

Abstract
Here we report the isolation, kinetic characterization, and X-ray structure determination of a cooperative E. coli aspartate transcarbamoylase (ATCase) without regulatory subunits. The native ATCase holoenzyme consists of six catalytic chains organized as two trimers bridged non-covalently by six regulatory chains organized as three dimers, c6r6. Dissociation of the native holoenzyme produces catalytically active trimers, c3, and nucleotide-binding regulatory dimers, r2. By introducing specific disulfide bonds linking the catalytic chains from the upper trimer site specifically to their corresponding chains in the lower trimer prior to dissociation, a new catalytic unit, c6, was isolated consisting of two catalytic trimers linked by disulfide bonds. Not only does the c6 species display enhanced enzymatic activity compared to the wild-type enzyme, but the disulfide bonds also impart homotropic cooperativity, never observed in the wild-type c3. The c6 ATCase was crystallized in the presence of phosphate and its X-ray structure determined to 2.10 Å resolution. The structure of c6 ATCase liganded with phosphate exists in a nearly identical conformation as other R-state structures with similar values calculated for the vertical separation and planar angles. The disulfide bonds linking upper and lower catalytic trimers predispose the active site into a more active conformation by locking the 240’s loop into the position characteristic of the high-affinity R state. Furthermore, the elimination of the structural constraints imposed by the regulatory subunits within the holoenzyme provides increased flexibility to the c6 enzyme enhancing its activity over the wild-type holoenzyme (c6r6) and c3. The covalent linkage between upper and lower catalytic trimers restores homotropic cooperativity so that a binding event at one or so active sites stimulates binding at the other sites. Reduction of the disulfide bonds in the c6 ATCase results in c3 catalytic subunits that display similar kinetic parameters to wild-type c3. This is the first report of an active c6 catalytic unit that displays enhanced activity and homotropic cooperativity.
Escherichia coli aspartate transcarbamoylase (ATCase, EC 2.1.3.2) catalyzes the committed step of the pyrimidine nucleotide biosynthesis pathway: the condensation of carbamoyl phosphate (CP) and L-aspartate (Asp) to form N-carbamoyl-L-aspartate (CA) and phosphate (Pi). The end products of the pyrimidine biosynthetic pathway, CTP and UTP in the presence of CTP, allosterically inhibit the enzyme (1,2). Conversely ATP, an end product of the purine biosynthetic pathway, allosterically activates the enzyme (1).
The quaternary structure of the E. coli enzyme is a dodecamer composed of two trimeric catalytic subunits (Mr = 34,000/chain) and three dimeric regulatory subunits (Mr = 17,000/chain). The six active sites are located at the interface between adjacent catalytic chains and six allosteric sites are located on each of the regulatory chains (3–6). The two catalytic trimers are bridged non-covalently by the three regulatory dimers. The enzyme exists in two different structural and functional states that have been characterized (7); the low-activity, low-affinity T state, and the high-activity, high-affinity R state (8–10). The conversion of the enzyme from the T to R state occurs upon Asp binding to the holoenzyme in the presence of CP. Upon the allosteric transition, several tertiary changes occur such as the reorganization of the 80’s and 240’s loops of the catalytic chains. Specific interchain interactions of the side chains of the 240’s loop in the T and R states have been identified as important contributors to stabilizing the T and R states (11).
The complex quaternary structure of the E. coli enzyme has previously been manipulated to study its catalytic properties. Treatment of the holoenzyme with heat or mercurials, such as p-hydroxymercuribenzoate or neohydrin, results in the dissociation of the catalytic and regulatory subunits (12–16). The isolated regulatory dimer binds nucleotide effectors, but is catalytically inactive. The isolated trimeric catalytic subunit is approximately 50% (16,17) more active than the native enzyme, but is noncooperative and does not exhibit heterotropic effects (1). A complex of a single catalytic trimer, c3, and three regulatory dimers, 3r2, can be formed by mixing a very dilute solution of c3 with a large excess of r2 (18). The c3r6 complex displays Michaelis-Menten kinetics similar to c3, but it shows a much lower Km for aspartate, a significantly lower Vmax, much stronger substrate inhibition (19), and it is not activated by the substrate analog, succinate, in the presence of a saturating concentration of CP (20). These results suggest that the active sites in the c3r6 complex exist in only one functional state. A similar complex was formed between a catalytic trimer and the zinc-binding domain of the regulatory chain, which consists of 70 amino acids. This complex displayed properties similar to the R functional state of the holoenzyme, increased thermal stability compared to c3, hyperbolic dependence of initial velocity on aspartate concentration, and 50% increase in aspartate affinity indicating that substantial changes occur at the active site of the catalytic trimer when it interacts with the zinc-containing polypeptide of the regulatory chain (21–23). The native enzyme exists in two forms with the low-affinity, low-activity T state being due to constraints imposed by quaternary structure that must be overcome, upon ligand binding, by conformational changes to the R state. The c3r6 and the c3-(zn)3 complexes do not have these same constraints and therefore ligand binding is more favorable. Furthermore, it was suggested that the two c3 subunits of the native enzyme, while not in direct contact, are both required for the observed homotropic effects of the native enzyme because the homotropic interactions arise from substrate binding at sites on different c3 subunits (13,24–26). However, cooperativity has been observed in a relatively inactive c3 subunit with the active site mutation R105A.
Other quaternary structures of ATCase have been investigated also. A quaternary structure in which the holoenzyme is lacking a single regulatory dimer (c6r4) was isolated as a less stable component observed as an intermediate in the assembly and dissociation of the native enzyme. The c6r4 complex exhibits characteristic sigmoidal saturation behavior and CTP inhibition, but the allosteric effects are reduced by about one-third in comparison to the wild-type enzyme suggesting that the allosteric regulation does not require the intact c6r6 structure (13,24–26).
Stabilization of the quaternary state of the holoenzyme has also been achieved with cross-linking reagents. Tartryl diazide was used to react with the side chains of lysine residues within subunits to form amide bonds. When this reaction is performed in the absence of ligands, the T-functional state of the enzyme is stabilized, whereas in the presence of substrate analogs, the R-functional state of the enzyme is stabilized (27,28). The results of the cross-linking showed that the derivatives obtained in the presence or absence of substrate analogs differed greatly in substrate affinity by up to 20-fold (28). These cross-linking studies however used a nonspecific bifunctional reagent. The R state of ATCase has also been specifically stabilized via formation of disulfide bonds after introduction of Cys residues at positions in the 240’s loop of the catalytic chains (29). The T- to the R- state transition involves a reorientation of 240’s loop. In the T state the 240’s loops of one upper and lower catalytic chain are far apart, but become stacked and close together in the R state. This conformational change facilitates the domain closure in the catalytic chains, forming the high-activity and high-affinity active sites characteristic of the R state. A mutation at position 241 in the 240’s loop from Ala to Cys, as well as a mutation at position 47 from a Cys to an Ala provided the possibility to selectively form disulfide bonds between Cys241 from the upper and lower catalytic chains which would lock the 240’s loop into the position characteristic for the R-quaternary state (29). The aspartate saturation curve (performed under non-reducing conditions) of the C47A/A241C holoenzyme is hyperbolic and exhibited high activity, and the enzyme activity was not affected by CTP or ATP (29) indicating that the enzyme is held in the R-functional state.
