

Abstract
ETS proteins are a family of transcription factors that play important roles in the development of cancer. The Ewing’s sarcoma EWS/ETS fusion oncoproteins control a number of cancer-relevant phenotypes in that disease. We recently demonstrated that EWS/FLI, the most common EWS/ETS fusion in Ewing’s sarcoma, regulates a portion of its target genes, including the critical target NR0B1, via GGAA-containing microsatellites in their promoters. Given the unusual nature of microsatellites as EWS/FLI response elements, we sought to elucidate the mechanism of EWS/FLI activity at these sites. We found that the ability to bind GGAA microsatellites is shared by multiple ETS family members from distinct phylogenetic subfamilies. Importantly, however, only EWS/ETS-containing fusions are capable of mediating transcriptional activation via these elements, highlighting a neomorphic function of the Ewing’s sarcoma fusion proteins. Additional analysis revealed that the GGAA microsatellite binds EWS/FLI with an affinity that is 2 to 3 orders of magnitude lower than previously identified high-affinity consensus/redundant binding sites. The stoichiometry of this interaction is 2 protein molecules for each DNA molecule, suggesting that EWS/FLI binds these elements as a homodimer. The isolated FLI ETS domain bound microsatellite sequences in a nearly identical fashion to full-length EWS/FLI, thus indicating that residues required for homodimeric binding are localized to the ETS domain. These data suggest a new paradigm for an ETS family member binding to DNA at cancer-relevant genetic loci and highlight emergent properties of EWS/FLI that are required for the development of Ewing’s sarcoma.
Keywords: EWS/FLI, ETS, Ewing’s sarcoma, microsatellites
Introduction
ETS proteins are a family of transcription factors that play important roles in the development of cancer. The ETS family is defined by the presence of a highly conserved ETS-type DNA-binding domain.1 ETS domains generally bind DNA at 9- to 10-bp sequences that contain a “GGAA/T core” motif surrounded by bases that dictate the affinity of the interaction.1-3 In vitro binding site selection studies have identified ACCGGAAGT as a high-affinity site that binds many different ETS family members as monomers.1-4 The ability of different family members to bind to the same sequence in vitro highlights an important issue in understanding ETS protein function: how do different ETS proteins regulate different genes?
Genome-wide binding site identification using chromatin immunoprecipitation (ChIP) recently provided one answer to this question. Hollenhorst et al. found 2 distinct classes of ETS target binding sequences: high-affinity/redundant sites and divergent/specific sites.5 The in vivo high-affinity sites identified were nearly identical to the high-affinity site identified through in vitro selection and were bound by multiple different ETS family members in vivo. These sites likely allow adjacent genes to be regulated by any one of a number of different ETS family members. In contrast to these redundant sites, divergent sites consist of partial ETS binding sites in immediate proximity to (or overlapping with) partial binding sites for non-ETS family members. Divergent sites may only allow for gene regulation when both transcription factors are present and bind DNA as heterodimers.
Ewing’s sarcoma serves as an excellent model in which to study gene regulation by ETS family members. Approximately 85% of Ewing’s sarcoma tumors contain a recurrent chromosomal translocation, t(11;22)(q24;q12), that encodes EWS/FLI.6,7 The remaining cases contain other translocations that encode similar fusion proteins (EWS/ERG, EWS/FEV, EWS/ETV1, and EWS/ETV4), collectively referred to here as EWS/ETS fusions.8-12 EWS/ETS proteins are capable of inducing the oncogenic transformation of NIH3T3 cells, while wild-type EWS or wild-type FLI cannot.13-15 Furthermore, ongoing EWS/FLI expression is required for the transformed phenotype of Ewing’s sarcoma cells.16,17 Thus, EWS/FLI is a key oncoprotein in Ewing’s sarcoma, and understanding the mechanistic basis for its activity should provide insights into ETS protein function in the context of tumorigenesis.
EWS is an RNA-binding protein of uncertain normal function, while FLI is an ETS family member normally involved in hematopoietic development.18,19 Wild-type FLI contains a weak transcriptional activation domain in its amino terminus and an ETS-type DNA-binding domain and a second transcriptional activation domain in its carboxyl terminus.20,21 As a consequence of the (11;22) translocation, the amino terminal transcriptional activation domain of FLI is replaced by the amino terminus of EWS.6 Because the EWS portion of the fusion functions as a strong transcriptional activation domain when fused to heterologous DNA-binding domains, one model for EWS/FLI function suggests that the fusion protein acts as a much stronger transcriptional activator than wild-type FLI.20,22
We recently identified unusual EWS/FLI response elements in the promoters of genes involved in Ewing’s sarcoma development, such as NR0B1 and GSTM4: GGAA-containing microsatellites.23-27 GGAA microsatellites contain multiple consecutive copies of the GGAA core motif, with occasional single base changes, insertions, or deletions. These elements are unusual as they do not conform to the previously described “high-affinity” or “divergent” ETS binding sites, and they demonstrate a unique length dependence to their function.23,24,28,29 For example, DNA binding and transcriptional activity of EWS/FLI was not observed with 1, 2, or 3 consecutive GGAA motifs but only became apparent when 4 or more consecutive motifs were present.24 Furthermore, as the number of GGAA motifs increased, transcriptional activity also increased, both in model reporter assays and at bona fide target genes.24,28,29 These findings suggest that EWS/FLI may interact with GGAA microsatellites in a manner that is mechanistically distinct from other previously described ETS response elements. We therefore sought to understand the mechanistic basis for EWS/FLI transcriptional activity via GGAA microsatellites to understand the basis of oncogenesis mediated by these elements.
Results
The ETS family includes 27 members in humans that can be phylogenetically grouped based on the sequence of their ETS DNA-binding domains.30 Interestingly, the ETS portion of the 5 known EWS/ETS fusion proteins belongs to only 2 subfamilies, the ERG family (including FLI, ERG, and FEV) and the PEA3 family (including ETV1 and ETV4). To determine if transcriptional activity mediated through GGAA microsatellites is specific for EWS/FLI, or is a more general property of all EWS/ETS fusions, we performed reporter assays with a luciferase construct that contained the NR0B1 microsatellite and a minimal SV40-based promoter.24 As previously demonstrated, EWS/FLI induced robust luciferase activity (Fig. 1A).24 Similarly, expression of any of the alternate EWS/ETS fusions (EWS/ERG, EWS/FEV, EWS/ETV1, or EWS/ETV4) induced strong luciferase activity (Fig. 1A). Thus, transcriptional activity through GGAA microsatellites is a general property of all known EWS/ETS fusions.
