

Abstract
Leptin is a 16-kd hormone that mediates a range of metabolic effects by using a transduction pathway from the long form of the leptin receptor, OB-RL, through Janus kinase–signal transducer and activator of transcription (Jak-Stat) signaling components. Leptin is produced by hepatic stellate cells (HSCs) but only following their “activation.” Because activation of stellate cells is a central event in the fibrotic response to liver injury, we hypothesized that leptin may directly stimulate fibrogenesis in activated stellate cells via OB-RL. We analyzed leptin receptors and their signaling partners in a stellate cell line (HSC-T6) as well as in primary stellate cell isolates. We also examined the effect of leptin on stellate cell expression of α2(I) collagen messenger RNA (mRNA) levels by ribonuclease protection analysis (RPA). Finally, we examined the role of leptin in in vivo fibrogenesis by inducing a wounding response in ob/ob mice, which lack functional leptin. HSC-T6 and culture-activated stellate cells expressed OB-RL. Scatchard analysis verified specific binding of leptin to HSCs, with an association constant (Kd) equal to 660 ± 5.8 pmol/L. Exposure of HSCs to leptin resulted in significant increases in α2(I) collagen mRNA expression. Transient transfection with a promoter reporter construct showed a 3-fold increase in α2(I) collagen transgene activity. Leptin stimulated activation of Stat3 in activated HSCs. Finally, lean animals, but not ob/ob littermates, had significant fibrosis as assessed by picrosirius red staining and abundant α–smooth muscle actin staining. In conclusion, these results indicate that leptin is profibrogenic in activated HSCs and can signal via the Jak-Stat pathway. Up-regulation of leptin signaling in liver injury could contribute to enhanced fibrogenesis, particularly in states in which leptin levels are high.
Leptin, a 16-kd peptide product of the ob gene,1 is a potent adipocyte-derived hormone that plays a key role in the control of energy balance and food in-take.2–4 Leptin receptors, initially found primarily in central nervous tissues such as the hypothalamus,5 have also been localized to other tissues, including the liver.6 Leptin signaling is mediated by the leptin receptor (OB-R), a member of the hematopoietin receptor family that is most closely related to the signal-transducing subunits of the interleukin 6–type cytokine receptors.5,7,8 In humans and rodents, at least 2 predominant forms of OB-R are detected. Both isoforms have identical extracellular domains and ligand-binding affinity but differ in the intracellular domains, which represent alternative splice products. The major OB-R short form (OB-RS), a 34–amino acid cytoplasmic domain, is found in many organs. However, despite normal ligand-binding activity, OB-RS is considered incapable of signaling.9,10 In contrast, the long form (OB-RL) is the signaling-competent receptor isoform and contains a 302–amino acid cytoplasmic domain.9–11 Reverse-transcription polymerase chain reaction (RT-PCR) and ribonuclease protection analysis (RPA) show that various peripheral organs, including the liver, have detectable levels of messenger RNA (mRNA) encoding OB-RL,9 implying that leptin has the potential to stimulate liver cells. Homozygous mutations resulting in leptin deficiency in mice (i.e., ob/ob mouse) and mutations for leptin signaling (i.e., db/db mice) are both associated with obesity7,12 and provide useful models to study the role of leptin insufficiency in vivo. However, the cellular sources and targets of leptin outside the central nervous system and adipose tissue as well as the relative distribution of long-form versus short-form receptor are not clearly established.
The homology of the OB-R to class I cytokine receptors implicates the Janus kinase–signal transducer and activator of transcription (Jak-Stat) proteins as signal transducers for leptin receptors.13,14 Typically, Jak proteins are associated constitutively with membrane-proximal sequences of the receptor intracellular domain and phosphorylate the receptor on ligand binding. The phosphorylated intracellular domain then provides a binding site for a Stat protein, which is activated on binding the phosphorylated receptor intracellular domain. The activated Stat proteins then translocate to the nucleus and stimulate transcription.15 Two published reports show that Stat3 and Stat5 are stimulated in COS cells by OB-R.8,10 As observed for Stat protein activation, the naturally occurring mouse OB-RS is incapable of stimulating transcription.10
We previously explored the production of leptin by hepatic stellate cells (HSCs). This resident perisinusoidal cell type is the primary storage site for vitamin A in normal liver but undergoes a characteristic “activation” in liver injury, leading to increased proliferation, fibrogenesis, and contractility.16 In liver injury, stellate cells are the primary source of extracellular matrix. We previously showed that only activated rat HSCs but not quiescent or freshly isolated HSCs expressed leptin protein and mRNA.17 Hepatocytes or Kupffer cells did not express leptin. More recently, a role for leptin in accelerating wound healing has emerged.18 Further, leptin levels are reportedly higher in patients with alcoholic cirrhosis regardless of body mass index.19
In this study, our aims were to determine whether leptin has the capacity to augment fibrosis by increasing α2(I) collagen gene expression and to determine whether HSCs have OB-R signaling capabilities in response to leptin. By using CCl4, a well-described liver-injury model in rodents known to produce liver fibrosis, we determined in ob/ob mice and their lean littermates whether the absence of leptin prohibited the development of fibrosis when compared with the wild-type lean littermates.
