After removal of solvents, the residue was purified using the same method as the general procedure using Sephadex-DEAE A-25 resin and HPLC to get compound 20 (3.9 mg, 31%) as a white solid. 1H NMR (D2O) δ 7.25 (d, J = 8.1 Hz, 1H), 5.97 (d, J = 6.3 Hz, 1H), 5.83 (d, J = 8.4 Hz, 1H), 4.40 (m, 2H), 4.22 (m, 3H), 3.81 (s, 3H); 31P NMR (D2O) δ -8.82 (d, J = 20.2 Hz), -11.0 (d, J = 20.2 Hz), -22.40 (t, J = 19.5 Hz); HRMS-EI found 511.9873 (M – H+)-. C10H17N3O15P3 requires 511.9857; purity > 98% by HPLC (System A: 17.2 min).
P1,P3-Di(uridine-5′-)α,β-methylenetriphosphate triethylammonium salt (22)
Compound 10 (3 mg, 0.006 mmol) and UMP (1.6 mg, 0.006 mmol) were converted to the corresponding tributylammonium salts by ion exchange. A solution of compound 10, tributylammonium salt, and N,N′-diisopropylcarbodiimide (DIC, 2.8 μL, 0.018 mmol) in DMF (0.06 mL) was stirred for 3 h at rt. Then a solution of the UMP tributylammonium salt in DMF (0.04 mL) and a solution of MgCl2 (1.7 mg, 0.018 mmol) in DMF (0.02 μL) were added. After stirring the reaction mixture at rt for 16 h, the solvent was removed and water (0.6 mL) was added. The resulting solid was removed by filtration, and the filtrate was purified by semipreparative HPLC as described above to obtain 22 (1.2 mg, 27 %) as a white solid.
1H NMR (D2O) δ 8.03 (d, J = 8.1 Hz, 1H), 7.98 (d, J = 8.1 Hz, 1H), 6.02-5.94 (m, 4H), 4.43-4.35 (m, 2H), 4.32-4.17 (m, 3H), 2.37 (t, J = 20.1 Hz, 1H); 31P NMR (D2O) δ 17.35 (s), 7.42 (d, J = 27.6 Hz), -10.93 (d, J = 27.6 Hz; HRMS-EI found 751.0009 (M + 2Na+ – 3H+)-. C19H24N4Na2O19P3 requires 751.0043; purity > 99% by HPLC (System B: 9.3 min).
P1-Uridine-5′- P3-cyclohexyl-triphosphate triethylammonium salt (25)
Compound 1 (4 mg, 0.010 mmol) was exchanged to the tributylammonium salt form. A solution of compound 1, tributylammonium salt, and DIC (4.6 μL, 0.030 mmol) in DMF (0.08 mL) was stirred for 3 h at rt. Then a solution of cyclohexyl phosphate tributylammonium salt in DMF (0.08 mL) and a solution of MgCl2 (2.8 mg, 0.030 mmol) in DMF (0.04 mL) were added. After stirring the reaction mixture at rt for 16 h, the solvent was removed and water (0.4mL) was added. The resulting solid was removed by filtration, and the filtrate was purified by semipreparative HPLC as described above to obtain 25 (1.35 mg, 18%) as a white solid.
1H NMR (D2O) δ 7.92 (d, J = 8.1 Hz, 1H), 7.39 (dd, J = 8.2, 8.7 Hz, 2H), 7.26 (d, J = 8.7 Hz, 2H), 7.19 (t, `J = 8.2 Hz, 1H), 5.94 (d, J = 4.5 Hz, 1H), 5.92 (d, J = 8.7 Hz, 1H), 4.38-4.32 (m, 1H), 4.30-4.18 (m, 4H); 31P NMR (D2O) δ -11.22 (d, J = 19.1 Hz), -15.71 (d, J = 19.9 Hz), -23.07 (t, J = 19.1 Hz); HRMS-EI found 558.9928 (M – H+)-. C15H18N2O15P3 requires 558.9920; purity > 99% by HPLC (System B: 8.9 min).
P1-Uridine-5′- P3-phenyl-triphosphate triethylammonium salt (26)
Compound 1 (4 mg, 0.010 mmol) and sodium phenyl phosphate dibasic dihydrate (5.0 mg, 0.020 mmol) were cation exchanged to the tributylammonium salts. A solution of compound 1, tributylammonium salt, and DIC (4.6 μL, 0.030 mmol) in DMF (0.08 mL) was stirred for 3 h at rt. Then a solution of phenyl phosphate tributylammonium salt in DMF (0.08 mL) and a solution of MgCl2 (2.8 mg, 0.030 mmol) in DMF (0.04 mL) were added. After stirring the reaction mixture at rt for 16 h, the solvent was removed and water (0.4 mL) was added. The resulting solid was removed by filtration, and the filtrate was purified by semipreparative HPLC as described above to obtain 26 (1.24 mg, 16%) as a white solid.