Figure 1.
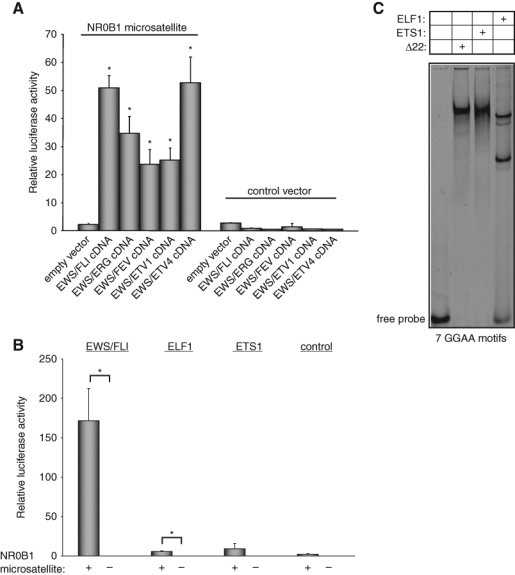
The GGAA microsatellite in the NR0B1 promoter is responsive to all EWS/ETS fusions but not to ETS1 and ELF1. Luciferase assay in HEK 293EBNA cells cotransfected with the 102-bp NR0B1 microsatellite luciferase reporter vector (or control vector that did not contain the NR0B1 microsatellite) and (A) the indicated EWS/ETS (or empty control) cDNA expression vectors or (B) ELF1 and ETS1 cDNA expression vectors. All proteins were expressed at equal levels (data not shown). Error bars indicate standard deviations; asterisks indicate P < 0.05. (C) EMSA with radiolabeled probe harboring 7 consecutive GGAA motifs and recombinant Δ22, ETS1, or ELF1 proteins. The upper band in the ELF1 lane is a nonspecific band as determined by competition with a high-affinity site containing DNA duplex(I) (data not shown).
We next asked whether transcriptional activation through GGAA microsatellites is a property shared by ETS family members not involved in Ewing’s sarcoma. We tested ETS proteins from 2 additional subfamilies, the ELF and ETS families, in the same luciferase reporter assay. We found that neither ELF1 nor ETS1 were capable of stimulating high-level transcriptional activity through the microsatellite (Fig. 1B). To determine whether the failure of transcriptional activity was due to the lack of ability of these proteins to bind the GGAA microsatellite, we performed electrophoretic mobility shift assays (EMSAs) using recombinant ELF1 and ETS1 proteins. Interestingly, both proteins bound a microsatellite-derived DNA duplex harboring 7 GGAA motifs in a manner similar to that observed for the EWS/FLI-derived protein Δ22 (Δ22 is described in more detail below) (Figs. 1C, 2A). We previously found that wild-type FLI bound the GGAA microsatellite in EMSAs but also was unable to transcriptionally activate through these elements (Fig. 2 B and D).24 Taken together, these data demonstrate that although a wide variety of ETS family members from different phylogenetic subfamilies share the ability to bind GGAA microsatellites, transcriptional activation using microsatellites is an emergent property of EWS/ETS fusion proteins.
Figure 2.
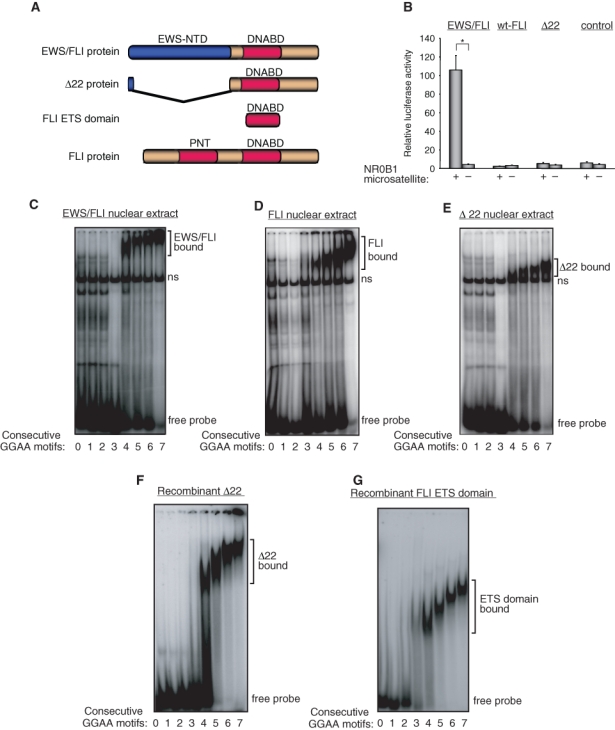
EWS portion of EWS/FLI is necessary for transcriptional activity but is dispensable for binding to GGAA repeats. (A) Schematic representation of EWS/FLI, Δ22, FLI ETS domain, and wild-type FLI proteins. EWS-NTD indicates the EWS amino terminal domain, DNABD indicates the FLI ETS DNA-binding domain, and PNT indicates the FLI pointed domain. (B) Luciferase assay in HEK 293EBNA cells cotransfected with the 102-bp NR0B1 microsatellite luciferase reporter vector and either EWS/FLI, wild-type FLI (wt-FLI), Δ22, or empty control cDNA expression vectors. Error bars indicate standard deviations; asterisks indicate P < 0.05. (C-G) EMSA with DNA duplexes containing the indicated number of consecutive GGAA motifs and EWS/FLI nuclear extract (C), wild-type FLI nuclear extract (D), Δ22 nuclear extract (E), recombinant Δ22 protein (F), and recombinant FLI ETS domain protein (G). The positions of specific protein-bound complexes and unbound “free” probe are indicated. A nonspecific band present in the nuclear extracts is indicated by “ns.”
EWS/FLI has 2 primary differences to wild-type FLI: the loss of the amino terminus of FLI and the gain of the EWS amino terminus (Fig. 2A). To determine the contribution of each of these differences to transcriptional activity, we tested a previously described mutant protein, called Δ22, which contains only the portion of FLI that is retained in the EWS/FLI fusion (Fig. 2A).14 We found that Δ22 was inactive in luciferase reporter assays (Fig. 2B) but that Δ22 from nuclear extracts, or purified recombinant Δ22 protein, bound microsatellite sequences in a pattern that was identical to that of EWS/FLI and wild-type FLI (Fig. 2 C-F) (Supp. Fig. S1 A and B).24 This result indicated that the EWS portion of the fusion is absolutely required for transcriptional activity but does not contribute to its DNA-binding function.