Materials and Methods
Materials
HSC-T6 cells, an immortalized rat HSC cell line, exhibits an activated HSC phenotype.20 Quiescent and activated HSC cell lysates were from isolated rat HSCs as described in detail elsewhere.21,22 Dulbecco’s modified Eagle medium, fetal bovine serum, penicillin-streptomycin, amphotericin B, trypsin/ethylenedia-minetetraacetic acid, β-mercaptoethanol, acrylamide, molecular weight markers, LipofectaminePLUS, TRIzol reagent, and agarose were purchased from Gibco-BRL (Rockville, MD). Plastic tissue culture ware was purchased from Falcon Division, Becton Dickinson Co. (Mountain View, CA). Phenylmethylsulfonyl fluoride, β-glycerophosphate, sodium orthovanadate, antipain, leupeptin, aprotinin, chymostatin, pepstatin, and rat-recombinant leptin were purchased from Sigma Chemical Co. (St. Louis, MO). Dimethyl sulfoxide was from Fisher Scientific (Pittsburgh, PA). Luciferase reporter vectors were purchased from Promega (Madison, WI). The mouse α2(I) collagen complementary DNA (cDNA) was provided by Dr. Benoit de Cromrugghe (M. D. Anderson Hospital and Tumor Institute, Houston, TX). Horseradish peroxidase–labeled anti-goat, anti-rabbit, and anti-mouse immunoglobulin G; protein A and G agarose beads; and antibodies to Stat3, phosphorylated Stat3 (pStat3), and leptin receptor antibody (OB-RL [H-300], OB-R [K-20]) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). 125I-Rat leptin (specific activity, 135 μCi/μg) was purchased from Linco Research Inc. (St. Charles, MO). T7 RNA polymerase was purchased from Ambion (Austin, TX).
HSC Isolation
HSC-T6 and activated rat HSCs were cultured as described elsewhere.20–22 All rats received humane care, and the Institutional Animal Care and Use Committees of the University of Maryland, Duke University, and Mount Sinai School of Medicine approved the RSC isolation protocol. In brief, after in situ perfusion of the liver with 20 mg/dL pronase (Boehringer Mannheim, Indianapolis, IN) followed by collagenase (Crescent Chemical, Hauppauge, NY), dispersed cell suspensions were layered on a discontinuous density gradient of 8.2% and 15.6% Accudenz (Accurate Chemical and Scientific, Westbury, NY). The resulting upper layer consisted of more than 95% stellate cells. Cells were placed in modified medium 199 OR containing 20% serum (10% horse/10% calf) (Flow Laboratories, Naperville, IL). The purity of stellate cells was assessed by immunolocalization of desmin in the monolayer as well as by intrinsic autofluorescence. The viability of all cells was verified by phase-contrast microscopy as well as the ability to exclude propidium iodide. Cell viability of cultures used for study was greater than 95%.
HSC Primary and Cell Line Culture and Treatment With Recombinant Leptin
Subconfluent cells in culture (75%) were washed twice with phosphate-buffered saline and serum-starved for 16 hours with 0.1% fetal bovine serum and 1% penicillin-streptomycin in Dulbecco’s modified Eagle medium. To determine the optimal concentration of rat-recombinant leptin to be used in these experiments, dose-response and time-course studies were initially performed with the following concentrations: 25, 50, 100, 150, and 300 ng/mL added to cell cultures for 15, 30, 60, and 90 minutes and 3, 6, 9, 12, and 24 hours. Purchased recombinant leptin was routinely tested for lipopolysaccharide contaminant levels with a limulus lysate assay (Associated of Cape Cod, Fal-mouth, MA) and was found to contain less than 0.02 pmol/L. The optimal concentration of recombinant leptin was determined to be 100 ng/mL; this concentration was used in all subsequent experiments. This concentration has been used in other published work and is physiologically relevant in obesity.23–25
Analysis of α2(I) Collagen and OB-RL mRNA by RT-PCR
Total RNA was isolated from HSC-T6 cells and culture-activated HSCs as described by Chomczynski and Sacchi.26 For each reaction, 2 μg of total RNA was reverse-transcribed to cDNA using RETROscript First Strand Synthesis Kit for RT-PCR (Ambion) as recommended by the manufacturer. The resulting cDNA was subsequently subjected to 30 cycles of PCR. PCR products were quantitated to confirm that they were in the linear range of amplification. PCR products were quantified by Quantum RNA 18S internal standards (Ambion). The primers encoding the leptin receptor (common to both the long and short forms), yielding an amplified fragment of 233 base pairs, consisted of a forward primer 5′ ATATGGAGCCATTACCTAAGAACC and a reverse primer 5′ GAAATCACAAGACGGGGTGT-GAGT3′ from mouse (#U46135). Primers encoding the long form of the rat and murine OB-RL included a forward primer 5′ GATGATGGAATGAAGTGG-CTTAG3′ and a reverse primer 5′ GTAAAAAGATG-CTCAAATGTTTC3′ (#D84550). A portion of the glyceraldehyde-3-phosphate dehydrogenase cDNA encoding a region common to murine, rat, and human species was used to normalize mRNA expression: 5′ AC-CACAGTCCATGCCATCAC′3 and 5′TCCACCA-CCCTGTTGCTGTA3′ (#AF106860). The conditions for RT-PCR to detect OB-RL were as follows. cDNA synthesis and predenaturation were at 42°C for 60 minutes followed by 94°C for 10 minutes. PCR amplification sequence was performed for 35 cycles at 94°C for 30 seconds, 55°C for 30 seconds, and 72°C for 1 minute. After amplification, each sample was applied to 1.2% agarose/ethidium bromide gel. The resolved PCR products were photographed under UV illumination.
Detection of α2(I) Collagen mRNA by RPA
A 533–base pair fragment of the α2(I) collagen gene with the T7 promoter was constructed by RT-PCR. Subsequently, a 533-nucleotide antisense riboprobe was transcribed with T7 RNA polymerase (MAXIscript kit; Ambion) by using [α32P]uridine triphosphate (specific activity, 800 Ci/mmol) (Amersham Pharmacia Biotech, Piscataway, NJ). RPAs were performed using the RPAIII kit (Ambion). Gel-purified riboprobes (105 cpm for α2(I) collagen gene and 3 × 104 cpm for β-actin) were hybridized with 10 μg total cellular RNA extracted from either HSC-T6, primary cultured HSCs, or yeast transfer RNA at 42°C for 18 hours followed by ribonuclease A/T1 digestion at 37°C for 30 minutes. Protected fragments were heat denatured and separated on 3.5% denaturing urea/polyacrylamide gels. A rat β-actin antisense probe was used as a control. Radioactive signals were recorded and quantitated by using the BioRad Imaging System and Quantity One quantitation software (Life Sciences Group, Hercules, CA). The experiments were performed 3 times; each experiment used newly prepared total RNA from new tissue cultures. Each α2(I) collagen signal was normalized to the β-actin signal from the same sample, and the normalized values were expressed relative to signals in the untreated HSC-T6 or primary cultured HSC (control) cells, respectively.