The results from the experiments presented here describe the properties of a new quaternary structure of E. coli ATCase produced by treatment of the disulfide-linked C47A/A241C holoenzyme with p-hydroxymercuribenzoate. This procedure resulted in the dissociation of catalytic and regulatory subunits, producing a disulfide linked c6 catalytic unit and r2 regulatory subunits. This disulfide linked c6 catalytic species provides a means to investigate a hexameric ATCase without any regulatory subunits to impose quaternary structure constraints, thus allowing the investigation of the enzyme’s homotropic cooperativity. Furthermore, the X-ray structure of the c6 ATCase was determined providing structural evidence to explain the enhanced catalytic activity and restoration of homotropic cooperativity observed for this species.
EXPERIMENTAL PROCEDURES
Materials
L-aspartate ATP, CTP, carbamoyl phosphate dilithium salt, N-carbamoyl-L-aspartate, 2-mercaptoethanol, potassium dihydrogen phosphate, sodium EDTA, uracil, and DEAE Sephadex A50 were purchased from Sigma-Aldrich. Carbamoyl phosphate dilithium salt was purified before use by precipitation from 50% (v/v) ethanol and was stored dessicated at −20°C. Enzyme grade ammonium sulfate, Tris, and electrophoresis grade acrylamide were from ICN Biomedicals (Costa Mesa, CA). Antipyrine and diacetylmonoxime were obtained from Fisher.
Overexpression and Purification of the C47A/A241C Holoenzyme and Catalytic Subunits
The C47A/A241C holoenzyme was overexpressed from plasmid pEK613 (29) and purified to homogeneity as described previously (30) under non-reducing conditions. The purity of the enzyme was checked by SDS PAGE (31), non-reducing SDS PAGE (in which 2-mercaptoethanol is omitted from the sample buffer), and nondenaturing PAGE (32,33).
The C47A/A241C holoenzyme was dissociated into catalytic and regulatory subunits with pHMB as described previously described (34). The C47A/A241C c6 was isolated from the mixture by anion-exchange chromatography employing DEAE Sephadex A50. The r2 dimers eluted with 0.23 M KCl, and the C47A/A241C c6 eluted with 0.5 M KCl. The C47A/A241C c6 was dialyzed extensively against 0.1 M Tris-acetate pH 8.3 to remove KCl and pHMB before kinetic tests were performed.
The disulfide bonds in the C47A/A241C c6 were reduced with 20 mM 2-mercaptoethanol to form C47A/A241C c3. The complete reduction from C47A/A241C c6 to C47A/A241C c3 was verified by non-reducing SDS PAGE and non-denaturing PAGE.
The concentrations of the wild-type holoenzyme and catalytic subunit were determined using extinction coefficients of 0.59 and 0.72 cm2 mg−1 respectively (16). The concentration of the C47A/A241C c6 and c3 were determined by the BioRad version of the Bradford dye binding assay using wild-type holoenzyme and catalytic subunit as standard (35).
Aspartate Transcarbamoylase Assay
The ATCase activity was measured at 25 °C colorimetrically by reaction of the carbamoyl-L-aspartate formed employing a solution composed of a mixture of 50 mL 0.5% (w/v) antipyrine in 50% (v/v) sulfuric acid and 25 mL 0.4% (w/v) in 5% acetic acid, as previous described (36). Aspartate and PALA saturation curves were performed in duplicate. Assays were performed in 50 mM Tris-acetate buffer, pH 8.3 in the presence of a saturating concentration of CP (4.8 mM). For the PALA activation curve, the Asp concentration was held constant at 7 mM. Data analysis of the steady-state kinetics was carried out as described previously (37). Fitting of the experimental data to theoretical equations was accomplished by non-linear regression. The data were analyzed using an extension of the Hill equation that included a term for substrate inhibition. If the fit to the Hill equation gave a Hill coefficient of 1 or less, the experimental data were fit to the Michaelis-Menten equation with an additional term for substrate inhibition (38).
Crystallization, data collection, and structure refinement
A sample of purified C47A/A241C c6 at a concentration of 10 mg/mL was sent to the Hauptman Woodward Institute for high-throughput crystallization screening (39). The results of the screen of 1536 under-oil conditions indicated buffer conditions in which the C47A/A241C c6 enzyme formed well-defined crystals. Crystals of C47A/A241C c6 were obtained by the hanging-drop vapor diffusion method under conditions suggested by the HWI crystal screen. Single crystals were obtained by mixing 10 mg/mL of C47A/A241C c6 in 100 mM Tris-acetate pH 8.3, in a 1:1 ratio (v/v), with crystallization buffer and suspended over a well containing 1 mL of the same buffer. Crystallization buffer consisted of 40% PEG 1000, 100 mM NH4H2PO4, and 100 mM HEPES pH 7.5. The crystals grew over one week at 20 °C and were approximately 0.2 × 0.2 × 0.2 mm3.
The crystals were soaked in a cryoprotectent solution containing 20% PEG 400 and 80% crystallization well buffer before freezing in liquid nitrogen. X-ray diffraction data were collected at Brookhaven National Synchrotron Light Source (Brookhaven National Laboratory, New York), beamline X29. C47A/A241C c6 crystallized in space group P4332 with unit cell dimensions a = b = c = 142.5 Å and diffracted to 2.10 Å resolution.
The initial model for the C47A/A241C c6 structure was derived from the coordinates of the RPALA structure with all of the water molecules, ligands, and regulatory chains removed (40). Molecular replacement was performed utilizing Phaser within the CCP4 Suite (41). The structure was solved with one catalytic chain in the asymmetric unit, which gives a Matthew’s coefficient of 3.55 and 65% solvent.
The coordinates from the molecular replacement were subjected to rigid-body refinement and simulated annealing within PHENIX, reducing the Rfactor/Rfree to 0.228/0.242. Further model rebuilding and structure refinement was performed using COOT (42) and PHENIX (43), respectively. Two molecules of phosphate were fit into the difference density in the active site. Water molecules were added to the structure using PHENIX (43) on the basis of the Fo–Fc electron density maps at or above the 3.0 σ level. After completion of the refinement the final Rfactor/Rfree was 0.177/0.205. The model was checked for errors using PROCHECK (44). The details of data processing and refinement statistics are given in Table 2.
Table 2.
X-ray Data Collection and Refinement Statistics for the ATCase C6 Structure
| Data collection | |
| Space Group | P4332 |
| Cell Dimensions: | |
| a = b = c (Å) 141.78 | |
| α, β, γ (°) 90, 90, 90 | |
| Resolution (Å) | 50.00–2.10 |
| Rmergea (%) | 0.077 (0.535) |
| Average (I/σ) | 8.9 |
| Completeness (%) | 100.0 (100.0) |
| Total Reflections | 1,221,103 |
| Unique reflections | 28,952 |
| Redundancy | 42.2 (43.0) |
| Refinement | |
| Resolution (Å) | 30.00–2.10 |
| Reflections | 28,904 |
| Rfactor/Rfree | 0.177/0.205 |
| RMS deviations: | |
| Bonds (Å) | 0.006 |
| Angles (°) | 0.916 |
| Mean B value (Å2) | 32.0 |
| Total number waters | 222 |
Values in parentheses are for the highest resolution shell
Protein Data Bank Deposition
The coordinate and structure factors for the C47A/A241C c6 complex with phosphate have been deposited in the RCSB Protein Data Bank under accession code 3NPM.
RESULTS AND DISCUSSION
Isolation of C47A/A241C c6 from the C47A/A241C Holoenzyme
Under non-reducing conditions the C47A/A241C holoenzyme can be isolated with three sets of disulfide bridges between the Cys241 residues in the upper and lower catalytic subunits (29). In this cross-linked form, the enzyme did not exhibit cooperativity for aspartate, displayed a slightly higher maximal velocity than the wild-type enzyme and was almost insensitive to the allosteric effectors (29). The unliganded C47A/A241C holoenzyme was determined to be in the R-quaternary structure by small-angle X-ray scattering (29). Thus, the loss of cooperativity and lack of allosteric effector response can be attributed to the stabilization of the R-quaternary structure of the C47A/A241C holoenzyme by the formation of the interchain disulfide bridges.