To define the molecular requirements for DNA binding, we next asked whether the isolated FLI ETS domain (Fig. 2A) was sufficient to bind the GGAA microsatellite or whether sequences outside the ETS domain were also required. We purified recombinant FLI ETS domain and tested it for microsatellite binding by EMSA. We found that the isolated FLI ETS domain efficiently bound GGAA-repetitive elements in a manner that was similar to full-length EWS/FLI, wild-type FLI, and Δ22 (Fig. 2G) (Supp. Fig. S1C).24
We previously proposed a series of hypotheses to explain the characteristics of EWS/FLI binding to GGAA microsatellites, including the slower mobility observed for EWS/FLI-bound bands in EMSA experiments with increasing numbers of consecutive GGAA motifs.23 It is known that ETS family members induce a bend in bound target DNA and that the inherent ability of DNA to adopt a bent conformation may increase the affinity of ETS family members for that target DNA.31,32 One hypothesis was that microsatellite sequences with 4 or more consecutive GGAA motifs might adopt a bent conformation that would be energetically favorable for EWS/FLI binding. We tested this hypothesis by designing multiple synthetic 36-bp DNA probes containing 4 consecutive GGAA motifs. The consecutive GGAA motifs were located in different positions in each probe, starting close to the 5′ end of the top strand and shifting by 2 bp in the 3′ direction with each probe (Supplementary Table S1). If a bend were present at baseline, or induced by Δ22 binding, we would anticipate an altered mobility in EMSAs based on the location of the GGAA motifs (i.e., a bend close to the end of the probe would have a faster mobility, while a bend close to the middle of the probe would have a slower mobility). We found that none of the probes demonstrated a bent conformation, either in the absence or presence of Δ22 (Supp. Fig. S2). Similar results were obtained using the pBend vector (data not shown).33 Thus, significant DNA conformational effects mediated by GGAA microsatellites are unlikely to explain their ability to bind ETS family members.
A second hypothesis we proposed was that EWS/FLI might bind to GGAA microsatellites as homodimers.23 In this hypothesis, the length dependence of DNA binding and associated transcriptional activity would be related to the ability of multiple EWS/FLI molecules to bind simultaneously to the DNA target and thus would be dependent on avoiding steric hindrance between adjacent bound protein molecules. To test this hypothesis, we evaluated the molecular characteristics of EWS/FLI-derivative binding to GGAA-repetitive sequences. We used recombinant Δ22 protein because this derivative could be readily purified and it bound DNA with a pattern that was nearly identical to that of EWS/FLI (Fig. 2).24 In contrast, we were unsuccessful at preparing highly purified active preparations of recombinant full-length EWS/FLI, presumably because its disordered characteristics precluded efficient refolding of the recombinant protein.34
The binding of ETS family members to high-affinity DNA sites produces a very characteristic DNAse I footprint in which approximately 14 bp of DNA are protected from digestion, and a highly reproducible DNAse I hypersensitive site appears on the negative strand adjacent to the GGAA (TTCC on the negative strand) core motif.35 Purified recombinant Δ22 protein produced an identical DNAse I footprint pattern on the high-affinity ACCGGAAGT site (Fig. 3A). However, when GGAA-repetitive elements of increasing lengths were analyzed, the pattern was somewhat different (Fig. 3B). For example, on a DNA fragment containing 4 consecutive GGAA motifs, the Δ22 protein protected 28 bp, which is approximately twice the length protected on the high-affinity site. Additionally, there was no evidence of a DNAse I hypersensitive site on the negative strand. As the number of GGAA motifs was increased, the length of the protected region increased by 4 bases for each additional motif included. These data suggested 1 of 2 possible interpretations. First, 2 or more Δ22 monomers bind DNA simultaneously. Second, Δ22 monomers do not have a preferential single binding site on a GGAA-repetitive region but rather occupy multiple positions across the microsatellite.
Figure 3.
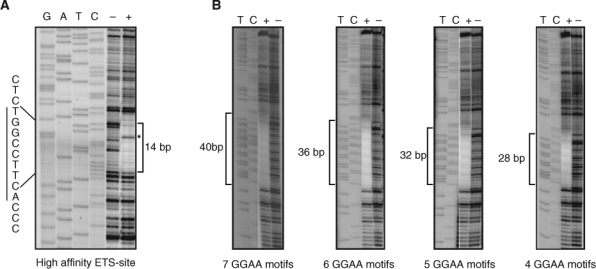
DNAse I footprinting assays reveal differences between high-affinity and GGAA-repetitive DNA sequences. (A) The positive control DNA duplex (I) with a single high-affinity ETS binding site shows an approximately 14-bp footprint (marked by a bracket) in the presence of Δ22 protein (marked by “+”) as compared to in the absence of protein (marked by “–”). The characteristic hypersensitive site is marked by an asterisk. Sequencing lanes are indicated by “GATC.” (B) DNA containing the indicated number of consecutive GGAA motifs demonstrates larger protected regions, as indicated by brackets. The DNA probes contained the following protected sequences (labeled strand indicated): AGCT(TTCC)4-7TGCAGCCC.