α2(I) Collagen Gene Transactivation by Leptin in HSCs
The α2(I) collagen promoter containing −2,000 to +54 base pairs was subcloned into the pGL3 luciferase reporter vector (pGL3-1009).27 Transfections were performed with LipofectaminePLUS according to manufacturer’s specifications. After serum starvation for 16 hours, 2 μg of the reporter construct was added to 80% confluent cell cultures along with 0.5 μg of the internal control plasmid pSV-βgal and the transfection reagents. The empty pGL3 vector served as a negative control; the pGL3 vector containing an SV40 enhancer served as a positive control. After leptin exposure (100 ng/mL) for the designated times, cell lysate luciferase activity was determined using a Monolight 3010 Luminometer (Analytical Luminescence Laboratory, San Diego, CA). Preliminary transfection studies were also performed with 25, 50, 75, 125, and 150 ng/mL leptin to determine the lowest effective concentration. Results were expressed in relative light units per microgram protein quantitated by the method of Lowry et al.28
Scatchard Analysis of Leptin Binding to HSCs
HSC cell monolayers in 6-well culture plates were incubated with 125I-leptin at 62.5 pmol/L or saturating concentrations of unlabeled leptin (5 μmol/L) for 10, 20, 30, 40, 50, 60, 75, 90, and 105 minutes and 2, 4, 8, 12, 24, 36, and 48 hours. Cells were incubated at 37°C in a humidified atmosphere of 5% CO2/95% humidified air. Binding experiments were performed 24 to 48 hours after plating as follows. Cells were washed once with Krebs-Ringer medium (pH 7.4) containing 20 mmol/L HEPES and 1% bovine serum albumin and then incubated in duplicate at 25°C with 125I-labeled leptin in 300 μL of the same buffer. Controls for nonspecific binding of labeled leptin contained unlabeled leptin at the times indicated previously.
Incubations were terminated by rapid removal of the incubation medium and addition of 500 μL ice-cold buffer. The cells were washed with 6 changes of buffer and then lysed in 1% Triton X-100, 10% (vol/vol) glycerol, 25 mmol/L HEPES, pH 7.5, and 0.1% bovine serum albumin at 4°C for 40 minutes. Cell-bound radioactivity was determined. Data are shown in Scatchard plots29 according to analysis by a modified Gauss-Newton nonlinear least-square program using SigmaPlot (SPSS Science, Chicago, IL) computer software.30 Protein was measured by the method of Lowry et al.,27 and specific binding is expressed per microgram of protein.
Stat3 Phosphorylation Assays
Cell lysates prepared from HSC-T6 or primary HSCs were treated with 100 ng/mL leptin in 0.1% serum-free media for 1, 3, 6, 9, 12, and 24 hours. Cells were washed twice with ice-cold phosphate-buffered saline. Protein was extracted on ice in radioimmunoprecipitation assay buffer that included 50 mmol/L Tris-HCl, pH 7.5, 1% Triton X-100, 10% glycerol, 137 mmol/L NaCl, 2 mmol/L ethylenediaminetetraacetic acid, 25 mmol/L β-glycerophosphate, 1 mmol/L sodium orthovanadate, and 1 mmol/L phenylmethylsulfonyl fluoride. Additional protease inhibitors were included in all subsequent procedures in radioimmunoprecipitation assay buffer. Cell extracts were passed through a 20-gauge sterile syringe and centrifuged at 6000g for 10 minutes and protein content assayed.28 Control studies were performed as indicated except for the absence of leptin treatment. For each time point, un-phosphorylated Stat3 protein was assayed as a control by immunoblot analysis as described subsequently. Leptin-treated HSC lysate supernatants were subsequently treated with 1 μg of anti-pStat3 antibody and incubated for 90 minutes at 4°C. Immunoprecipitation was performed with protein G agarose bead conjugate and incubated at 4°C for 16 hours. Agarose beads were washed extensively with radioimmunoprecipitation assay buffer containing protease inhibitors. The resultant beads were mixed in 20 μL Laemmli loading buffer and subjected to 10% sodium dodecyl sulfate/polyacrylamide gel electrophoresis.31 Transfer to nitrocellulose membrane was performed at 4°C for 16 hours and reacted with secondary antibodies as indicated in the figure legends. The immunocomplexes were visualized by enhanced chemiluminescence reaction (Amersham, Arlington Heights, MA). All experiments were performed in triplicate.
Immunoprecipitation of pStat3 and the Leptin Receptor, OB-R
Lysates, quiescent and culture-activated rat HSCs and HSC-T6 cells, were extracted, quantitated, electrophoresed, and transblotted as previously described. Immunodetection of OB-RL, OB-R, Stat3, and pStat3 was performed with polyclonal antibody for the respective protein (H-300, K-20, Stat3, and pStat3 antibodies) at a dilution of 1:250 for OB-R antibodies and 1:1,000 for Stat3 and pStat3, respectively. After subsequent washes, the blots were exposed to 1:1,000 dilution of anti-rabbit immunoglobulin G peroxidase conjugate for 1 hour. Immunocomplexes were also visualized by enhanced chemiluminescence (Amersham) and performed in triplicate. pStat3 activity was compared with pStat3 activity in untreated HSCs and quantitated using the BioRad Imaging System and Quantity One quantitation software. All experiments were performed in triplicate.