The regulatory and catalytic subunits of ATCase interact only by noncovalent interactions, which can be disturbed in a variety of ways such as by heat treatment (1) as well as by treatment of the holoenzyme with certain mercurials (1,14,16). In principle, treatment of the C47A/A241C holoenzyme with a mercurial, such as pHMB, should dissociate the regulatory subunits and produce an ATCase species containing six catalytic chains organized in two trimers linked by disulfide bridges, the C47A/A241C c6 enzyme.
When the C47A/A241C holoenzyme was treated with pHMB the c6r6 species disappears (see Figure 1A, compare Lanes 3 and 4), yet by non-reducing SDS PAGE no detectable alteration occurred (see Figure 1B, compare Lanes 3 and 4). After treatment of the C47A/A241C holoenzyme with pHMB, the mixture was separated by anion-exchange chromatography utilizing DEAE Sephadex A50. The separation yielded regulatory subunits (r2) and a species that did not migrate as either c6r6 or c3 on non-denaturating PAGE (see Figure 1A, compare Lanes 5 and 6). This species migrated at the same position as the C47A/A241C c2 species observed on the non-reducing SDS PAGE (see Figure 1B, compare Lanes 5 and 6 to the upper band of Lane 3), with an estimated molecular mass of 68 KDa. In this species disulfide bonds linked the two catalytic subunits together to form a hexamer, c6.
Figure 1.

Characterization of c6 by gel electrophoresis. Non-denaturating (A) and non-reducing SDS PAGE (B). The two gels were composed of 7.5% and 12% acrylamide, respectively, and only differed in that the non-reducing gel did not have 2-mercaptoethanol added to either gel or loading buffer. Lane 1, wild-type E. coli ATCase holoenzyme; Lane 2, E. coli ATCase catalytic subunit (c3); Lane 3, C47A/A241C holoenzyme; Lane 4, C47A/A241C after treatment with pHMB; Lane 5–6, Pure C47A/A241C c6 at a low and high concentration, respectively.
Steady-state Kinetics
Shown in Figure 2 are the Asp saturation curves for the wild-type holoenzyme and C47A/A241C c6 and Table 1 provides a complete summary of the kinetic parameters. Most notable was the almost 3-fold enhanced activity of the C47A/A241C c6 as compared to the wild-type holoenzyme, with maximal velocities of 55.6 and 19.3 mmol•h−1•mg−1 respectively. The [Asp]0.5 values were similar, 17.9 mM for the C47A/A241C c6 as compared to 13.5 mM for the wild-type holoenzyme. The higher [Asp]0.5 value for the C47A/A241C c6 was unexpected since the [Asp]0.5 value for the wild-type and C47A/A241C c3 are 10.4 mM and 8.7 mM respectively. Even more unexpected was that the C47A/A241C c6 exhibited substantial cooperativity for aspartate (nH = 1.7) even in the absence of the regulatory subunits. The higher specific activity of the C47A/A241C c6 cannot be accounted for by the mutations themselves since the C47A/A241C c6r6 and the C47A/A241C c3 have specific activities of 25.6 and 21.5 mmol•h−1•mg−1, respectively. Thus, the disulfide bridges holding the two catalytic trimers together in the C47A/A241C c6 impart not only a higher specific activity onto the C47A/A241C c6, but also homotropic cooperativity. Previous to this study the only catalytic chain based species of ATCase that exhibited Asp cooperativity was the catalytic subunit (c3) of the R105A mutant enzyme. This relatively inactive mutant exhibited a Hill coefficient of 2.0 (17).
Figure 2.
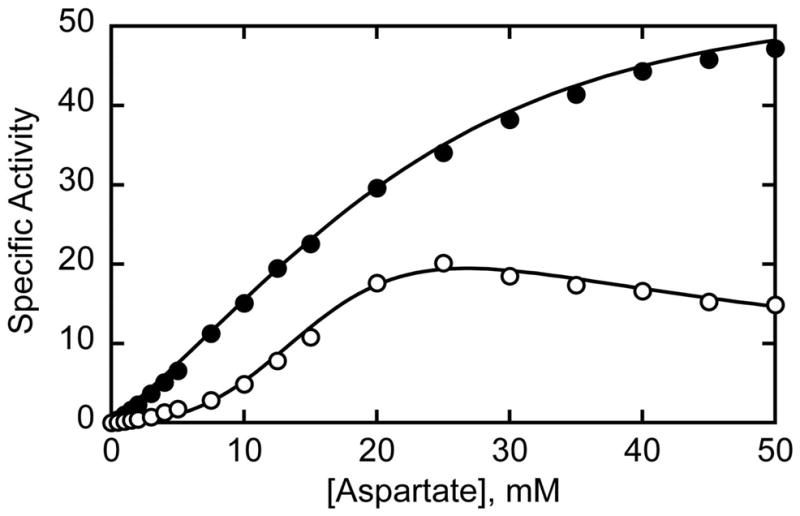
Aspartate saturation curves for the wild-type and mutant enzymes. Specific activity (mmol•hr−1•mg−1) versus the concentration of Asp for the C47A/A241C c6 (filled circles) and wild-type holoenzyme (c6r6) (open circles) enzymes. The assays were performed at 25° C at saturating concentrations of CP (4.8 mM) in 50 mM Tris-acetate buffer, pH 8.3.
Table 1.
Kinetic parameters for wild-type and mutant forms of ATCasea
| Enzyme | Form | Vmax | [Asp]0.5 | nH |
|---|---|---|---|---|
| mmol h−1 mg−1 | mM | |||
| Wild-type | Holoenzyme | 19.3 ± 0.5 | 13.5 ± 0.9 | 2.6 ± 0.2 |
| C47A/A241COXc | Holoenzyme | 25.6 ± 0.9 | 2.3 ± 0.2 | 1b |
| C47A/A241CREDd | Holoenzyme | 25.4 ± 2.3 | 10.8 ± 0.8 | 1.3 ± 0.1 |
| C47A/A241C | C6e | 55.6 ± 1.8 | 17.9 ± 0.8 | 1.7 ± 0.2 |
| Wild-type | C3 | 22.9 ± 2.1 | 10.4 ± 0.8 | 1b |
| C47A/A241C | C3f | 21.5 ± 1.9 | 8.7 ± 1.9 | 1b |
Data were determined from the Asp saturation curves (Figure 2), and are the average of 3 independent determinations. Colorimetric assays were performed at 25° C, in 50 mM Tris acetate buffer, pH 8.3, and at saturating levels of CP (4.8 mM)
No cooperativity observed, data were fit to the Michaelis-Menten equation with an additional term for substrate inhibition.
Disulfide-linked C47A/A241C holoenzyme
Reduced C47A/A241C holoenzyme
A part of catalytic subunits linked by disulfides
c3 after reduction of the disulfide linked c6.
Tests of Homotropic Cooperativity
Homotropic cooperativity in ATCase is due to a shift from a low-activity low-affinity T state to a high-activity high-affinity R state. Not only does the natural substrate Asp, in the presence of a saturating concentration of CP induce the T to R transition, but also substrate analogs such as succinate, in the presence of a saturating concentration of CP, or the bisubstrate analog PALA. At low Asp concentrations, much less than the [Asp]0.5, the enzyme is essentially in the T state and the binding of succinate or PALA to one or more active sites shifts the entire enzyme to the R state. Thus under these conditions these inhibitors can activate the enzyme. To test the homotropic cooperativity of the C47A/A241C c6, the activity of the enzyme was measured as a function of the PALA concentration at 7 mM Asp.