To distinguish between these interpretations, we evaluated the stoichiometry of EWS/FLI-derived protein binding to GGAA motif containing DNA using an EMSA approach. Both recombinant Δ22 and recombinant FLI ETS domain bind DNA with similar characteristics but with different electrophoretic mobilities due to their different sizes (Figs. 2, 4A). When both proteins are mixed together and then bound to DNA duplex (I) probe containing a single high-affinity site,24 2 separate DNA-bound complexes are resolved, one containing Δ22 and one containing the FLI ETS domain (Fig. 4A). In contrast, when these proteins are mixed and bound to probes containing 4, 5, 6, or 7 consecutive GGAA motifs, an intermediate mobility band appears, consistent with the presence of both proteins binding the same target DNA simultaneously (Fig. 4A). Interestingly, heterodimers were preferentially bound over homodimers, as demonstrated by the lack of homodimer bands for either of the 2 proteins. The intermediate mobility band was further confirmed by performing EMSAs with a fixed amount of Δ22 protein in the presence of increasing concentrations of the FLI ETS domain protein (Fig. 4B). In this instance, in addition to the preferential appearance of the heterodimeric band, a small amount of homodimeric FLI ETS domain was also noted, suggesting that the FLI ETS domain may have a higher affinity for GGAA microsatellite DNA than Δ22 protein. This possibility was further supported by titration experiments in which the amount of either Δ22 or FLI ETS domain protein used in EMSA experiments was varied (Supp. Fig. S3). In these experiments, the FLI ETS domain was found to induce electrophoretic mobility shifts at lower protein concentrations than those found for Δ22 protein. The appearance of the intermediate mobility band in mixing experiments was dependent on the intact DNA-binding function of each partner, as no such heterodimeric complex was observed when the same experiment was performed using Δ22 protein in conjunction with a mutant FLI ETS domain that could not bind DNA (R2L2 mutant) (data not shown).36,37 These data suggest that 2 protein molecules bind and interact with each DNA molecule. Furthermore, these data suggest that the slower mobility noted with increasing numbers of consecutive GGAA motifs in EMSA experiments is not due to greater numbers of bound EWS/FLI molecules.
Figure 4.
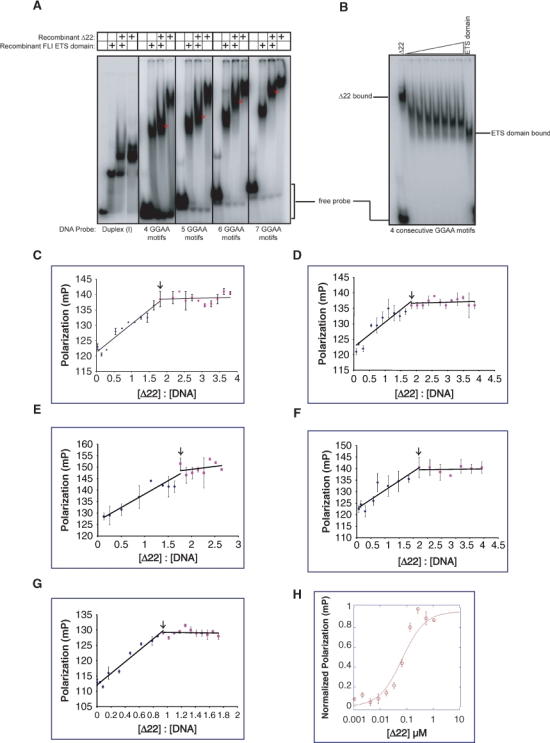
EWS/FLI derivatives bind GGAA-repetitive DNA in a dimeric fashion. (A) EMSA with DNA duplexes containing either the high affinity ETS binding site [DNA duplex (I)] or the indicated number of consecutive GGAA motifs, with either recombinant Δ22 and/or recombinant FLI ETS domain proteins. The positions of intermediate mobility dual-protein bound bands are denoted by an asterisk in each panel. Sequences of the DNA probes used are provided in the Materials and Methods section. (B) EMSA with probe containing 4 consecutive GGAA motifs, a fixed amount of recombinant Δ22 protein, and increasing amounts of recombinant FLI ETS domain protein. (C-F) Fluorescence polarization results of recombinant Δ22 protein binding to a fluorescein-labeled DNA probe containing 4, 5, 6, or 7 consecutive GGAA motifs (C, D, E, and F, respectively), plotted as change in polarization versus molar ratio of protein:DNA. The inflection point at a ratio of approximately 2 is indicated by an arrow. Error bars indicate standard error of means for 2 independent experiments. (G) Fluorescence polarization results of recombinant Δ22 protein binding to a fluorescein-labeled DNA probe containing a single high-affinity ETS binding site [DNA duplex (I)]. The inflection point at a ratio of approximately 1 is indicated by an arrow. Error bars indicate standard error of means for 2 independent experiments. (H) Fluorescence polarization results of recombinant Δ22 protein bound to a fluorescein-labeled oligonucleotide probe harboring 4 consecutive GGAA motifs. The Kd was determined to be approximately 70 nM. Error bars indicate standard error of the means of 3 independent experiments.
To evaluate the stoichiometry of binding using an independent approach, we performed fluorescence polarization assays. A DNA probe containing 4 consecutive GGAA motifs was fluorescein labeled and used in binding reactions with recombinant Δ22 protein. Fluorescence polarization was assessed at protein:DNA molar ratios ranging from 0 to 4. We observed a linear increase in polarization until the protein:DNA ratio was approximately 2 (1.81), and no further increases were observed at higher ratios (Fig. 4C). Evaluation of DNA targets containing 5, 6, or 7 consecutive GGAA motifs also showed inflection points at molar ratios of approximately 2 (Fig. 4 D-F). As expected, DNA containing a single high-affinity ETS binding site showed an inflection point at a molar ratio of approximately 1 (Fig. 4G). These data are consistent with a stoichiometry of 2 protein monomers per DNA target for probes containing 4, 5, 6, or 7 consecutive GGAA motifs.
During the course of these experiments, we noted that the affinity of EWS/FLI-derived proteins appeared to be significantly lower for GGAA microsatellite sequences than for the high-affinity site. To better define this observation, we used fluorescence polarization to determine the Kd of Δ22 protein for a DNA probe containing 4 consecutive GGAA motifs. Using this approach, the Kd was approximately 70 nM (Fig. 4H). This value is approximately 2 to 3 orders of magnitude lower than the 0.01 to 0.3 nM affinity reported for ETS1 derivatives on a similar high-affinity site.38 Experiments designed to determine the affinity of Δ22 on the high-affinity site using fluorescence polarization were unsuccessful because the higher affinity interaction required quantities of DNA that were below the limit of sensitivity of the assay. Limitations in determining affinities of less than 1 nM by fluorescence polarization, due to instrumental constraints, have been previously noted.39
To estimate the relative affinities of EWS/FLI-derived protein for the high-affinity and microsatellite sites, we used competitive EMSA. We first radiolabeled a DNA duplex containing 4 consecutive GGAA motifs and found that the presence of just 10-fold excess unlabeled DNA duplex (I) containing the high-affinity ETS binding sequence was sufficient to effectively compete for Δ22 binding (Supp. Fig. S4). We next radiolabeled duplex (I) and found that even a 9,000-fold excess of unlabeled DNA containing 4 consecutive GGAA motifs was unable to compete for binding Δ22 protein (Supp. Fig. S4). While highly accurate affinities were not determined using this approach, these data indicate that the affinity of EWS/FLI-derived proteins for GGAA-repetitive sequences may be approximately 3 orders of magnitude lower than the affinity for high-affinity sites.