CCl4 Treatment of ob/ob Mice and Their Lean Littermates and Histologic Analysis of Liver Specimens
Male ob/ob mice and their lean littermates were purchased from Jackson Laboratories (Bar Harbor, ME). All animals received humane care, and the experimental protocol was approved by the Institutional Care and Use Committee of the University of Maryland. Mice were allowed free access to a laboratory chow diet and were housed for 1 week before CCl4 injections. Mice were given an intraperitoneal injection of 50% CCl4 (1 mL/kg) with olive oil twice weekly for 8 weeks. Control mice were also injected with 0.9% sterile saline and olive oil. All mice were killed for histologic analysis. Liver tissues were fixed with 10% buffered formalin and embedded with paraffin. Picrosirius red stain was used to detect collagen fibrils as described elsewhere.32
To detect α–smooth muscle actin (SMA) in the liver sections, formalin-fixed and paraffin-embedded tissue sections were deparaffinized and immunohistochemical staining using a mouse monoclonal anti-SMA antibody (American Research Products, Inc., Belmont, MA) was performed. After rinsing the primary antibody, the sections were incubated with a biotinylated mouse anti-mouse immunoglobulin G F(ab′) fragment, followed by incubation with the avidin-biotin complex solution using a VECTOR Mouse on Mouse Immunodetection kit (Vector Laboratories Inc., Burlingame, CA).
Statistical Analysis
Results are expressed as the mean ± SEM. Quantitative data from RPA, transfection analysis, and Stat3 phosphorylation assays were subjected to an unpaired 2-tailed Student’s t test. The differences were considered significant at P < .05. All experiments were performed in triplicate on 3 separate occasions.
Results and Discussion
To determine whether leptin was fibrogenic toward stellate cells, we first examined its effect on α2(I) collagen mRNA expression by RPAs as shown in Fig. 1. Data from primary cultured HSCs (Fig. 1A) and HSC-T6 cells (Fig. 1B) confirm that leptin increased α2(I) collagen mRNA when compared with untreated (control) cultures. A 4-fold increase in leptin-treated primary HSC and HSC-T6 α2(I) collagen mRNA was observed, and this effect was significant at all time points through 24 hours.
Fig. 1.
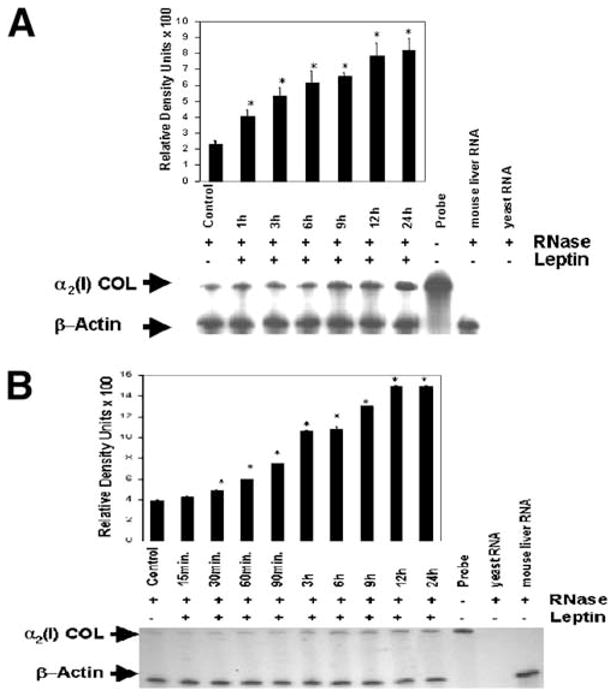
RPA of the α2(I) collagen mRNA from HSC-T6 cells and primary cultured HSCs. β-actin, 304 nucleotides; α2(I)COL (α2(I) collagen), 533 nucleotides. (A) Primary HSCs were isolated from normal rats and allowed to grow in culture in standard serum-containing medium. After 3 days, serum-containing medium was exchanged with serum-free medium; cells were exposed to 100 ng/mL leptin in SF medium, total cellular RNA was isolated at the indicated time points, and RPA was performed as described in Materials and Methods. Results with primary cultured HSCs include a representative histogram of densitometric analysis of autoradiography shown. (B) A representative autoradiogram and histogram results from RPA from HSC-T6 cells. RPA from primary cultured HSCs and HSC-T6 cells was performed separately in triplicate in 3 independent experiments. Statistical analysis of RPA products was performed by Student’s t test. All treatment data were compared with controls (untreated cells), *P < .05. Error bars represent SEM.
To further explore transcriptional activation of the α2(I) collagen gene by leptin, transient transfection experiments were performed in HSC-T6 cells followed by treatment with leptin. The results of this experiment are shown in Fig. 2. Addition of 100 ng/mL leptin resulted in a 6-fold increase in α2(I) collagen transgene activation at 6 to 9 hours in HSC-T6; however, this effect was not observed with the empty vector pGL3 lacking the collagen promoter (Fig. 2B). Compared with untreated transfected cells (Fig. 2, white bars), the α2(I) collagen transgene activation was significantly increased (P < .05) for all time intervals for the leptin-treated cells (Fig. 2, black bars).
Fig. 2.

Transient transfection of the α2(I) collagen promoter in HSC-T6 cells exposed to leptin. Transient transfections with HSC-T6 were performed with 2 μg of the α2(I) collagen promoter (pGL3-1009) and 0.5 μg of pSV-βgal. Leptin treatment (100 ng/mL) is as in Fig. 1 and Materials and Methods. Luciferase activity was determined and results shown as relative light units/microgram protein. The pSV-βgal reporter gene was cotransfected to normalize for transfection efficiency. Untreated pGL3-1009 (□) represents transgene activity of the α2(I) collagen promoter in cells not exposed to leptin for each treatment time indicated. The transfected pGL3 empty vector (B) was used as a negative control; the pGL3-SV40 enhancer vector (P,  ) served as a positive control. All assays were performed in triplicate. Data shown represent the average of 3 independent experiments ± SEM. Data analysis was performed by Student’s t test, where *P < .05 was significant and compares collagen transgene expression between untreated cells (□) and leptin-treated cells (■) normalized to total protein assessed by Lowry assay25 for each time point indicated in hours.