1H NMR (D2O) δ 8.02 (d, J = 8.7 Hz, 1H), 6.07-6.00 (m, 2H), 4.49-4.40 (m, 2H), 4.35-4.15 (m, 4H), 4.01-3.77 (m, 4H), 2.06-1.96 (m, 2H), 1.79-1.69 (m, 2H), 1.59-1.14 (m, 6H); 31P NMR (D2O) δ -11.5 (br, 2H), -23.1 (br, 1H); HRMS-EI found 565.0405 (M – H+)-. C15H24N2O15P3 requires 565.0390; purity > 99% by HPLC (System B: 9.8 min).
P1-Uridine-5′- P3-[1]glucose-1′-triphosphate triethylammonium salt (27)
Compound 1 (10 mg, 0.025 mmol) and α-D-glucose-1-phosphate disodium salt hydrate (10 mg, 0.038 mmol) were exchanged to the tributylammonium salts. A solution of compound 1, tributylammonium salt, and DIC (12 μL, 0.075 mmol) in DMF (0.10 mL) was stirred for 3 h at rt. Then a solution of the α-D-glucose-1-phosphate tributylammonium salt in DMF (0.10 mL) and a solution of MgCl2 (7 mg, 0.075 mmol) in DMF (0.05 μL) were added. After stirring the reaction mixture at rt for 16 h, the solvent was removed and water (0.4 mL) was added. The resulting solid was removed by filtration, and the filtrate was purified by semipreparative HPLC as described above to obtain 27 (1.8 mg, 9%) as a white solid.
1H NMR (D2O) δ 7.97 (d, J = 7.8 Hz, 1H), 6.01 (d, J = 4.5 Hz, 1H), 5.99 (d, J = 7.8 Hz, 1H), 5.63 (dd, J = 7.2, 3.6 Hz, 1H), 4.45-4.37 (m, 2H), 4.32-4.22 (m, 3H), 3.97-3.73 (m, 4H), 3.52 (dt, J = 9.9, 3.3 Hz, 1H), 3.45 (t, J = 9.6 Hz); 31P NMR (D2O) δ -11.27 (d, J = 19.8 Hz), -12.65 (d, J = 19.8 Hz), -22.70 (d, J = 19.8 Hz); HRMS-EI found 645.0100 (M – H+)-. C15H24N2O20P3 requires 645.0135; purity > 99% by HPLC (System B: 8.8 min).
(S)-Methanocarba-uridine-3′, 5′-cyclic diphosphate triethylammonium salt (9) and (S)-Methanocarba-uridine-5′-glucose-1′-triphosphate triethylammonium salt (28)
To a solution of compound 7 (2.6 mg, 0.0036 mmol) in DMF (2 mL), 1,1′-carbonyldiimidazole (3.0 mg, 0.018 mmol) was added. The reaction mixture was stirred at rt for 5 h. Then, 5% triethylamine solution in 1/1 water/methanol (1 mL) was added and stirring was continued at rt for an additional 2 h. After removal of the solvent, the residue was dried under high vacuum and dissolved in DMF (2 mL). Glucose-1-monophosphate tributylammonium salt (4 mg, 0.006 mmol) in DMF (0.2 mL) was added to this mixture. The reaction mixture was stirred at rt for 2 d. After removal of the solvent, the residue was purified using the same method as the general procedure using Sephadex-DEAE A-25 resin and HPLC to get compound 28 (0.8 mg, 23%) and 9 (1.1 mg, 51%) additionally. Compound 28: 1H NMR (D2O) δ 7.79 (d, J = 8.1 Hz, 1H), 5.86 (d, J = 8.1 Hz, 1H), 5.64 (dd, J = 3.3, 7.2 Hz, 1H), 4.62 (m, 1H), 4.15 (t, J = 6.3 Hz, 2H), 3.86 (m, 4H), 3.50 (m, 3H), 2.39 (t, J = 6.3 Hz, 1H), 1.88 (dd, J = 4.8, 9.9 Hz, 1H), 1.72 (t, J = 5.7 Hz, 1H), 1.39 (m, 1H); 31P NMR (D2O) δ -10.87 (d, J = 21.4 Hz), -12.58 (d, J = 20.76 Hz); HRMS-EI found 655.0326 (M – H+)-. C17H26N2O19P3 requires 655.0343; purity > 98% by HPLC (System A: 16.9 min). Compound 9: 1H NMR (D2O) δ 7.81 (d, J = 8.1 Hz, 1H), 5.84 (d, J = 8.1 Hz, 1H), 5.30 (m, 1H), 4.08 (m, 3H), 2.63 (t, J = 6.3 Hz, 1H), 2.03 (m, 1H), 1.71 (t, J = 6.0 Hz, 1H), 1.50 (ddd, J = 2.1, 6.3, 8.4 Hz, 1H); 31P NMR (D2O) δ -9.55 (app d), -22.66 (app t); HRMS-EI found 395.0044 (M – H+)-. C11H13N2O10P2 requires 395.0045; purity > 98% by HPLC (System A: 12.0 min).