Discussion
The Ewing’s sarcoma–specific EWS/FLI protein (and presumably other EWS/ETS fusions as well) plays a key role in tumorigenesis through its ability to regulate target genes that mediate the transformed phenotype. However, until recently, the mechanisms by which gene-specific regulation occurs have been largely unknown. Recent work has suggested that ETS family members bind target DNA via 1 of 2 mechanisms: by binding as monomers to high-affinity sequences or by cooperative binding as heterodimers with other transcription factors to divergent sites.5 The work presented here now provides a third mechanism as well: by binding as homodimers or higher order oligomers to highly repetitive low-affinity sequences.
We found that DNA binding to microsatellite-derived sequences is a property shared by at least 7 different ETS family members, including those of the ERG, PEA3, ELF, and ETS families. This result is consistent with our additional finding that the EWS portion of EWS/FLI is dispensable for microsatellite binding. Multiple experimental approaches demonstrated that the stoichiometry of the interaction is 2 protein molecules to 1 DNA molecule. The 2:1 protein:DNA ratio was observed for microsatellite sequences that included 4, 5, 6, or 7 consecutive GGAA motifs. Thus, changes in mobility noted in EMSA with increasing numbers of consecutive motifs do not appear to be due to binding of additional EWS/FLI molecules. One possible interpretation of the EMSA data is that different numbers of consecutive GGAA motifs allow for different configurations of EWS/FLI binding, thus leading to differences in mobility by EMSA. For example, 4 consecutive GGAA motifs may allow for binding of 2 EWS/FLI molecules in close proximity to one another, while 5, 6, or 7 consecutive motifs may allow for EWS/FLI to bind DNA with greater spacing between each protein monomer. DNA with greater than 7 consecutive repeats was not tested. We previously demonstrated increased transcriptional activity from the full-length NR0B1 promoter harboring 25 total GGAA repeats (with the greatest stretch consisting of 11 consecutive repeats) as compared with smaller regions containing up to 7 consecutive repeats.23 Based on this finding, we hypothesize that longer repetitive regions might have the ability to bind larger numbers of protein molecules. Such binding could occur via at least 2 distinct configurations. First, EWS/FLI may bind as higher order oligomers in which the number of molecules is directly proportional to the number of consecutive GGAA repeats present on the DNA target (a “head-to-tail” configuration). In this case, even or odd numbers of molecules may bind. Alternately, EWS/FLI may only bind as homodimers. In this latter configuration, additional EWS/FLI molecules might only bind when there is a sufficient length and/or spacing of GGAA repeats to allow additional homodimers to bind. In this latter case, only even numbers of EWS/FLI molecules could bind. Indeed, bound homodimers might not interact explicitly with adjacent homodimers. Additional study will be required to assess EWS/FLI binding to longer repetitive GGAA target sequences.
Our demonstration that the isolated ETS DNA-binding domain from FLI is itself sufficient to mediate binding to microsatellite-derived sequences indicates that residues required for homotypic interactions are present in the ETS domain itself. Such an interpretation is consistent with a prior study that detected homotypic interactions between EWS/FLI molecules that were mediated by the carboxyl terminus of the FLI portion, which included the FLI ETS DNA-binding domain.40 This prior study also demonstrated that oligomerization did not occur upon binding to an EWS/FLI response element in the TGFBR2 promoter. When taken in concert with our current study, these data suggest that homodimeric/oligomeric binding may be highly specific to GGAA microsatellite sequences.
The current study was not designed to provide detailed structural information on how EWS/FLI homotypic interactions facilitate DNA binding to suboptimal/low-affinity sites (such as GGAA microsatellites). We speculate that these interactions might be similar to interactions between ETS1 and PAX5 in which PAX5 residues interact with the ETS domain of ETS1 and induce a conformational change that allows for binding to a divergent DNA target site that includes a suboptimal ETS binding site.41 Protein-protein interactions between the ETS domain and an adjacent N-terminal portion from ETS1 were also noted in studies evaluating ETS binding sites in the stromolysin promoter.42 In this latter study, however, mutant forms of ETS1 that were unable to form homodimers were still capable of binding DNA as monomers. In contrast, in the current study, ETS1 was able to bind GGAA microsatellite DNA (presumably as a dimer) but was unable to transcriptionally activate through this element. These data suggest that transcriptional activity is not simply a function of bringing a transcriptional activation domain to a promoter or enhancer element but is also dependent on the sequence or context of the DNA-binding site.
In support of this notion, we demonstrated that although all ETS family members tested were able to bind GGAA microsatellite sequences, only EWS-containing fusion proteins were able to mediate transcriptional activation through these elements. This indicates that EWS/ETS fusion proteins have emergent properties that are dependent on their EWS domain. Prior work demonstrated that the EWS portion of EWS/FLI functions as a strong activation domain when fused to the GAL4 heterologous DNA-binding domain, while the amino terminus of wild-type FLI (which is lost in the fusion protein) is a relatively weak activation domain.13,22 This suggested that EWS/FLI may be a stronger transcriptional activator than wild-type FLI. This hypothesis was supported by additional experiments that demonstrated that the oncogenic transforming function of FLI-based fusions was dependent on the presence of a strong transcriptional activation domain.22 However, that same report was unable to show a significant difference in transcriptional activity between full-length EWS/FLI and wild-type FLI in reporter assays. One interpretation of these data is that the amino terminal EWS domain functions as a strong transcriptional activation domain in some promoter contexts but not in others. Our data demonstrate that GGAA microsatellites are one such context. Thus, EWS/ETS fusion proteins have emergent properties at GGAA microsatellites but not at other response elements.