) served as a positive control. All assays were performed in triplicate. Data shown represent the average of 3 independent experiments ± SEM. Data analysis was performed by Student’s t test, where *P < .05 was significant and compares collagen transgene expression between untreated cells (□) and leptin-treated cells (■) normalized to total protein assessed by Lowry assay25 for each time point indicated in hours.
The data in Figs. 1 and 2 strongly suggest that leptin elicits a biologic effect in stellate cells and prompted us to characterize leptin binding and possible signaling via the leptin receptor. To investigate whether saturable leptin binding occurs in stellate cells, binding assays were performed in HSCs. Saturable, specific binding was documented (Fig. 3A) with an association constant (Kd) for leptin-HSC binding calculated to be 660 ± 5.8 pmol/L (Fig. 3B). This Kd value is comparable to other leptin-binding cells, including HepG2 human and rat hepatoma cells.5,6,33 These analyses indicate that there are ~4,000 leptin-binding sites per cell.
Fig. 3.

Specific binding of 125I-leptin to HSCs. (A) Time course showing leptin binding to HSCs. Cells were cultured as monolayers and incubated with 62.5 pmol/L 125I-leptin for time indicated in minutes (◆). To determine specific leptin-HSC binding, an identical assay was performed with excess unlabeled leptin (5 μmol/L) (●). Bound radioactive ligand was measured and standardized to microgram protein assayed.28 (B) Scatchard analysis.29 Incubation with labeled leptin was performed for 2 hours with cell monolayers at 4°C. Cell-associated radioactivity was determined by scintillation counting. Data for Kd are expressed as mean ± SEM. The plots represent duplicate experiments performed twice. The details of these studies are described in Materials and Methods.
Because OB-RS is abundant in nonneural tissues but OB-RL is required for signal transduction, the data in Fig. 3 cannot discriminate whether leptin binds primarily to OB-RS or OB-RL or both in HSCs. However, the binding assays presented raise the possibility of autocrine signaling by leptin, a possibility made more likely because of extensive autocrine/cytokine signaling in stellate cells by a large number of cytokines and growth factors.34 For example, transforming growth factor β1 is both produced by activated HSCs and signals through transforming growth factor β receptor isoforms.35
We performed immunoprecipitation and RT-PCR to identify the long-form leptin receptor, OB-RL, in both primary cultured HSCs and HSC-T6 cells. As expected, antibodies against OB-R, which detect sequences common to both OB-RL and OB-RS, show the presence of receptor in all stellate cells studied (Fig. 4C). Because the antibody to detect all forms of OB-R is aimed at the extracellular portion of the receptor, multiple isoforms, including a soluble form OB-Re, may explain the multiple band forms detected in HSCs, representing the differential expression of these receptors as seen in other tissues.36,37 Figure 4A and B show OB-RL protein and a corresponding 361–base pair cDNA by RT-PCR for both primary cultured HSCs and HSC-T6 cells.
Fig. 4.
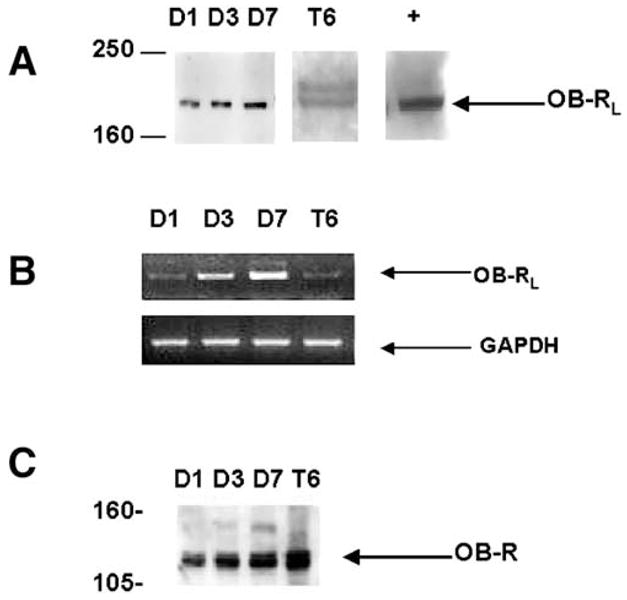
Detection of OB-RL in HSC-T6 cells and primary, culture-activated rat HSCs by immunoprecipitation and RT-PCR. (A) Immunoprecipitation of OB-RL. Whole-cell lysate was prepared from primary HSCs in culture for 1 day (D1), 3 days (D3), and 7 days (D7). OB-RL in HSC-T6 (T6) cells was detected by immunoprecipitation. An equal quantity of protein was immunoprecipitated with antibody against the cytoplasmic domain of the leptin receptor. Positive control was from mouse brain (+) and was supplied by Santa Cruz Biotechnology. (B) RT-PCR detecting mRNA for OB-RL. Details of reactions and primer pairs are described in Materials and Methods. Total RNA was isolated from primary HSCs and HSC-T6 in culture at times identical to immunoprecipitation analysis as previously described. The expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. (C) Immunoprecipitation of OB-R with antibody directed at the extracellular domain of the receptor common to all OB-R isoforms. Molecular weight is on the left in kilodaltons.
To determine whether stellate cell OB-RL signaling can occur via the Jak-Stat pathway in HSCs as it does in other tissues, we examined whether leptin elicits an increase in phosphorylation of Stat3, the major signal transducer of OB-RL. In both primary cultured HSCs (Fig. 5A) and HSC-T6 cells (Fig. 5B), pStat3 was detectable over the same interval in which effects on collagen gene expression were elicited. However, maximal phosphorylation of Stat3 occurred between 1 and 6 hours before returning to control pStat3 levels. The quantity of total Stat3 available for phosphorylation as shown did not change in response to leptin. Although not shown, Stat1 phosphorylation was also increased in HSCs exposed to leptin over the same time intervals indicated here.
Fig. 5.