5-Iodouridine-5′-glucose-1′-triphosphate triethylammonium salt (29)
Compound 3 (2 mg, 0.003 mmol) and α-D-glucose-1-phosphate disodium salt hydrate (2 mg, 0.005 mmol) were exchanged to the tributylammonium salts. A solution of compound 3, tributylammonium salt, and DIC (1.3 μL, 0.008 mmol) in DMF (0.04 mL) was stirred for 3 h at rt. Then a solution of the α-D-glucose-1-phosphate tributylammonium salt in DMF (0.04 mL) and a solution of MgCl2 (0.8 mg, 0.008 mmol) in DMF (0.02 μL) were added. After stirring the reaction mixture at rt for 16 h, the solvent was removed and water (0.2 mL) was added. The resulting solid was removed by filtration, and the filtrate was purified by semipreparative HPLC as described above to obtain 29 (0.35 mg, 13%) as a white solid.
1H NMR (D2O) δ 8.26 (s, 1H), 6.01 (d, J = 4.8 Hz, 1H), 5.68 (dd, J = 3.0, 6.9 Hz, 1H), 4.49-4.40 (m, 2H), 4.36-4.25 (m, 3H), 4.03-3.80 (m, 4H), 3.62-3.47 (m, 2H); 31P NMR (D2O) δ -11.46 (d, J = 19.1 Hz), -12.60 (d, J = 19.1 Hz), -22.60 (t, J = 19.1 Hz); HRMS-EI found 770.9131 (M – H+)-. C15H23IN2O20P3 requires 770.9102; purity > 99% by HPLC (System B: 9.7 min).
3-Phenacyluridine-5′-glucose-1′-triphosphate triethylammonium salt (30)
Compound 5 (2 mg, 0.005 mmol) and α-D-glucose-1-phosphate disodium salt hydrate (2 mg, 0.010 mmol) were exchanged to the tributylammonium salts. A solution of compound 5, tributylammonium salt, and DIC (2 μL, 0.015 mmol) in DMF (0.04 mL) was stirred for 3 h at rt. Then, a solution of the α-D-glucose-1-phosphate tributylammonium salt in DMF (0.04 mL) and a solution of MgCl2 (1 mg, 0.015 mmol) in DMF (0.02 μL) were added. After stirring the reaction mixture at rt for 16 h, the solvent was removed and water (0.2 mL) was added. The resulting solid was removed by filtration, and the filtrate was purified by semipreparative HPLC as described above to obtain 30 (1.0 mg, 29%) as a white solid.
1H NMR (D2O) δ 8.08-8.04 (m, 3H), 7.80 (t, J = 7.2 Hz, 1H), 7.65 (t, J = 7.2 Hz, 2H), 6.17 (d, J = 7.8 Hz, 1H), 6.04 (d, J =4.5 Hz, 1H), 5.66 (dd, J = 3.6, 6.9 Hz), 5.55 (s, 2H), 4.49-4.43 (br, 2H), 4.37-4.29 (br, 3H), 4.01-3.77 (m, 4H), 3.60-3.50 (br, 1H), 3.48 (t, J = 9.1 Hz, 1H); 31P NMR (D2O) δ -11.24 (d, J = 25.4 Hz), -12.63 (d, J = 25.4 Hz), -22.65 (d, J = 25.8 Hz); HRMS-EI found 763.0554 (M – H+)-. C23H30N2O21P3 requires 763.0589; purity > 99% by HPLC (System B: 9.9 min).
Uridine-5′-glucose-1′-α,β-methylenetriphosphate triethylammonium salt (31)
Compound 10 (2.4 mg, 0.004 mmol) and α-D-glucose-1-phosphate disodium salt hydrate (3.6 mg, 0.012 mmol) were exchanged to the tributylammonium salts. A solution of compound 10, tributylammonium salt, and DIC (1.8 μL, 0.012mmol) in DMF (0.04 mL) was stirred for 3 h at rt. Then, a solution of the α-D-glucose-1-phosphate tributylammonium salt in DMF (0.04 mL) and a solution of MgCl2 (2.6 mg, 0.028 mmol) in DMF (0.02 μL) were added. After stirring the reaction mixture at rt for 16 h, the solvent was removed and water (0.4 mL) was added. The resulting solid was removed by filtration, and the filtrate was purified by semipreparative HPLC as described above to obtain 31 (2.7 mg, 80%) as a white solid.