These emergent properties have important implications for the development of Ewing’s sarcoma. We have previously shown that GGAA microsatellites serve as EWS/FLI response elements for at least 2 target genes that are important for Ewing’s sarcoma development, NR0B1 and GSTM4.24-26 We also noted that GGAA microsatellites are present in the promoters of a variety of additional EWS/FLI upregulated targets, including CAV1, which is also required for the transformed phenotype of Ewing’s sarcoma.24,43 Thus, EWS/FLI binding and transcriptional activation at these elements is of critical importance to the development of this tumor. Additional work is required to fully understand the mechanistic basis for the emergent properties of EWS/FLI at GGAA microsatellites with a particular focus on the transcriptional activation properties of the fusion protein at these loci. It may be possible to identify approaches to inhibit these emergent properties and, in doing so, to develop new therapeutic approaches for this highly malignant disease.
Materials and Methods
DNA Cloning and Protein Purification
The 3xFLAG-EWS/ETS, 3xFLAG-FLI expression constructs, and the NR0B1 microsatellite reporter construct were described previously.15,24 The 3xFLAG-ETS1 cDNA and the ELF1 cDNAs were cloned in retroviral expression plasmid pQCXIH (Clontech, Mountain View, CA), while 3xFLAG-Δ22 cDNA was cloned in retroviral expression plasmid pMSCV-Hygro (Clontech) using standard molecular biology approaches. The cDNAs expressing 6xHis-Δ22 and 10xHis-ETS domain of FLI (amino acid residues 270-371) were cloned in the bacterial expression vector pET28a (EMD Chemicals, Gibbstown, NJ). These constructs were expressed in BL21 cells and batch purified using Ni-NTA beads (Qiagen, Valencia, CA). Cloning and purification of recombinant ETS1 and ELF1 have been described previously.44,45
Electrophoretic Mobility Shift Assay (EMSA)
Nuclear extracts were prepared from HEK293 cells as described earlier.24 Twenty micrograms of nuclear extract protein were used per 20 µL reaction volume. Recombinant proteins were prepared from bacteria transformed with pET28a (EMD Chemicals) expression plasmids containing the various cDNAs as described above. Reactions contained 0.67 µM of Δ22 or FLI ETS domain proteins, 1.5 µM of ELF1 protein, or 9 µM of ETS1 protein per 20 µL reaction volume. For the gradient assay (Fig. 4B), a fixed amount of 6xHis-Δ22 (2.67 µM) was titrated with 0 to 1.67 µM 10xHis-FLI ETS domain protein. Each reaction contained 5 nM of [32P]-labeled probes containing 0 to 7 consecutive GGAA motifs and 1x Gel Shift Binding Buffer (Promega Corporation, Madison, WI). DNA duplex (I) (containing a high-affinity ETS binding site) was used as control for monomeric protein binding and as specific unlabeled competitor, while DNA duplex (II) (containing a variant ETS motif that does not bind to EWS/FLI) was used as a nonspecific unlabeled competitor for protein binding.24 Sequences of the consecutive GGAA motifs harboring probes used in Figure 2C to G and Supplementary Figures S1 and S3 were described previously.24 For Figures 1C and 4 and Supplementary Figure S4, sequences of probes were as follows:
4 GGAA motifs: 5′-tgcaggaaggaaggaaggaaagct-3′;
5 GGAA motifs: 5′-tgcaggaaggaaggaaggaaggaaagct-3′;
6 GGAA motifs: 5′-tgcaggaaggaaggaaggaaggaaggaaagct-3′;
7 GGAA motifs: 5′-tgcaggaaggaaggaaggaaggaaggaaggaaagct-3′.
Luciferase Assays
HEK 293EBNA cells were cultured as described previously.26 Cells were transfected with firefly luciferase reporter, Renilla luciferase control, and cDNA expression plasmids. Firefly luciferase activity was normalized to Renilla luciferase activity to control for transfection efficiency and is reported in figures as “relative luciferase activity.” Two-tailed Student t tests were used for statistical comparisons.
DNAse I Footprinting
DNA probes containing 4 to 7 GGAA motifs, and the positive control DNA duplex (I), were cloned into pBluescript II KS(+) vector (Stratagene, La Jolla, CA). An approximately 200-bp fragment was PCR amplified using an M13 forward primer and a [32P]-labeled T3 reverse primer. The resulting amplicons were used in 25 µL binding reactions with recombinant Δ22 protein. After being allowed to equilibrate for 30 minutes at room temperature, the reaction mixtures were incubated with DNAse I (1.7 ng/µL in 0.25 mM CaCl2 final concentration) for 1 minute at room temperature. Reactions were stopped by the addition of EDTA and SDS to a final concentration of 15 mM and 1%, respectively. After precipitating the DNA overnight in ethanol, the mixtures were dried, resuspended in formamide-containing dye, boiled for 5 minutes, and then cooled on ice. Samples were applied to a 12% polyacrylamide-sequencing gel and electrophoresed for 4.5 hours at 40 W. The gel was dried for an hour and exposed to a phosporimager screen overnight.
Fluorescence Polarization
Fluorescein-labeled DNA duplexes were gel purified using a nondenaturing 20% polyacrylamide gel. Fluorescence polarization was performed in 96-well format using a Tecan fluorometer (Männedorf, Switzerland) with the same substrates and buffer conditions as used for EMSA except that the probes were fluorescein labeled instead of radiolabeled. Recombinant Δ22 protein concentrations were varied in individual wells and mixed with DNA at a final concentration that was at least 10-fold below the expected Kd. Samples were incubated at room temperature for at least 30 minutes prior to measuring polarization. Polarization values (mP) were measured and plotted as a function of Δ22 protein concentration. Data points were fit using ΔP=((ΔPtotal*( [Δ22]/Kd)) / (1+([Δ22]/Kd))).39 The free and total protein concentrations were assumed to be equal because the DNA concentration is at least 10-fold lower than the Kd. The total fluorescence intensities were conserved across all protein concentrations tested, indicating that the characteristics of the probe were stable across different protein concentrations. The affinity plots and curve fits were performed using the KaleidaGraph program (Synergy Software, Reading, PA).
When using fluorescence polarization to evaluate binding stoichiometry, polarization measurements were performed following the procedure described earlier.47 DNA was mixed in binding buffer solution at a concentration 20-fold above the determined Kd. Protein was added to the DNA solution in 96-well plate format at different final molar ratios and incubated for at least 25 minutes before measuring polarization. Polarization values were then plotted as a function of the concentration ratio of Δ22 protein versus DNA substrate. The Δ22 protein:DNA ratio in which an inflection in the data occurs represents the binding stoichiometry, as this is the point at which DNA has been saturated by protein and polarization values change minimally as protein concentration was increased.