Immunoprecipitation of pStat3 in stellate cells exposed to leptin. One microgram of polyclonal antibodies to pStat3 was added to (A) primary cultured HSCs or (B) HSC-T6 as described in detail in Materials and Methods. Equal protein loading28 was confirmed based on expression of total, unphosphorylated Stat3 (STAT3 in figures). Lanes 2 to 6 indicate leptin exposure for the times indicated in hours for either primary HSCs (A) or cell culture model (B). The corresponding histograms compare pStat3 from leptin-treated HSCs with untreated HSCs and are from 3 independent experiments performed in triplicate. Data analysis comparing pStat3 in leptin-treated cells for each time point vs. untreated (Lane 1, control [C]) HSCs was by Student’s t test. *P < .05.
Histology of liver sections examined for collagen fibril deposition in which CCl4 was administered to either ob/ob mice or their lean littermates is shown in Fig. 6. Figure 6A shows significant fibrosis in the lean littermates of the ob/ob mice, which is noticeably absent in the identically treated ob/ob animals (Fig. 6B). As anticipated, both the lean littermates and the ob/ob mice treated with vehicle only (saline/olive oil) (Fig. 6C and D) also show no significant picrosirius red staining, indicating a lack of type I collagen deposition outside of central and portal blood vessels.
Fig. 6.
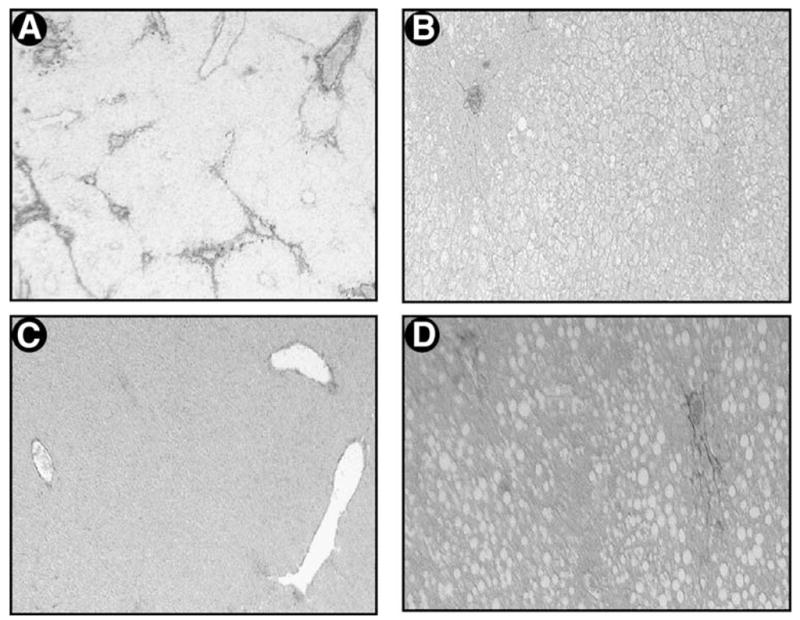
Histopathologic analysis for type I collagen in the liver after CCl4 administration in ob/ob mice and their lean littermates. Representative photomicrographs of (A) CCl4-treated male lean littermates of ob/ob mice and (B) CCl4-treated ob/ob mice. Simultaneous treatment with vehicle (saline and olive oil) is also shown in C for the lean littermates and D for ob/ob mice. These photomicrographs represent 3 independent studies in which 4 male mice were included in each treatment group (picrosirius red stain, original magnification 100×).
Figure 7 is a high-power photomicrograph of immunohistochemical staining for α-SMA of liver sections as indicated in Fig. 6. α-SMA, an indicator of activated HSCs in liver, is strongly detected in Fig. 7A, which shows liver sections stained from the lean littermates of the ob/ob animals treated with CCl4. Neither the control animals (Fig. 7C and D) nor the CCl4-treated ob/ob (Fig. 7B) mice show significant α-SMA–positive cells.
Fig. 7.
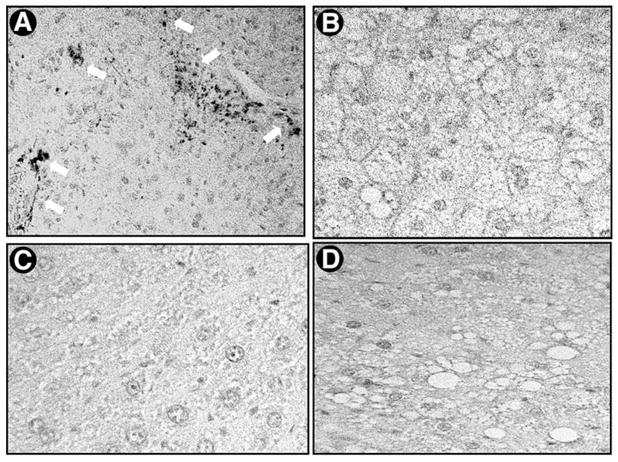
Expression of α-SMA in the liver after administration of CCl4 in ob/ob mice and their lean littermates. The expression and localization of α-SMA were detected by immunohistochemical staining as described in detail in Materials and Methods. Representative micrographs from 3 individual experiments are shown (original magnification 400×). (A) CCl4-treated lean littermates showing α-SMA–positive cells (white arrows), (B) CCl4-treated ob/ob mice, (C) vehicle-treated lean littermates, and (D) vehicle-treated ob/ob mice. Primary antibody dilution was 1:400; the experimental design and resultant liver sections used for α-SMA staining were as in Fig. 6.