1H NMR (D2O) δ 8.07 (d, J = 7.8 Hz, 1H), 6.06-5.99 (m, 2H), 5.70-5.63 (m, 1H), 4.49-4.43 (m, 2H), 4.35-4.27 (m, 1H), 4.27-4.21 (m, 2H), 4.00-3.77 (m, 4H), 3.60-3.43 (m, 2H), 2.43 (t, J = 19.8 Hz, 2H); 31P NMR (D2O) δ 17.32 (d, J = 9.1 Hz), 8.27 (dd, J = 9.1, 26.7 Hz), -12.23 (d, J = 26.7 Hz); HRMS-EI found 643.0348 (M – H+)-. C16H26N2O19P3 requires 643.0343; purity > 99% by HPLC (System B: 9.2 min).
N4-Methoxycytidine-5′-glucose-1′-triphosphate triethylammonium salt (32)
Compound 12 (4.9 mg, 0.009 mmol) and α-D-glucose-1-phosphate disodium salt hydrate (8.4 mg, 0.028 mmol) were exchanged to the tributylammonium salts. A solution of compound 12, tributylammonium salt, and DIC (4.3 μL, 0.028 mmol) in DMF (0.1 mL) was stirred for 3 h at rt. Then, a solution of the α-D-glucose-1-phosphate tributylammonium salt in DMF (0.1 mL) and a solution of MgCl2 (2.6 mg, 0.028 mmol) in DMF (50 μL) were added. After stirring the reaction mixture at rt for 16 h, the solvent was removed and water (1 mL) was added. The resulting solid was removed by filtration, and the filtrate was purified by semipreparative HPLC as described above to obtain 32 (0.62 mg, 10%) as a white solid.
1H NMR (D2O) δ 7.28 (d, J = 8.4 Hz, 1H), 5.99 (d, J = 5.4 Hz, 1H), 5.86 (d, J = 8.4 Hz, 1H), 5.65 (dd, J = 3.6, 7.2 Hz, 1H), 4.45-4.36 (m, 2H), 4.30-4.15 (m, 3H), 3.97-3.77 (m, 4H), 3.82 (s, 3H), 3.57-3.50 (m, 1H), 3.47 (t, J = 9.3 Hz, 1H); 31P NMR (D2O) δ -11.27 (d, J = 19.1 Hz), -12.65 (d, J = 19.1 Hz), -22.71 (t, J = 19.1 Hz); HRMS-EI found 674.0387 (M – H+)-. C16H27N3 O20P3 requires 674.0401; purity > 99% by HPLC (System B: 9.3 min).
N4-Methoxycytidine-5′-phenyl-triphosphate triethylammonium salt (33)
Compound 12 (6.9 mg, 0.011 mmol) and sodium phenyl phosphate dibasic dihydrate (8.3 mg, 0.033 mmol) were exchanged to the tributylammonium salts. A solution of compound 12 tributylammonium salt and DIC (5.0 μL, 0.033 mmol) in DMF (0.07 mL) was stirred for 3 h at rt. Then, a solution of the phenyl phosphate tributylammonium salt in DMF (0.07 mL) and a solution of MgCl2 (3.1 mg, 0.033 mmol) in DMF (0.07 mL) were added. After stirring the reaction mixture at rt for 16 h, the solvent was removed and water (1 mL) was added. The resulting solid was removed by filtration, and the filtrate was purified by semipreparative HPLC as described above to obtain 33 (0.9 mg, 14%) as a white amorphous solid.
1H NMR (D2O) δ 7.44-7.36 (m, 2H), 7.31-7.17 (m, 4H), 5.92 (d, J = 6.0 Hz, 1H), 5.77 (d, J = 8.4 Hz, 1H), 4.38-4.28 (m, 2H), 4.24-4.15 (m, 3H); HRMS-EI found 588.0186 (M – H+)-. C16H22N3 O15P3 requires 588.0199; purity > 99% by HPLC (System B: 10.3 min).