Supplementary Material
Acknowledgments
We thank Michelle Kinsey for critical reading of the article and Barbara Graves, Aykut Üren, Peter Hollenhorst, Charles Meeker, and members of the Lessnick laboratory for helpful discussions and reagents.
Footnotes
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
C.A.E. was supported by the Utah Research Access for Minorities Program (URAMP) at the University of Utah. This work was supported by funds awarded to S.L.L. from the Terri Anna Perine Sarcoma Fund, the Liddy Shriver Sarcoma Initiative, the Sunbeam Foundation, the Huntsman Cancer Institute/Huntsman Cancer Foundation, and the American Cancer Society (RSG0618801MGO). We also acknowledge NIH support to the Huntsman Cancer Institute (P30 CA042014).
Supplementary material for this article is available on the Genes & Cancer Web site at http://ganc.sagepub.com/supplemental.
References
- 1. Seth A, Watson DK. ETS transcription factors and their emerging roles in human cancer. Eur J Cancer 2005;41:2462-78 [DOI] [PubMed] [Google Scholar]
- 2. Sharrocks AD. The ETS-domain transcription factor family. Nat Rev Mol Cell Biol 2001;2:827-37 [DOI] [PubMed] [Google Scholar]
- 3. Nye JA, Petersen JM, Gunther CV, Jonsen MD, Graves BJ. Interaction of murine ets-1 with GGA-binding sites establishes the ETS domain as a new DNA-binding motif. Genes Dev 1992;6:975-90 [DOI] [PubMed] [Google Scholar]
- 4. Mao X, Miesfeldt S, Yang H, Leiden JM, Thompson CB. The FLI-1 and chimeric EWS-FLI-1 oncoproteins display similar DNA binding specificities. J Biol Chem 1994;269:18216-22 [PubMed] [Google Scholar]
- 5. Hollenhorst PC, Shah AA, Hopkins C, Graves BJ. Genome-wide analyses reveal properties of redundant and specific promoter occupancy within the ETS gene family. Genes Dev 2007;21:1882-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992;359:162-5 [DOI] [PubMed] [Google Scholar]
- 7. Turc-Carel C, Aurias A, Mugneret F, Lizard S, Sidaner I, Volk C, et al. Chromosomes in Ewing’s sarcoma. I: an evaluation of 85 cases of remarkable consistency of t(11;22)(q24;q12). Cancer Genet Cytogenet 1988;32:229-38 [DOI] [PubMed] [Google Scholar]
- 8. Sorensen PH, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ, Denny CT. A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat Genet 1994;6:146-51 [DOI] [PubMed] [Google Scholar]
- 9. Jeon IS, Davis JN, Braun BS, Sublett JE, Roussel MF, Denny CT, et al. A variant Ewing’s sarcoma translocation (7;22) fuses the EWS gene to the ETS gene ETV1. Oncogene 1995;10:1229-34 [PubMed] [Google Scholar]
- 10. Kaneko Y, Yoshida K, Handa M, Toyoda Y, Nishihira H, Tanaka Y, et al. Fusion of an ETS-family gene, EIAF, to EWS by t(17;22)(q12;q12) chromosome translocation in an undifferentiated sarcoma of infancy. Genes Chromosomes Cancer 1996;15:115-21 [DOI] [PubMed] [Google Scholar]
- 11. Peter M, Couturier J, Pacquement H, Michon J, Thomas G, Magdelenat H, et al. A new member of the ETS family fused to EWS in Ewing’s tumors. Oncogene 1997;14:1159-64 [DOI] [PubMed] [Google Scholar]
- 12. Urano F, Umezawa A, Hong W, Kikuchi H, Hata J. A novel chimera gene between EWS and E1A-F, encoding the adenovirus E1A enhancer-binding protein, in extraosseous Ewing’s sarcoma. Biochem Biophys Res Commun 1996;219:608-12 [DOI] [PubMed] [Google Scholar]
- 13. May WA, Lessnick SL, Braun BS, Klemsz M, Lewis BC, Lunsford LB, et al. The Ewing’s sarcoma EWS/FLI-1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than FLI-1. Mol Cell Biol 1993;13:7393-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. May WA, Gishizky ML, Lessnick SL, Lunsford LB, Lewis BC, Delattre O, et al. Ewing’s sarcoma 11;22 translocation produces a chimeric transcription factor that requires the DNA-binding domain encoded by FLI1 for transformation. Proc Natl Acad Sci U S A 1993;90:5752-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Braunreiter CL, Hancock JD, Coffin CM, Boucher KM, Lessnick SL. Expression of EWS-ETS fusions in NIH3T3 cells reveals significant differences to Ewing’s sarcoma. Cell Cycle 2006;5:2753-9 [DOI] [PubMed] [Google Scholar]
- 16. Smith R, Owen LA, Trem DJ, Wong JS, Whangbo JS, Golub TR, et al. Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing’s sarcoma. Cancer Cell 2006;9:405-16 [DOI] [PubMed] [Google Scholar]
- 17. Owen LA, Lessnick SL. Identification of target genes in their native cellular context: an analysis of EWS/FLI in Ewing’s sarcoma. Cell Cycle 2006;5:2049-53 [DOI] [PubMed] [Google Scholar]
- 18. Law WJ, Cann KL, Hicks GG. TLS, EWS and TAF15: a model for transcriptional integration of gene expression. Brief Funct Genomic Proteomic 2006;5:8-14 [DOI] [PubMed] [Google Scholar]
- 19. Spyropoulos DD, Pharr PN, Lavenburg KR, Jackers P, Papas TS, Ogawa M, et al. Hemorrhage, impaired hematopoiesis, and lethality in mouse embryos carrying a targeted disruption of the Fli1 transcription factor. Mol Cell Biol 2000;20:5643-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. May WA, Lessnick SL, Braun BS, Klemsz M, Lewis BC, Lunsford LB, et al. The Ewing’s sarcoma EWS/FLI-1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than FLI-1. Mol Cell Biol 1993;13:7393-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rao VN, Ohno T, Prasad DD, Bhattacharya G, Reddy ES. Analysis of the DNA-binding and transcriptional activation functions of human Fli-1 protein. Oncogene 1993;8:2167-73 [PubMed] [Google Scholar]