As seen in Figure 3, PALA did not activate the C47A/A241C holoenzyme but did activate the C47A/A241C c6 species, suggesting that the sigmoidal Asp saturation curve (see Figure 2) was indeed the result of homotropic cooperativity. The extent of the activation is not large, but is what would be expected for an enzyme with a relatively low Hill coefficient. These data confirm that the disulfide bridges between the two catalytic subunits of the C47A/A241C c6 are directly responsible for the homotropic linkage between the six active sites in this enzyme. Furthermore, the reduction of the disulfide bonds of the C47A/A241C c6 to produce catalytic subunits (c3), eliminated the homotropic cooperativity and reduced the specific activity and [Asp]0.5 (Table 1).
Figure 3.
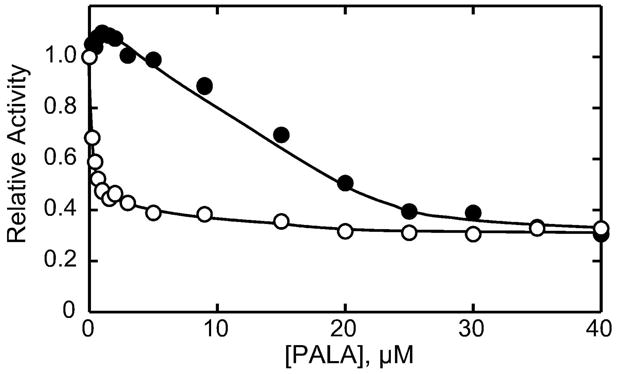
Activity of the C47A/A241C c6 and c6r6 enzymes as a function of PALA concentration. Assays were performed at 25° C at saturating concentrations of CP (4.8 mM) in 50 mM tris-acetate buffer, pH 8.3 for the C47A/A241C c6 (filled circles) and C47A/A241C c6r6 (open circles) enzymes. The Asp concentration was held constant at 8 mM for the mutant catalytic subunit and 7 mM Asp was used for the mutant holoenzyme.
Overall Description of the Structure
The catalytic subunit of wild-type ATCase, c3, does not display homotropic cooperativity due to the lack of constraints imposed by the regulatory subunits in the transition from the T to R state. It has been postulated that in order to observe homotropic cooperativity, both catalytic trimers must be present (45–47). The steady-state kinetic results of the C47A/A241C c6 displayed homotropic cooperativity and enhanced enzymatic activity. To understand how these disulfide bonds restore the homotropic cooperativity and enhance the activity of the C47A/A241C c6, X-ray crystallography was employed. Data quality crystals were obtained of the C47A/A241C c6 with Pi bound that diffracted to a maximal resolution of 2.10 Å.
To evaluate the quaternary conformation of the c6_Pi structure, the vertical separation between the upper and lower catalytic subunits was computed and compared to the vertical separations of known T- and R-state structures. The vertical separation for the T-state holoenzyme is approximately 45.6 Å (48) and for the R-state holoenzyme is approximately 56.4 Å (40). The difference between the T- and R-state vertical separations corresponds to the 11 Å vertical expansion of the enzyme during the T to R transition. For the c6_Pi structure, the vertical separation was computed as 56.0 Å, similar to R-state structures. Thus, even in the absence of regulatory subunits, the disulfide bonds in the c6_Pi structure stabilize the expanded R-state quaternary structure. In addition, the planar angle of the CP and Asp domains was computed. The planar angle is defined as the angle formed between the centers of gravity of the CP and Asp domains and a hinge point (49). The T-state holoenzyme active site is more open with a planar angle of approximately 135° and the R-state holoenzyme active site is more closed with a planar angle of approximately 127°. The planar angle computed for the catalytic chain of the c6_Pi structure was 127.7°, comparable to other R-state structures and the c3_PALA structure.
Disulfide Bonds
In order to verify the presence of the disulfide bonds between catalytic subunits in the crystal, one of the crystals was washed with well buffer, dissolved in 50 mM Tris pH 8.3, and the protein solution was characterized by non-reducing SDS PAGE. As seen in Figure 4 (Lanes 2 and 3), the band corresponding to the C47A/A241C c6 before and after crystallization was essentially the same, and indicates that the disulfide bridge linking the two catalytic chains was intact within the crystal.
Figure 4.
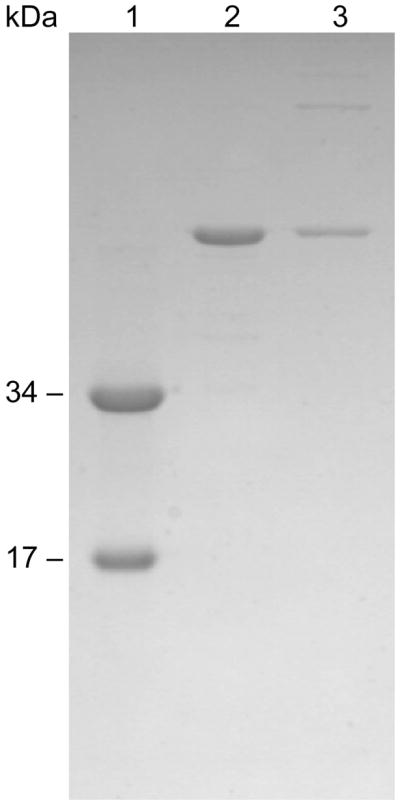
Non-reducing 12% SDS PAGE of dissolved crystals of the C47A/A241C c6 enzyme. A non-reducing SDS PAGE of a single crystal of the C47A/A241C c6 enzyme washed and dissolved in 100 mM Tris-acetate buffer pH 8.3. Lane 1, wild-type E. coli ATCase holoenzyme; Lane 2, C47A/A241C c6 solution before crystallization; Lane 3, C47A/A241C c6 crystal dissolved in buffer.
In addition, the disulfide bonds were clearly observed in the electron density maps generated for the c6_Pi structure. The initial model for refinement contained Ala at position 241, but the positive density observed in the Fo–Fc electron density map indicated the presence of a Cys residue at this position. COOT (42) was used to mutate and auto-fit a Cys residue into the positive density at position 241. Following refinement utilizing PHENIX (43), the resulting Fo–Fc electron density map showed positive density extending from the CB atom of Cys241 in the opposite direction of the refined SG atom suggesting that this residue exists in two alternate conformations. The alternate conformation was added to the coordinate files using COOT (42) and the resulting residue fit into the density appropriately. When symmetry atoms are generated, Cys241 from a catalytic chain in the upper catalytic trimer forms a disulfide bond with Cys241 from the corresponding chain in the lower catalytic trimer. The asymmetric unit of the c6_Pi structure contains a single C47A/A241C catalytic chain, and the c6 species is generated by the symmetry operations of space group P4332. There are three disulfide bonds in the C47A/A241C c6 species linking Cys241 from the 240’s loop of C1, C2, and C3 from the upper catalytic timer to Cys241 from the 240’s loop of C4, C5, and C6 from the lower catalytic trimer respectively. The dihedral angle between the Cβ-Sγ-Sγ′-Cβ′ was measured as 71.6° which is slightly less than the ideal disulfide bond angle of 90°. The disulfide bond distance was measured as 2.2 Å, also close to the ideal value. Figure 5 shows the location, geometry, and electron density for a disulfide bond between the C1 and C4 catalytic chains.
Figure 5.