Pharmacological analyses
UDP was purchased from Sigma (St. Louis, MO). Myo-[3H]inositol (20 Ci/mmol) was obtained from American Radiolabeled Chemicals (St. Louis, MO). Measurement of intracellular calcium ion concentration in response to nucleotides was performed as described.4
Assay of PLC activity stimulated by P2Y2, P2Y4, and P2Y6 receptors
Stable cell lines expressing the human P2Y2, P2Y4, or P2Y6 receptor in 1321N1 human astrocytoma cells were generated as described.26 Agonist-induced [3H]inositol phosphate production was measured in 1321N1 cells plated to 20,000 cells/well on 96-well plates two d prior to assay. Sixteen h before the assay, the inositol lipid pool of the cells was radiolabeled by incubation in 100 μL of serum-free inositol-free Dulbecco's modified Eagle's medium, containing 1.0 μCi of myo-[3H]inositol. No changes of medium were made subsequent to the addition of [3H]inositol. On the day of the assay, the cells were challenged with 25 μL of a fivefold concentrated solution of receptor agonists in 200 mM HEPES (N-(2-hydroxyethyl)-piperazine-N′-2-ethanesulfonic acid), pH 7.3 in HBSS, containing 50 mM LiCl for 30 min at 37°C. Incubations were terminated by aspiration of the drug-containing medium and addition of 450 μL of ice-cold 50 mM formic acid. [3H]Inositol phosphate accumulation was quantified using scintillation proximity assay methodology as previously described in detail.26
Data Analysis
Agonist potencies (EC50 values) were determined from concentration-response curves by non-linear regression analysis using the GraphPad software package Prism (GraphPad, San Diego, CA). All experiments examining the activity of newly synthesized molecules also included full concentration effect curves for the cognate agonist of the target receptor: UTP for the P2Y2 receptor, UTP for the P2Y4 receptor, and UDP for the P2Y6 receptor. Each concentration of drug was tested in triplicate assays, and concentration effect curves for each test drug were repeated in at least three separate experiments with freshly diluted molecule. The results are presented as mean ± SEM from multiple experiments or in the case of concentration effect curves from a single experiment carried out with triplicate assays that were representative of results from multiple experiments.
Stability of Nucleotide Derivatives
UTP was purchased from Sigma (St. Louis, MO). Up3U 19 was synthesized by the procedure previously reported.21 HCl buffer (pH 1.2) solution was prepared as follows. Concentrated HCl (7 mL, 12 N) was added to 2.0 g of sodium chloride and diluted with water to a volume of 1000 mL. Membranes were prepared as follows. P2Y1 receptor-expressing astrocytoma cells were grown to 80% confluence and then harvested. The cells were homogenized and the nuclear fraction was removed by centrifuging at 100 × g for 5 min. The pellet was resuspended in 50 mM tris(hydroxymethyl)aminomethane (Tris) hydrochloride buffer (pH 7.4). The suspension was homogenized with a polytron homogenizer (Brinkmann) for 10 s and was ultracentrifuged at 20,000 × g for 20 min at 4°C. The resultant pellets were resuspended in Tris buffer (pH 7.4), and the membrane was stored at -80° C until the experiments. The protein concentration of the membrane preparation was measured by Bradford assay.34 The membrane preparation was diluted with 4 mL of Dulbecco's Phosphate-Buffered Saline (pH 7.4) and homogenized before use for the stability check. The final protein concentration was 14.9 μg/mL.
Ten μL of a 2 mg/mL aqueous solution of each nucleotide derivative was mixed with 90 μL of HCl buffer (pH 1.2) or the membrane suspension and incubated at 37°C. At regular intervals, a 6 μL aliquot of the above mixture was mixed with 54 μL of water or 5 mM TBAP. Then, 50 μL of the mixure was injected to the HPLC.
AUC of compounds were measured using a Hewlett–Packard 1100 HPLC equipped with a Zorbax Eclipse 5 μm XDB-C18 analytical column (250 × 4.6 mm; Agilent Technologies Inc, Palo Alto, CA). Mobile phase: linear gradient solvent system: 5 mM TBAP-CH3CN from 80:20 to 40:60 in 6.5 min; the flow rate was 1 mL/min (System C) or System B. Peaks were detected by UV absorption with a diode array detector at 254, 275, and 280 nm and AUC were calculated based on the peak at 254 nm.
Molecular Modeling
P2Y6 - Compound 7 complex
Molecular modeling was based on our previously published model of the complex between UDP and the rhodopsin-based P2Y6 receptor, obtained through MonteCarlo conformational searches and molecular dynamics in a hydrated lipid bilayer.13
Here, compound 7 was sketched from the docked UDP, and subsequently subjected to a series of energy minimizations allowing flexibility to increasingly larger portions of the model, followed by a conformational search. In particular, the minimizations were performed on the cyclopropane moiety of the ligand, the ribose moiety of the ligand, and the entire ligand. The final minimization and the conformational search were performed on the entire ligand plus all the P2Y6 residues and the water molecules located within a radius of 5 Å. All of the atoms within an additional shell of a radius of 3 Å were also taken into account in the calculations as a frozen environment.
All the calculations were conducted using MacroModel, version 9.6, as implemented in the Schrödinger package, with the OPLS_2005 force field and water as implicit solvent (GB/SA model).35 The minimizations were based on the Polak-Ribier conjugate gradient algorithm and were performed with a threshold on the gradient of 0.05 KJ/Mol. The conformational search was based on 1000 steps of extended Monte Carlo Multiple Minimum (MCMM) torsional sampling. The sampling regarded the torsional angles of residues and ligand, as well as the rotation and translation of the ligand.