- 22. Lessnick SL, Braun BS, Denny CT, May WA. Multiple domains mediate transformation by the Ewing’s sarcoma EWS/FLI-1 fusion gene. Oncogene 1995;10:423-31 [PubMed] [Google Scholar]
- 23. Gangwal K, Lessnick SL. Microsatellites are EWS/FLI response elements: genomic “junk” is EWS/FLI’s treasure. Cell Cycle 2008;7:3127-32 [DOI] [PubMed] [Google Scholar]
- 24. Gangwal K, Sankar S, Hollenhorst PC, Kinsey M, Haroldsen SC, Shah AA, et al. Microsatellites as EWS/FLI response elements in Ewing’s sarcoma. Proc Natl Acad Sci U S A 2008;105:10149-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Luo W, Gangwal K, Sankar S, Boucher KM, Thomas D, Lessnick SL. GSTM4 is a microsatellite-containing EWS/FLI target involved in Ewing’s sarcoma oncogenesis and therapeutic resistance. Oncogene 2009;28:4126-32 [DOI] [PubMed] [Google Scholar]
- 26. Kinsey M, Smith R, Lessnick SL. NR0B1 is required for the oncogenic phenotype mediated by EWS/FLI in Ewing’s sarcoma. Mol Cancer Res 2006;4:851-9 [DOI] [PubMed] [Google Scholar]
- 27. Kinsey M, Smith R, Iyer AK, McCabe ER, Lessnick SL. EWS/FLI and its downstream target NR0B1 interact directly to modulate transcription and oncogenesis in Ewing’s sarcoma. Cancer Res 2009;69:9047-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Garcia-Aragoncillo E, Carrillo J, Lalli E, Agra N, Gomez-Lopez G, Pestana A, et al. DAX1, a direct target of EWS/FLI1 oncoprotein, is a principal regulator of cell-cycle progression in Ewing’s tumor cells. Oncogene 2008;27:6034-43 [DOI] [PubMed] [Google Scholar]
- 29. Guillon N, Tirode F, Boeva V, Zynovyev A, Barillot E, Delattre O. The oncogenic EWS-FLI1 protein binds in vivo GGAA microsatellite sequences with potential transcriptional activation function. PLoS ONE 2009;4:e4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Laudet V, Hanni C, Stehelin D, Duterque-Coquillaud M. Molecular phylogeny of the ETS gene family. Oncogene 1999;18:1351-9 [DOI] [PubMed] [Google Scholar]
- 31. Pio F, Kodandapani R, Ni CZ, Shepard W, Klemsz M, McKercher SR, et al. New insights on DNA recognition by ets proteins from the crystal structure of the PU.1 ETS domain-DNA complex. J Biol Chem 1996;271:23329-37 [DOI] [PubMed] [Google Scholar]
- 32. Szymczyna BR, Arrowsmith CH. DNA binding specificity studies of four ETS proteins support an indirect read-out mechanism of protein-DNA recognition. J Biol Chem 2000;275:28363-70 [DOI] [PubMed] [Google Scholar]
- 33. Zwieb C, Kim J, Adhya S. DNA bending by negative regulatory proteins: Gal and Lac repressors. Genes Dev 1989;3:606-11 [DOI] [PubMed] [Google Scholar]
- 34. Lee KA. Ewing’s family oncoproteins: drunk, disorderly and in search of partners. Cell Res 2007;17:286-8 [DOI] [PubMed] [Google Scholar]
- 35. Graves BJ, Gillespie ME, McIntosh LP. DNA binding by the ETS domain. Nature 1996;384:322. [DOI] [PubMed] [Google Scholar]
- 36. Bailly RA, Bosselut R, Zucman J, Cormier F, Delattre O, Roussel M, et al. DNA-binding and transcriptional activation properties of the EWS-FLI-1 fusion protein resulting from the t(11;22) translocation in Ewing’s sarcoma. Mol Cell Biol 1994;14:3230-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lessnick SL, Dacwag CS, Golub TR. The Ewing’s sarcoma oncoprotein EWS/FLI induces a p53-dependent growth arrest in primary human fibroblasts. Cancer Cell 2002;1:393-401 [DOI] [PubMed] [Google Scholar]
- 38. Pufall MA, Lee GM, Nelson ML, Kang HS, Velyvis A, Kay LE, et al. Variable control of Ets-1 DNA binding by multiple phosphates in an unstructured region. Science 2005;309:142-5 [DOI] [PubMed] [Google Scholar]
- 39. LiCata VJ, Wowor AJ. Applications of fluorescence anisotropy to the study of protein-DNA interactions. Methods Cell Biol 2008;84:243-62 [DOI] [PubMed] [Google Scholar]
- 40. Spahn L, Siligan C, Bachmaier R, Schmid JA, Aryee DN, Kovar H. Homotypic and heterotypic interactions of EWS, FLI1 and their oncogenic fusion protein. Oncogene 2003;22:6819-29 [DOI] [PubMed] [Google Scholar]
- 41. Garvie CW, Hagman J, Wolberger C. Structural studies of Ets-1/Pax5 complex formation on DNA. Mol Cell 2001;8:1267-76 [DOI] [PubMed] [Google Scholar]
- 42. Lamber EP, Vanhille L, Textor LC, Kachalova GS, Sieweke MH, Wilmanns M. Regulation of the transcription factor Ets-1 by DNA-mediated homo-dimerization. Embo J 2008;27:2006-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tirado OM, Mateo-Lozano S, Villar J, Dettin LE, Llort A, Gallego S, et al. Caveolin-1 (CAV1) is a target of EWS/FLI-1 and a key determinant of the oncogenic phenotype and tumorigenicity of Ewing’s sarcoma cells. Cancer Res 2006;66:9937-47 [DOI] [PubMed] [Google Scholar]
- 44. Lee GM, Pufall MA, Meeker CA, Kang HS, Graves BJ, McIntosh LP. The affinity of Ets-1 for DNA is modulated by phosphorylation through transient interactions of an unstructured region. J Mol Biol 2008;382:1014-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hollenhorst PC, Chandler KJ, Poulsen RL, Johnson WE, Speck NA, Graves BJ. DNA specificity determinants associate with distinct transcription factor functions. PLoS Genetics 2009;5:e100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hoffmann KM, Williams D, Shafer WM, Brennan RG. Characterization of the multiple transferable resistance repressor, MtrR, from Neisseria gonorrhoeae. J Bacteriol 2005;187:5008-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.