Disulfide linkages between upper and lower catalytic subunits. Stereoview of the 240’s loop from a single catalytic chain from the upper catalytic trimer, maroon, and a single catalytic chain from the lower catalytic trimer, gold. These two catalytic chains are covalently linked via a disulfide bond formed between the side chains of Cys241 residues in the upper and lower catalytic chains. The refined coordinates of residue Cys241 from both chains are overlaid on the 2Fo–Fc electron density map (blue) contoured at 1.0 σ. The Cys241 residue exists in two alternate conformations, each with 50% occupancy. The sulfur atoms involved in the bonds are green for one conformation and orange for the other. One position of the disulfide bond is outlined with maroon carbons and the other position with gold carbons.
Active Site Ligands
The C47A/A241C c6 sample that was sent for high-throughput crystallization was in 50 mM Tris buffer at pH 8.3. This very simple buffer was utilized to attempt to crystallize the enzyme in the absence of active site ligands. However, the crystallization screen produced well-defined crystals only in buffers containing phosphate at high concentrations, specifically 0.10 M NH4H2PO4. Analysis of the 2Fo–Fc electron density map indicated two Pi molecules in the area of the active site at 100% occupancy. The interactions of these Pi molecules with the active site residues are shown in Figure 6 overlaid on the 2Fo–Fc electron density map. As in the RS-S_Pi structure, one Pi is positioned in the active site in a similar position as the phosphate group of CP, or phosphonate group of PALA binds and makes identical interactions with active site residues. The second Pi is approximately 4.3 Å away and makes fewer hydrogen-bonding interactions with active site residues. When the active sites of the c6_Pi and RS-S_Pi structures are superimposed, as shown in Figure 7, both Pi molecules occupy identical positions and suggests the path that Pi takes as it dissociates from the active site (50).
Figure 6.

Active site interactions with Pi ligands. Stereoview of the interactions between the active site residues and the phosphate molecules generated by POVScript+ The refined coordinates of the backbone and side chains are overlaid on the 2Fo-Fc electron density map (grey) shown contoured at 1.5σ. The two phosphate molecules are overlaid on the composite omit map (magenta) contoured at 1.0σ. Residues labeled with (*) indicate they are donated from an adjacent catalytic chain.
Figure 7.

Superposition of the active sites from the c6_Pi and RS-S_Pi structures. Stereoview of the active site superposition of the c6_Pi structure complexed with Pi1 and Pi2 (white carbons) and the RS-S_Pi structure complexed with Pi1 and Pi2 (blue carbons and Pi). This figure was drawn with PyMOL.
Comparisons to Other ATCase Structures
The c6_Pi structure reported here was compared to other R-state structures to determine the structural basis for enhanced enzyme activity and homotropic cooperativity. Table 3 shows the values for the computed planar angles and vertical separations of the compared structures. First, the vertical separations were compared and indicated that the c6_Pi structure has a slightly smaller vertical separation, which may be explained by the covalent bond holding the catalytic trimers slightly closer together than other R-state structures. The planar angle for the c6_Pi structure is slightly more closed than the PALA-liganded R-state structure even though there are only two Pi molecules bound at the active site. The R236_PAM structure contains two catalytic chains in the asymmetric unit, one liganded with PAM and the other unliganded, providing a glimpse into the unliganded R-state active sites. Notably the active site bound to PAM is more open than that of the c6_Pi structure, with a computed planar angle of 128.4° which may be more appropriate for Asp binding and to promote catalysis. The unliganded active site is more closed than that of the c6_Pi structure, with a computed planar angle of 125.7° representing the active site immediately before substrate binding or immediately after product release. The two different conformations of the active site suggest that in the R state, the active site can open and close to bind substrates, promote catalysis, and release products without reverting back to the T state. The planar angle of the c6_Pi structure falls between that of the PAM-bound and unliganded R-state active sites. These results suggest that in the absence of regulatory subunits C47A/A241C c6 can bind substrates and release products without full closure of the active sites between reactions, enhancing catalytic efficiency. A crystal structure of C47A/A241C c6 in the unliganded form would provide decisive details concerning how fully the active site closes between each catalytic cycle, however the c6_Pi structure displays the active site in the process of product release.
Table 3.
Comparisons of ATCase X-ray Structures
| Structure | Planar Anglea (°) | Vertical Separation (Å) |
|---|---|---|
| c6_Pi | 127.7 | 56.0 |
| RS-S_Pi | 128.0 ± 0.5 | 56.4 |
| RPALA | 128.1 ± 0.6 | 57.9 |
| c3_PALA | 128.6 ± 0.2 | – |
| R236_PAM | 128.4b, 125.7c | 57.4 |
| TCTP | 136.4 ±0.3 | 47.3 |
The planar angle was calculated for each catalytic chain in the asymmetric unit and the average and standard deviation were calculated. For structures containing only one catalytic chain in the asymmetric unit, a single value is reported.
The planar angle calculated for the catalytic chain in the asymmetric unit bound to PAM
The planar angle calculated for the catalytic chain in the asymmetric unit that is unliganded
Local Conformational Changes Associated with the Allosteric Transition
The allosteric transition of ATCase from the low-activity, low-affinity T state to the high-activity, high-affinity R state involves both global and local changes in structure. It has been shown that the binding of PALA to just one of the active sites of the ATCase holoenzyme is sufficient to induce the global structural transition from the T to the R state (51). Once the global transformation has occurred, there remain local conformational changes, such as the repositioning of the 240’s loop in the catalytic chains, that do not take place until substrates bind to each of the high-affinity active sites created by the quaternary structure change (52). Forming disulfide bonds between Cys241 of the upper and lower catalytic chains stabilize the 240’s loop in the position characteristic of its final, closed position. The X-ray structure of the C47A/A241C holoenzyme in the presence of Pi is in the R structure and shows that the 240’s loop is in an identical conformation as the 240’s loop from the RPALA structure indicating that the disulfide bonds position this loop in its final, closed conformation. After the regulatory chains are dissociated from the holoenzyme to yield C47A/A241C c6, the 240’s loop remains in the same conformation, as shown in the tube diagram in Figure 8. Figure 8 compares a catalytic chain from the upper and lower catalytic trimers of the c6_Pi and RPALA structures. These structures are nearly identical with an overall RMS deviation of 0.71 Å. The width of the tube is proportional to the RMS deviation of the α-carbon backbone between the c6_Pi and RPALA structures, with RMS deviations exceeding 1.0 Å highlighted in blue. The only regions highlighted in blue represent the flexible N and C-termini. The 240’s loops of both structures are nearly identical indicating that the disulfide bonds of the c6_Pi structure predispose the active site into a more active conformation identical to that of the RPALA structure.
Figure 8.
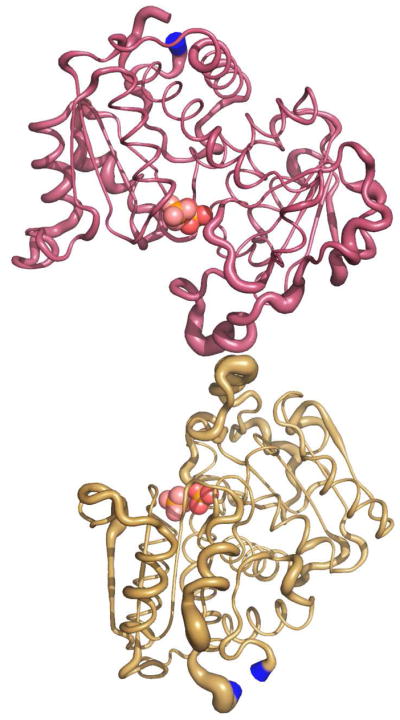
Comparison of the c6_Pi and RPALA structures. Comparison of one catalytic chain from the upper catalytic trimer, C1 maroon, and the corresponding catalytic chain from the lower catalytic trimer, C4 gold, from the c6_Pi and RPALA structures. The width of the tube is proportional to the RMS deviation between the α-carbon positions of the c6_Pi and RPALA structures. Regions colored in blue represent a RMS deviation greater than 1.0 Å. The two Pi molecules bound at the active site are shown as spheres distinguishable by the different coloring of the oxygen atoms. This figure was drawn using PyMOL.