Several recent reports have implicated leptin as a critical hormone in the development of liver fibrogenesis. Specifically in animal models with leptin deficiency, cirrhosis failed to develop in response to liver injury due to either thioacetamide or methionine/choline deficiency.38,39 In these animal studies, leptin was shown to have only a permissive role in liver fibrosis, acting primarily by enhancing transforming growth factor β1 activity. Importantly, these studies did not establish a direct fibrogenic effect of leptin on stellate cells, instead postulating an indirect effect through sinusoidal endothelial cells. Our data show a direct role of leptin in liver fibrosis. A leptin-associated increase in α2(I) collagen gene expression was observed by RPA in vitro in HSCs, as was leptin-increased α2(I) collagen gene promoter activity. Mature collagen in liver sections of the ob/ob lean littermates (Fig. 6A) as detected by picrosirius red staining confirmed the results of the in vitro data presented here. Moreover, the presence of α-SMA by immunolocalization confirms the importance of leptin as a profibrogenic factor in liver fibrosis. Without the presence of leptin, no α-SMA positive cells were detected (Fig. 7A) and no significant picrosirius red staining was present in the ob/ob animals treated with CCl4 (Fig. 6B). Taken together, these data implicate a direct role for leptin on HSCs in the genesis of liver fibrosis. The immunohistochemical studies, along with data suggesting increased leptin and leptin receptor production during the stellate cell activation process, also raises speculation that leptin may be involved in the activation of HSCs. Further work will be aimed at determining whether leptin is directly involved in the activation of HSCs.
Our data do not exclude the possibility that leptin signaling leads to increased transforming growth factor β1 but are consistent with a growing body of evidence from other organ systems implicating leptin itself as a profibrogenic hormone. For example, wound healing in ob/ob mice, which lack circulating leptin, has impaired surgical scar formation.18 Diabetic ophthalmopathy and glomerulosclerosis, two conditions associated with longstanding diabetes mellitus, show that leptin can induce cell proliferation as well as augment collagen gene expression.40
A direct role of leptin is further substantiated by our findings in both HSC-T6 cells and primary cultured HSCs that the long-form of the leptin receptor, OB-RL, is expressed. The long form of the receptor is widely implicated in leptin signal transduction, whereas very little evidence is available implicating a role for the short form, OB-RS, in the action of leptin.23 Further work regarding leptin signal transduction will need to be performed because Jak-Stat signaling usually results in events within minutes41 and not hours. Alternatively, Stat phosphorylation may result in the activation of other transcription factors responsible for the effect on collagen gene expression observed here. These characteristics of Jak-Stat signal transduction may account for the observation that collagen gene expression is still increasing at 12 and 24 hours (Fig. 1), whereas pStat3 activity returns to basal levels after 6 hours (Fig. 5) in both primary HSCs and the HSC-T6 cell line. Emerging evidence would suggest that leptin could act through other signaling cascades, including the family of mitogen-activated protein kinases and the stress-activated protein kinases.42,43 Mitogen-activated protein kinase or stress-activated protein kinase signaling could explain the persistent increase in collagen gene activation and expression by leptin in HSCs that we have observed here.
Our data provide convincing evidence, which provides novel mechanisms whereby leptin affects liver fibrogenesis. Other genes associated with liver fibrosis (e.g., tissue inhibitors of metalloproteinases) are currently being studied to determine whether leptin alters their expression and in turn acts as a profibrogenic substance in the liver microenvironment. These data have potential implications for clarifying mechanisms of fibrosis in diseases in which circulating leptin levels are elevated such as nonalcoholic steatohepatitis, type 2 diabetes mellitus, and alcoholic cirrhosis.19,44,45
Acknowledgments
Supported by a grant from the Alcohol Beverage Medical Research Foundation; Department of Medicine, University of Maryland; and grants AA 12933, DK 37340, DK 50574, and DK 54038.
Abbreviations
- OB-R
leptin receptor
- OB-RS
short-form leptin receptor
- OB-RL
long-form leptin receptor
- RT-PCR
reverse-transcription polymerase chain reaction
- RPA
ribonuclease protection analysis
- mRNA
messenger RNA
- Jak-Stat
Janus kinase–signal transducer and activator of transcription
- HSC
hepatic stellate cell
- cDNA
complementary DNA
- pStat3
phosphorylated Stat3
- SMA
smooth muscle actin
- Kd
association constant
References
- 1.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 2.Caro JF, Sinha MK, Kolaczynski JW, Zhang PL, Considine RV. Leptin: the tale of an obesity gene. Diabetes. 1996;45:1455–1462. doi: 10.2337/diab.45.11.1455. [DOI] [PubMed] [Google Scholar]
- 3.Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 4.Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science. 1995;269:546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- 5.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Kuropatwinski KK, White DW, Hawley TS, Hawley RG, Tartaglia LA, Baumann H. Leptin receptor action in hepatic cells. J Biol Chem. 1997;272:16216–16223. doi: 10.1074/jbc.272.26.16216. [DOI] [PubMed] [Google Scholar]
- 7.Lee GHL, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, Friedman JM. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996;379:632–635. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- 8.Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, Lakey ND, et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell. 1996;84:491–495. doi: 10.1016/s0092-8674(00)81294-5. [DOI] [PubMed] [Google Scholar]