P2Y6 - Compound 12, 26 or 33 complex
Molecular modeling was based on the same structure described in the above section. Compounds 12, 26 and 33 were sketched from the docked UDP, and subsequently subjected to a series of energy minimizations allowing flexibility to increasingly larger portions of the model, followed by a conformational search. All the conformational searches were performed on each entire ligand plus all the P2Y6 residues located within a radius of 6 Å from the initial ligand, UDP. All of the atoms within an additional shell of a radius of 4 Å were also taken into account in the calculations as a frozen environment.
All the calculations were conducted using MacroModel, version 9.7.1, as implemented in the Schrödinger package, with the OPLS_2001 force field.36 The minimizations were based on the Polak-Ribier conjugate gradient algorithm and were performed with a threshold on the gradient of 0.05 KJ/Mol. The conformational search was based on 100 steps of extended Monte Carlo Multiple Minimum (MCMM) torsional sampling. The sampling regarded the torsional angles of residues and ligand, as well as the rotation and translation of the ligand.
Residue indexing
To facilitate the comparison among receptors, residues are identified through the GPCR residue indexing system. In each TM, the index X.50, where X is the TM number, is assigned to the position hosting the most conserved residue among class A GPCRs. All other positions in the TM are numbered relatively to the X.50 position.37
Supplementary Material
Acknowledgements
Mass spectral measurements were performed by Dr. Noel Whittaker and NMR by Wesley White and Dr. Herman Yeh (NIDDK). We thank Dr. Andrei A. Ivanov (NIDDK) for helpful discussions and Rhonda Carter for technical assistance. This research was supported in part by the Intramural Research Program of the NIH, NIDDK and by an NIH extramural grant GM38213 from the National Institute of General Medical Sciences.
Abbreviations
- CDI
N,N-carbonyldiimidazole
- DCC
N,N′-dicyclohexylcarbodiimide
- DIC
N,N′-diisopropylcarbodiimide
- DMF
dimethylformamide
- HEK
human embryonic kidney
- HBSS
Hank's balanced salt solution
- HEPES
N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid
- HPLC
high performance liquid chromatography
- PLC
phospholipase C
- SAR
structure activity relationship
- TBAP
tetrabutylammonium dihydrogenphosphate
- TEAA
triethylammonium acetate
- UDP
uridine-5′-diphosphate
- UTP
uridine-5′-triphosphate
Footnotes
Presented in part at the 238th ACS National Meeting, Aug. 2009, Washington, DC, MEDI187.
Supporting Information Available. Time course of decomposition/hydrolysis of selected pyrimidine nucleotide derivatives in acidic solution and in the presence of cell membranes and stick representation of Fig. 2A. This material is available free of charge via the Internet at http://pubs.acs.org.
Reference List
- 1.Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol. Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Costanzi S, Mamedova L, Gao ZG, Jacobson KA. Architecture of P2Y nucleotide receptors: structural comparison based on sequence analysis, mutagenesis, and homology modeling. J. Med. Chem. 2004;47:5393–5404. doi: 10.1021/jm049914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malmsjö M, Hou M, Pendergast W, Erlinge D, Edvinsson L. Potent P2Y6 receptor mediated contractions in human cerebral arteries. BMC Pharmacol. 2003;3:4. doi: 10.1186/1471-2210-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balasubramanian R, Ruiz de Azua I, Wess J, Jacobson KA. Activation of distinct P2Y receptor subtypes stimulates insulin secretion and cytoprotection in MIN6 mouse pancreatic β cells. Biochem. Pharmacol. 2010;79:1317–132. doi: 10.1016/j.bcp.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim SG, Gao ZG, Soltysiak KA, Chang TS, Brodie C, Jacobson KA. P2Y6 nucleotide receptor activates PKC to protect 1321N1 astrocytoma cells against tumor necrosis factor-induced apoptosis. Cell. Mol. Neurobiol. 2003;23:401–418. doi: 10.1023/A:1023696806609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mamedova LK, Wang R, Besada P, Liang BT, Jacobson KA. Attenuation of apoptosis in vitro and ischemia/reperfusion injury in vivo in mouse skeletal muscle by P2Y6 receptor activation. Pharmacol. Res. 2008;58:232–239. doi: 10.1016/j.phrs.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korcok J, Raimundo LN, Du X, Sims SM, Dixon SJ. P2Y6 nucleotide receptors activate NF-κB and increase survival of osteoclasts. J. Biol. Chem. 2005;280:16909–16915. doi: 10.1074/jbc.M410764200. [DOI] [PubMed] [Google Scholar]