Influence of Structure on Enzyme Activity
The C47A/A241C c6 enzyme displays enhanced activity compared to the wild-type holoenzyme and its catalytic subunit c3. Wild-type c3 displays enhanced activity compared the wild-type holoenzyme that can be attributed to the lack of structural constraints imposed by the bridging regulatory subunits. C47A/A241C c6 is not associated with regulatory subunits, but displays enhanced activity compared to wild-type c3. The X-ray structure reveals that the c6_Pi structure is in the R-quaternary structure with disulfide bonds locking the 240’s loop into the position that has been shown to facilitate domain closure to form the high-activity and high-affinity active sites. Hence, the increase in activity of the C47A/A241C c6 over wild-type c3 may be explained by the disulfide bonds stabilizing the position of the 240’s loop in such a way that predisposes the active site into its high-activity form even in the absence of substrates. The c3_Apo and c3_PALA structures revealed the major differences between the unliganded and liganded forms of c3 respectively (53,54). The binding of substrates facilitates the conformational changes in wild-type c3 to form the R-like active sites comparable to those of the RPALA structure. If the disulfide bonds of the C47A/A241C c6 predispose the active sites in their high-activity form in the absence of substrates, this can explain the observed increase in activity over wild-type c3. Additional structural evidence is necessary to explain the nearly 3-fold increase in activity compared to the wild-type holoenzyme. Although the disulfide bonds in the C47A/A241C holoenzyme also stabilize the 240’s loops in the positions characteristic of high-affinity R-state, this species does not display the same enhanced activity because there are structural constraints imposed by the bridging regulatory subunits that stiffen the structure and impede catalytic turnover. In the holoenzyme (c6r6) form, the disulfide bonds stabilize the R-allosteric state but do not increase the specific activity. Dissociation of the regulatory subunits relieves these structural constraints, resulting in a more flexible molecule capable of increased catalytic activity compared to the wild-type holoenzyme.
Cooperativity without Regulatory Subunits
The unregulated wild-type catalytic subunit, c3, does not exhibit homotropic cooperativity for Asp, yet it is known to exist in two different conformations resembling the catalytic trimers of either the T or R state of the holoenzyme depending on the absence or presence of PALA (53,54). Prior to the determination of the c3, c3_Apo (53) and c3_PALA (54) structures, it was assumed that, because wild-type c3 displays a higher specific activity than c6r6 and lacks cooperativity, that the unliganded c3 would resemble the high-activity, high-affinity catalytic subunits of the R-state holoenzyme. Instead the results suggest that c3 behaves similarly to the holoenzyme with respect to the existence of both low-affinity and high-affinity active sites dependent upon the binding of ligands. Yet, wild-type c3 does not display homotropic cooperativity, even when associated with regulatory chains. The association of wild-type c3 with full-length regulatory chains (c3r6) (19) or the zinc-binding domain of the regulatory chain (c3(zn)3) (21–23) influences substrate affinity, but does not restore homotropic cooperativity. These results suggest that both catalytic trimers are required for homotropic effects because homotropic interactions arise from substrate binding at sites on different c3 subunits (45–47). The interactions of the catalytic and regulatory subunits do not impart cooperativity unless both catalytic trimers are present in the same molecule.
However, there is an example of a cooperative catalytic subunit of ATCase, the R105A c3 enzyme, which is relatively inactive. The side chain of Arg105 makes important interactions with active site ligands, particularly with the phosphate group and carbonyl oxygen of CP (55), so when this residue is mutated to Ala, the R105A c3 displays a substantial reduction in CP affinity (17). The binding of CP to the wild-type holoenzyme induces conformational changes that create the binding pocket for Asp, yet the enzyme remains in the T-quaternary structure. It is the binding of Asp that induces the transition to the R state and cooperativity is observed. However, when interactions are weakened between catalytic and regulatory subunits, such as in the D236A holoenzyme, the binding of CP alone is sufficient to induce the allosteric transition, and thus the D236A holoenzyme does not display cooperativity for Asp. If we consider ATCase c3 to behave similarly to the holoenzyme, yet without the same structural constraints, then the binding of CP to wild-type c3 may induce the conformational change in the active sites to resemble those in the c3_PALA or RPALA structures. As yet a X-ray structure of c3 bound to CP has not been reported. This proposal predicts that the ATCase c3 displays the same activity before and after Asp binds, resulting in no Asp cooperativity and hyperbolic kinetics as is observed experimentally. However, when the binding of CP is impinged, such as in the R105A c3, CP binds incorrectly and it is not until Asp binds, that the active site is transformed into the R-like conformation comparable to the c3_PALA active sites. Therefore the homotropic cooperativity of the R105A c3 is actually a result of poor substrate binding.
The C47A/A241C c6 ATCase is the first catalytic species to exhibit homotropic cooperativity without active-site mutations that drastically reduce the affinity for substrates. The C47A and A241C mutations are not responsible for the observed cooperativity because the C47A/A241C c3 subunit does not display cooperativity for aspartate. In fact, the disulfide-linked C47A/A241C holoenzyme is locked in the R-quaternary state, does not display cooperativity for aspartate, and is not activated by PALA. The dissociation of the regulatory subunits from the C47A/A241C holoenzyme results in a c6 catalytic unit shown to be in the R-quaternary structure by X-ray crystallography, displays cooperativity for Asp with a Hill coefficient of 1.7, and shows activation by PALA. Homotropic cooperativity arises when the binding of substrates to one active site of a multimeric enzyme influences the binding affinity of substates to the other active sites. To display cooperativity, the active sites of C47A/A241C c6 must exist in more than one conformation, corresponding to lower and higher affinity and/or activity conformations, dependent upon ligand binding. The c6_Pi structure is essentially identical to the RPALA structure, suggesting that the binding of Pi is sufficient to induce the change to the higher-affinity conformation, unlike the wild-type holoenzyme that remains in the low-affinity T state when bound to product molecules (56). By covalently linking the two catalytic trimers through specific disulfide bonds in the C47A/A241C c6 enzyme, the binding of Asp to the c6•CP complex induces the conformational change to the higher-affinity active site and this transition is transmitted through the molecule to the other active sites, increasing the affinity of the other active sites for Asp and resulting in homotropic cooperativity.
Acknowledgments
Data for the crystals with Pi bound were measured at Beamline X29 of the National Synchrotron Light Source. We thank Howard Robinson of Brookhaven National Laboratory for data collection and assistance with data processing.