- 8.Koizumi S, Shigemoto-Mogam Y, Nasu-Tada K, Shinozaki Y, Ohsawa K, Tsuda M, Joshi BV, Jacobson KA, Kohsaka S, Inoue K. UDP acting at P2Y6 receptors is a novel mediator of microglial phagocytosis. Nature. 2007;446:1091–1095. doi: 10.1038/nature05704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grbic DM, Degagné E, Langlois C, Dupuis AA, Gendron FP. Intestinal inflammation increases the expression of the P2Y6 receptor on epithelial cells and the release of CXC chemokine ligand 8 by UDP. J. Immunol. 2008;180:2659–2668. doi: 10.4049/jimmunol.180.4.2659. [DOI] [PubMed] [Google Scholar]
- 10.Markovskaya A, Crooke A, Guzmán-Aranguez AI, Peral A, Ziganshin AU, Pintor J. Hypotensive effect of UDP on intraocular pressure in rabbits. Eur. J. Pharmacol. 2008;579:93–97. doi: 10.1016/j.ejphar.2007.10.040. [DOI] [PubMed] [Google Scholar]
- 11.Bar I, Guns P-J, Metallo J, Wilkin F, Cammarata D, Boeynaems J-M, Bult H, Robaye B. Knock-out mice reveal a role for the P2Y6 receptor in macrophages, endothelial cells and vascular smooth muscle cells. Mol. Pharmacol. 2008;74:777–784. doi: 10.1124/mol.108.046904. [DOI] [PubMed] [Google Scholar]
- 12.Jacobson KA, Boeynaems JM. P2Y nucleotide receptors: Promise of therapeutic applications. Drug Disc. Today. doi: 10.1016/j.drudis.2010.05.011. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Costanzi S, Joshi BV, Maddileti S, Mamedova L, Gonzalez-Moa MJ, Marquez VE, Harden TK, Jacobson KA. Human P2Y6 receptor: molecular modeling leads to the rational design of a novel agonist based on a unique conformational preference. J. Med. Chem. 2005;48:8108–8111. doi: 10.1021/jm050911p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Besada P, Shin DH, Costanzi S, Ko HJ, Mathé C, Gagneron J, Gosselin G, Maddileti S, Harden TK, Jacobson KA. Structure activity relationship of uridine 5′-diphosphate analogues at the human P2Y6 receptor. J. Med. Chem. 2006;49:5532–5543. doi: 10.1021/jm060485n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El-Tayeb A, Qi A, Müller CE. Synthesis and structure-activity relationships of uracil nucleotide derivatives and analogues as agonists at human P2Y2, P2Y4, and P2Y6 receptors. J. Med. Chem. 2006;49:7076–7087. doi: 10.1021/jm060848j. [DOI] [PubMed] [Google Scholar]
- 16.Ko H, Carter RL, Cosyn L, Petrelli R, de Castro S, Besada P, Zhou Y, Cappellacci L, Franchetti P, Grifantini M, Van Calenbergh S, Harden TK, Jacobson KA. Synthesis and potency of novel uracil nucleotide analogues as P2Y2 and P2Y6 receptor agonists. Bioorg. Med. Chem. 2008;16:6319–6332. doi: 10.1016/j.bmc.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carter RL, Fricks IP, Barrett MO, Burianek LE, Zhou Y, Ko H, Das A, Jacobson KA, Lazarowski ER, Harden TK. Quantification of Gi-mediated inhibition of adenylyl cyclase activity reveals that UDP is a potent agonist of the human P2Y14 receptor. Mol. Pharmacol. 2009;76:1341–1348. doi: 10.1124/mol.109.058578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Das A, Ko H, Burianek LB, Barrett MO, Harden TK, Jacobson KA. Human P2Y14 receptor agonists: Truncation of the hexose moiety of uridine-5′-diphosphoglucose and its replacement with alkyl and aryl groups. J. Med. Chem. 2010;53:471–480. doi: 10.1021/jm901432g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melman A, Zhong M, Marquez VE, Jacobson KA. Synthesis of enantiomerically pure (S)-methanocarbaribo uracil nucleoside derivatives for use as antiviral agents and P2Y receptor ligands. J. Org. Chem. 2008;73:8085–8088. doi: 10.1021/jo801224j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zimmerman H. Extracellular metabolism of ATP and other nucleotides. Naunyn-Schmiedeberg's Arch Pharmacol. 2000;362:299–309. doi: 10.1007/s002100000309. [DOI] [PubMed] [Google Scholar]
- 21.Shaver SR, Rideout JL, Pendergast W, Douglass JG, Brown EG, Boyer JL, Patal RI, Redick CC, Jones AC, Picher M, Yerxa BR. Structure activity relationships of dinucleotides: Potent and selective agonists of P2Y receptors. Purinergic Signal. 2005;1:183–191. doi: 10.1007/s11302-005-0648-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yerxa BR, Sabater JR, Davis CW, Stutts MJ, Lang-Furr M, Jones AC, Cowlen M, Dougherty R, Boyer J, Abraham WM, Boucher RC. Pharmacology of INS37217 [P(1)-(uridine 5′)-P(4)- (2’-deoxycytidine 5′)tetraphosphate, tetrasodium salt], a next-generation P2Y2 receptor agonist for the treatment of cystic fibrosis. J. Pharmacol. Exp. Ther. 2002;302:871–880. doi: 10.1124/jpet.102.035485. [DOI] [PubMed] [Google Scholar]
- 23.Kogo S, Yamada K, Iwai Y, Osawa K, Hayakawa H. Process for producing di(pyrimidine nucleoside 5′-)polyphosphate. 2008 WO 2008/012949 A1.