Abbreviations
- ATCase
aspartate transcarbamoylase (EC 2.1.3.2, L-aspartate carbamoyltransferase)
- CP
carbamoyl phosphate
- PALA
N-(phosphonacetyl)-L-aspartate
- PAM
phosphonacetamide
- pHMB
p-hydroxymercury benzoic acid
- C47A/A241C holoenzyme
the mutant ATCase in which Cys47 was replaced by Ala and Ala241 was replaced by Cys
- c3
one catalytic subunit composed of three ATCase catalytic chains
- r2
one regulatory subunit composed of two ATCase regulatory chains
- c6
two catalytic chains of the C47A/A241C ATCase containing three disulfide linkages between Cys241 residues linking the upper and lower catalytic subunits
- nH
the Hill coefficient
- 80’s loop
a loop in the catalytic chain of ATCase comprised of residues 73-88
- 240’s loop
a loop in the catalytic chain of ATCase comprised of residues 230-245
- RS-S_Pi
the X-ray structure of the disulfide-linked C47A/A241C holoenzyme in the presence of Pi (PDB code 3PMU)
- RPALA
the X-ray structure of wild-type ATCase in the presence of PALA
- R236_PAM
the X-ray structure of D236A ATCase in the presence of PAM (PDB code 2A0F)
- c3_Apo
the X-ray structure of the unliganded wild-type c3 ATCase (PDB code 3CSU)
- c3_PALA
the X-ray structure of the PALA-liganded wild-type c3 ATCase (PDB code 1EKX)
- TCTP
the X-ray structure of wild-type ATCase in the T state the presence of CTP (PDB code 1ZA1)
Footnotes
This work was supported by National Institutes of Health (GM26237). Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886
X-ray coordinates have been deposited in the Protein Data Bank (3NPM).
References
- 1.Gerhart JC, Pardee AB. Enzymology of control by feedback inhibition. J Biol Chem. 1962;237:891–896. [PubMed] [Google Scholar]
- 2.Wild JR, Loughrey-Chen SJ, Corder TS. In the presence of CTP, UTP becomes an allosteric inhibitor of aspartate transcarbamylase. Proc Natl Acad Sci U S A. 1989;86:46–50. doi: 10.1073/pnas.86.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerhart JC, Pardee AB. The effect of the feedback Inhibitor CTP, on subunit interactions in aspartate transcarbamylase. Cold Spring Harbor Symp Quant Biol. 1963;28:491–496. [Google Scholar]
- 4.Gouaux JE, Stevens RC, Lipscomb WN. Crystal structures of aspartate carbamoyltransferase ligated with phosphonoacetamide, malonate and CTP or ATP at 2.8 Å resolution and neutral pH. Biochemistry. 1990;29:7702–7715. doi: 10.1021/bi00485a020. [DOI] [PubMed] [Google Scholar]
- 5.Honzatko RB, Lipscomb WN. Interactions of phosphate ligands with Escherichia coli aspartate carbamoyltransferase in the crystalline state. J Mol Biol. 1982;160:265–286. doi: 10.1016/0022-2836(82)90176-0. [DOI] [PubMed] [Google Scholar]
- 6.Ladjimi MM, Ghellis C, Feller A, Cunin R, Glansdorff N, Pierard A, Hervé G. Structure-function relationship in allosteric aspartate carbamoyltransferase from Escherichia coli: II. Involvement of the C-terminal region of the regulatory chain in homotropic and heterotropic interactions. J Mol Biol. 1985;186:715–724. doi: 10.1016/0022-2836(85)90391-2. [DOI] [PubMed] [Google Scholar]
- 7.Kantrowitz ER, Lipscomb WN. Escherichia coli aspartate transcarbamoylase: The molecular basis for a concerted allosteric transition. Trends Biochem Sci. 1990;15:53–59. doi: 10.1016/0968-0004(90)90176-c. [DOI] [PubMed] [Google Scholar]
- 8.Gerhart JC, Schachman HK. Allosteric interactions in aspartate transcarbamylase II. Evidence for different conformational states of the protein in the presence and absence of specific ligands. Biochemistry. 1968;7:538–552. doi: 10.1021/bi00842a600. [DOI] [PubMed] [Google Scholar]
- 9.Howlett GJ, Blackburn MN, Compton JG, Schachman HK. Allosteric regulation of aspartate transcarbamoylase. Analysis of the structural and functional behavior in terms of a two-state model. Biochemistry. 1977;16:5091–5099. doi: 10.1021/bi00642a023. [DOI] [PubMed] [Google Scholar]
- 10.Kantrowitz ER, Lipscomb WN. Escherichia coli aspartate transcarbamylase: The relations between structure and function. Science. 1988;241:669–674. doi: 10.1126/science.3041592. [DOI] [PubMed] [Google Scholar]
- 11.Ladjimi MM, Kantrowitz ER. A possible model for the concerted allosteric transition in Escherichia coli aspartate transcarbamylase as deduced from site-directed mutagenesis studies. Biochemistry. 1988;27:276–283. doi: 10.1021/bi00401a042. [DOI] [PubMed] [Google Scholar]
- 12.Yang YR, Kirschner MW, Schachman HK. Aspartate transcarbamoylase (Escherichia coli): preparation of subunits. Methods Enzymol. 1978;51:35–41. doi: 10.1016/s0076-6879(78)51007-0. [DOI] [PubMed] [Google Scholar]
- 13.Yang YR, Syvanen JM, Nagel GM, Schachman HK. Aspartate transcarbamoylase molecules lacking one regulatory subunit. Proc Natl Acad Sci U S A. 1974;71:918–922. doi: 10.1073/pnas.71.3.918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerhart JC, Schachman HK. Distinct subunits for the regulation and catalytic activity of aspartate transcarbamylase. Biochemistry. 1965;4:1054–1062. doi: 10.1021/bi00882a012. [DOI] [PubMed] [Google Scholar]
- 15.Subramani S, Schachman HK. The mechanism of dissociation of aspartate transcarbamoylase by p-mercuribenzoate. J Biol Chem. 1981;256:1255–1262. [PubMed] [Google Scholar]
- 16.Gerhart JC, Holoubek H. The purification of aspartate transcarbamylase of Escherichia coli and separation of its protein subunits. J Biol Chem. 1967;242:2886–2892. [PubMed] [Google Scholar]
- 17.Stebbins JW, Xu W, Kantrowitz ER. Three residues involved in binding and catalysis in the carbamyl phosphate binding site of Escherichia coli aspartate transcarbamylase. Biochemistry. 1989;28:2592–2600. doi: 10.1021/bi00432a037. [DOI] [PubMed] [Google Scholar]
- 18.Griffin JH, Rosenbusch JP, Blout ER, Weber K. Conformational changes in aspartate transcarbamylase: II. Circular dichroism evidence for the involvement of metal ions in allosteric interactions. J Biol Chem. 1973;248:5057–5062. [PubMed] [Google Scholar]
- 19.Mort JS, Chan WWC. Subunit interactions in aspartate transcarbamylase. Characterization of a complex between the catalytic and the regulatory subunits. J Biol Chem. 1975;250:653–660. [PubMed] [Google Scholar]
- 20.Blackburn MN, Schachman HK. Alteration of the allosteric properties of aspartate transcarbamoylase by pyridoxylation of the catalytic and regulatory subunits. Biochemistry. 1976;15:1316–1323. doi: 10.1021/bi00651a023. [DOI] [PubMed] [Google Scholar]