- 24.Kim HS, Ravi RG, Marquez VE, Maddileti S, Wihlborg A-K, Erlinge D, Malmsjö M, Boyer JL, Harden TK, Jacobson KA. Methanocarba modification of uracil and adenine nucleotides: High potency of Northern ring conformation at P2Y1, P2Y2, P2Y4 and P2Y11, but not P2Y6 receptors. J. Med. Chem. 2002;45:208–218. doi: 10.1021/jm010369e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ivanov MA, Antonova EV, Maksimov AV, Pigusova LK, Belanov EF, Aleksandrova LA. New N4-hydroxycytidine derivatives: synthesis and antiviral activity. Coll. Czech. Chem. Comm. 2006;71:1099–1106. [Google Scholar]
- 26.Bourdon DM, Wing MR, Edwards EB, Sondek J, Harden TK. Quantification of Isozyme-Specific Activation of Phospholipase C-β2 by Rac GTPases and Phospholipase C-ε by Rho GTPases in an Intact Cell Assay System. Methods in Enzymol. 2006;406:489–499. doi: 10.1016/S0076-6879(06)06037-X. [DOI] [PubMed] [Google Scholar]
- 27.Ko H, Das A, Carter RL, Fricks IP, Zhou Y, Ivanov AA, Melman A, Joshi BV, Kováč P, Hajduch J, Kirk KL, Harden TK, Jacobson KA. Molecular recognition in the P2Y14 nucleotide receptor: Probing the structurally permissive terminal sugar moiety of uridine-5′-diphosphoglucose. Bioorg. Med. Chem. 2009;17:5298–5311. doi: 10.1016/j.bmc.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan SW, Gallo SJ, Kim BK, Guo MJ, Blackburn GM, Zamecnik PC. P1,P4-dithio-P2,P3-monochloromethylene diadenosine 5′,5′′′-P1,P4-tetraphosphate: a novel antiplatelet agent. Proc. Natl. Acad. Sci. U. S. A. 1997;94:4034–4039. doi: 10.1073/pnas.94.8.4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eliahu SE, Camden J, Lecka J, Weisman GA, Sévigny J, Gélinas S, Fischer B. Identification of hydrolytically stable and selective P2Y1 receptor agonists. Eur. J. Med. Chem. 2009;44:1525–1536. doi: 10.1016/j.ejmech.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robaye B, Boeynaems JM, Communi D. Slow desensitization of the human P2Y6 receptor. Eur. J. Pharmacol. 1997;329:231–236. [PubMed] [Google Scholar]
- 31.Phillps JH, Brown DM, Grossman L. The efficiency of induction of mutations by hydroxylamine. J. Mol. Biol. 1966;21:405–419. doi: 10.1016/0022-2836(66)90015-5. [DOI] [PubMed] [Google Scholar]
- 32.Parandeh F, Abaraviciene SM, Amisten S, Erlinge D, Salehi A. Uridine diphosphate (UDP) stimulates insulin secretion by activation of P2Y6 receptors. Biochem. Biophys. Res. Commun. 2008;370:499–503. doi: 10.1016/j.bbrc.2008.03.119. [DOI] [PubMed] [Google Scholar]
- 33.Ohtani M, Suzuki JI, Jacobson KA, Oka T. Evidence for the possible involvement of P2Y6 receptor in Ca2+ mobilization and insulin secretion in mouse pancreatic islets. Purinergic Signalling. 2008;4:365–375. doi: 10.1007/s11302-008-9122-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal. Biochem. 1976;76:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 35.MacroModel, version 9.6. Schrödinger, LLC; New York, NY: 2008. [Google Scholar]
- 36.MacroModel, version 9.7. Schrödinger, LLC; New York, NY: 2009. [Google Scholar]
- 37.Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G-protein coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.