- 21.Markby DW, Zhou BB, Schachman HK. A 70-amino acid zinc-binding polypeptide from the regulatory chain of aspartate transcarbamoylase forms a stable complex with the catalytic subunit leading to markedly altered enzyme activity. Proc Natl Acad Sci U S A. 1991;88:10568–10572. doi: 10.1073/pnas.88.23.10568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peterson CB, Zhou BB, Hsieh D, Creager AN, Schachman HK. Association of the catalytic subunit of aspartate transcarbamoylase. Protein Sci. 1994;3:960–966. doi: 10.1002/pro.5560030611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou BB, Waldrop GL, Lum L, Schachman HK. A 70-amino acid zinc-binding polypeptide fragment from the regulatory chain of aspartate transcarbamoylase causes marked changes in the kinetic mechanism of the catalytic trimer. Protein Sci. 1994;3:967–974. doi: 10.1002/pro.5560030612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evans DR, Pastra-Landis SC, Lipscomb WN. An intermediate complex in the dissociation of aspartate transcarbamylase. Proc Natl Acad Sci U S A. 1974;71:1351–1355. doi: 10.1073/pnas.71.4.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evans DR, Pastra-Landis SC, Lipscomb WN. Isolation and properties of a species produced by the partial dissociation of aspartate transcarbamylase from Escherichia coli. J Biol Chem. 1975;250:3571–3583. [PubMed] [Google Scholar]
- 26.Subramani S, Schachman HK. Mechanism of disproportionation of asparate transcarbamoylase molecules lacking one regulatory subunit. J Biol Chem. 1980;255:8136–8143. [PubMed] [Google Scholar]
- 27.Enns CA, Chan WC. Conformational states of aspartate transcarbamylase stabilized with a cross linking reagent. J Biol Chem. 1979;254:6180–6186. [PubMed] [Google Scholar]
- 28.Enns CA, Chan WC. Stabilization of the relaxed state of aspartate transcarbamoylase by modification with a bifunctional reagent. J Biol Chem. 1978;253:2511–2513. [PubMed] [Google Scholar]
- 29.West JM, Tsuruta H, Kantrowitz ER. Stabilization of the R allosteric structure of E. coli aspartate transcarbamoylase by disulfide bond formation. J Biol Chem. 2002;277:47300–47304. doi: 10.1074/jbc.M209913200. [DOI] [PubMed] [Google Scholar]
- 30.Nowlan SF, Kantrowitz ER. Superproduction and rapid purification of E. coli aspartate transcarbamoylase and its catalytic subunit under extreme derepression of the pyrimidine pathway. J Biol Chem. 1985;260:14712–14716. [PubMed] [Google Scholar]
- 31.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 32.Davis BJ. Disc electrophoresis-II Method and application to human serum proteins. Ann NY Acad Sci. 1964;121:404–427. doi: 10.1111/j.1749-6632.1964.tb14213.x. [DOI] [PubMed] [Google Scholar]
- 33.Ornstein L. Disc electrophoresis. I. Background and theory. Ann NY Acad Sci. 1964;121:321–349. doi: 10.1111/j.1749-6632.1964.tb14207.x. [DOI] [PubMed] [Google Scholar]
- 34.Kantrowitz ER, Lipscomb WN. An essential arginine residue at the active site of aspartate transcarbamylase. J Biol Chem. 1976;251:2688–2695. [PubMed] [Google Scholar]
- 35.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 36.Pastra-Landis SC, Foote J, Kantrowitz ER. An improved colorimetric assay for aspartate and ornithine transcarbamylases. Anal Biochem. 1981;118:358–363. doi: 10.1016/0003-2697(81)90594-7. [DOI] [PubMed] [Google Scholar]
- 37.Silver RS, Daigneault JP, Teague PD, Kantrowitz ER. Analysis of two purified mutants of Escherichia coli aspartate transcarbamylase with single amino acid substitutions. J Mol Biol. 1983;168:729–745. doi: 10.1016/s0022-2836(83)80072-2. [DOI] [PubMed] [Google Scholar]
- 38.Pastra-Landis SC, Evans DR, Lipscomb WN. The effect of pH on the cooperative behavior of aspartate transcarbamylase from Escherichia coli. J Biol Chem. 1978;253:4624–4630. [PubMed] [Google Scholar]
- 39.Luft JR, Collins RJ, Fehrman NA, Lauricella AM, Veatch CK, DeTitta GT. A deliberate approach to screening for initial crystallization conditions of biological macromolecules. J Struct Biol. 2003;142:170–179. doi: 10.1016/s1047-8477(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 40.Jin L, Stec B, Lipscomb WN, Kantrowitz ER. Insights into the mechanism of catalysis and heterotropic regulation of E. coli aspartate transcarbamoylase based upon a structure of enzyme complexed with the bisubstrate analog N-phosphonacetyl-L-aspartate at 2.1 Å. Proteins: Struct Funct Genet. 1999;37:729–742. [PubMed] [Google Scholar]
- 41.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and Development of Coot. Acta Cryst. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 45.Chan WWC, Mort JS. A complex between the catalytic and regulatory subunits of aspartate transcarbamylase. J Biol Chem. 1973;248:7614–7616. [PubMed] [Google Scholar]
- 46.Endrenyi L, Chan MS, Wong JT. Interpretation of nonhyperbolic behavior in enzymic systems. II. Quantitative characteristics of rate and binding functions. Can J Biochem. 1971;49:581–598. doi: 10.1139/o71-085. [DOI] [PubMed] [Google Scholar]
- 47.Markus G, McClintock D, Bussel J. Conformational changes in aspartate transcarbamylase. 3 A functional model for allosteric behavior. J Biol Chem. 1971;246:762–771. [PubMed] [Google Scholar]
- 48.Kosman RP, Gouaux JE, Lipscomb WN. Crystal structure of CTP-ligated T state aspartate transcarbamoylase at 2.5 Å resolution: Implications for aspartate transcarbamoylase mutants and the mechanism of negative cooperativity. Proteins: Struct Funct Genet. 1993;15:147–176. doi: 10.1002/prot.340150206. [DOI] [PubMed] [Google Scholar]
- 49.Stieglitz K, Stec B, Baker DP, Kantrowitz ER. Monitoring the transition from the T to the R state in E. coli aspartate transcarbamoylase by X-ray crystallography: Crystal structures of the E50A mutant in four distinct allosteric states. J Mol Biol. 2004;341:853–868. doi: 10.1016/j.jmb.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 50.Mendes KR, Kantrowitz ER. The pathway of product release from the R state of aspartate transcarbamoylase. J Mol Biol. 2010 doi: 10.1016/j.jmb.2010.07.003. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Macol CP, Tsuruta H, Stec B, Kantrowitz ER. Direct structural evidence for a concerted allosteric transition in Escherichia coli aspartate transcarbamoylase. Nat Struc Biol. 2001;8:423–426. doi: 10.1038/87582. [DOI] [PubMed] [Google Scholar]
- 52.West JM, Tsuruta H, Kantrowitz ER. A fluorescent probe-labeled aspartate transcarbamoylase that monitors the allosteric conformational state. J Biol Chem. 2004;279:945–951. doi: 10.1074/jbc.M304018200. [DOI] [PubMed] [Google Scholar]
- 53.Beernink PT, Endrizzi JA, Alber T, Schachman HK. Assessment of the allosteric mechanism of aspartate transcarbamoylase based on the crystalline structure of the unregulated catalytic subunit. Proc Natl Acad Sci U S A. 1999;96:5388–5393. doi: 10.1073/pnas.96.10.5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Endrizzi JA, Beernink PT, Alber T, Schachman HK. Binding of bisubstrate analog promotes large structural changes in the unregulated catalytic trimer of aspartate transcarbamoylase: implications for allosteric regulation. Proc Natl Acad Sci U S A. 2000;97:5077–5082. doi: 10.1073/pnas.090087197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang J, Stieglitz KA, Cardia JP, Kantrowitz ER. Structural basis for ordered substrate binding and cooperativity in aspartate transcarbamoylase. Proc Natl Acad Sci U S A. 2005;102:8881–8886. doi: 10.1073/pnas.0503742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang J, Lipscomb WN. Products in the T-state of aspartate transcarbamylase: crystal structure of the phosphate and N-carbamyl-L-aspartate ligated enzyme. Biochemistry. 2004;43:6422–6426. doi: 10.1021/bi0302144. [DOI] [PubMed] [Google Scholar]