- 9.Ghilardi N, Ziegler S, Wiestner A, Stoffel R, Heim MH, Skoda RC. Defective STAT signaling by the leptin receptor in diabetic mice. Proc Natl Acad Sci U S A. 1996;93:6231–6235. doi: 10.1073/pnas.93.13.6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baumann H, Morella KK, White DW, Dembski M, Bailonn PS, Kim H, Lai C-F, et al. The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proc Natl Acad Sci U S A. 1996;93:8374–8378. doi: 10.1073/pnas.93.16.8374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaisse C, Halaas JL, Horvath CM, Darnell JE, Stoffel M, Friedman JM. Leptin activation of stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat Genet. 1996;14:95–97. doi: 10.1038/ng0996-95. [DOI] [PubMed] [Google Scholar]
- 12.Takaya K, Ogawa Y, Isse N, Okazaki T, Satoh N, Masuzaki H, Mori K, et al. Molecular cloning or rat leptin receptor isoform complementary DNAs—identification of a missense mutation in Zucker fatty (fa/fa) rats. Biochem Biophys Res Commun. 1996;225:75–83. doi: 10.1006/bbrc.1996.1133. [DOI] [PubMed] [Google Scholar]
- 13.Heldin CH. Dimerization of cell surface receptors in signal transduction. Cell. 1995;80:213–223. doi: 10.1016/0092-8674(95)90404-2. [DOI] [PubMed] [Google Scholar]
- 14.Kishimoto T, Taga T, Akira S. Cytokine signal transduction. Cell. 1994;76:253–262. doi: 10.1016/0092-8674(94)90333-6. [DOI] [PubMed] [Google Scholar]
- 15.Tartaglia LA. The leptin receptor. J Biol Chem. 1997;272:6093–6096. doi: 10.1074/jbc.272.10.6093. [DOI] [PubMed] [Google Scholar]
- 16.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 17.Potter JJ, Womack L, Mezey E, Anania FA. Transdifferentiation of rat hepatic stellate cells results in leptin expression. Biochem Biophys Res Commun. 1998;244:178–182. doi: 10.1006/bbrc.1997.8193. [DOI] [PubMed] [Google Scholar]
- 18.Frank S, Stallmeyer B, Kampfer H, Kolb N, Pfeilschifter J. Leptin enhances wound re-epithelialization and constitutes a direct function of leptin in skin repair. J Clin Invest. 2000;106:501–509. doi: 10.1172/JCI9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCullough AJ, Bugianesi E, Marchesini G, Kalhan SC. Gender-dependent alterations in serum leptin levels in alcoholic cirrhosis. Gastroenterology. 1998;115:947–953. doi: 10.1016/s0016-5085(98)70267-7. [DOI] [PubMed] [Google Scholar]
- 20.Vogel S, Piantedosi R, Frank J, Lalazar A, Rockey DC, Friedman SL, Blaner WS. An immortalized rat liver stellate cell line (HSC-T6): a new cell model for the study of retinoid metabolism in vitro. J Lipid Res. 2000;41:882–893. [PubMed] [Google Scholar]
- 21.Anania FA, Potter JJ, Rennie-Tankersley L, Mezey E. Effects of acetaldehyde on nuclear protein binding to the nuclear factor I consensus sequence in the α2(I) collagen promoter. Hepatology. 1995;21:1640–1648. [PubMed] [Google Scholar]
- 22.Friedman SL, Roll FJ. Isolation and culture of hepatic lipocytes, Kupffer cells, and sinusoidal endothelial cells by density gradient centrifugation with stractan. Anal Biochem. 1987;161:207–218. doi: 10.1016/0003-2697(87)90673-7. [DOI] [PubMed] [Google Scholar]
- 23.Han DC, Isono M, Chen S, Casaretto A, Hong SW, Wolf G, Ziyadeh FN. Leptin stimulates type I collagen production in db/db mesangial cells: glucose uptake and TGF-beta type II receptor expression. Kidney Int. 2001;59:1315–1323. doi: 10.1046/j.1523-1755.2001.0590041315.x. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi Y, Okimura Y, Mizuno I, Iida K, Takahashi T, Kaji H, Abe H, et al. Leptin induces mitogen-activated protein kinase-dependent proliferation of C3H10T1/2 cells. J Biol Chem. 1997;272:12897–12900. doi: 10.1074/jbc.272.20.12897. [DOI] [PubMed] [Google Scholar]
- 25.Tungtrongchitr R, Pongpaew P, Phonrat B, Tribunyatkul S, Viroonudomphol D, Supawan V, Jintaridhi P, et al. Serum leptin and lipid profiles in Thai obese and overweight subjects. Int J Vitam Nutr Res. 2001;71:74–81. doi: 10.1024/0300-9831.71.1.74. [DOI] [PubMed] [Google Scholar]
- 26.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 27.Anania FA, Womack L, Potter JJ, Mezey E. Acetaldehyde enhances murine α2(I) collagen promoter activity by Ca2+-independent protein kinase C activation in cultured rat hepatic stellate cells. Alcohol Clin Exp Res. 1999;23:279–284. [PubMed] [Google Scholar]
- 28.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 29.Scatchard G. The attraction of proteins for small molecules and ions. Ann N Y Acad Sci. 1949;51:660–673. [Google Scholar]
- 30.Press WH, Flannery BP, Teukolsky SA, Vetterling WT. Numerical Recipes C. Cambridge: Cambridge University Press; 1988. pp. 540–557. [Google Scholar]
- 31.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 32.Janqueira L, Bignolas G, Brentani R. Picrosirius staining plus polarization microscopy: a specific method for collagen detection in tissue sections. Histochem J. 1979;11:447–455. doi: 10.1007/BF01002772. [DOI] [PubMed] [Google Scholar]
- 33.Cohen B, Novick D, Rubinstein M. Modulation of insulin activities by leptin. Science. 1996;274:1185–1188. doi: 10.1126/science.274.5290.1185. [DOI] [PubMed] [Google Scholar]
- 34.Friedman SL. Cytokines and fibrogenesis. Semin Liver Dis. 1999;19:129–140. doi: 10.1055/s-2007-1007105. [DOI] [PubMed] [Google Scholar]
- 35.Gressner AM. Cytokines and cellular crosstalk involved in the activation of fat-storing cells. J Hepatol. 1995;22:28–36. [PubMed] [Google Scholar]
- 36.Mix H, Widjaja A, Jandl O, Cornberg M, Kaul A, Goke M, Beil W, et al. Expression of leptin and leptin receptor isoforms in the human stomach. Gut. 2000;47:481–486. doi: 10.1136/gut.47.4.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lammert A, Kiess W, Bottner A, Glasow A, Kratzsch J. Soluble leptin receptor represents the main leptin binding activity in human blood. Biochem Biophys Res Commun. 2001;283:982–988. doi: 10.1006/bbrc.2001.4885. [DOI] [PubMed] [Google Scholar]
- 38.Ikejima K, Honda H, Yoshikawa M, Hirose M, Kitamura T, Takei Y, Sato N. Leptin augments inflammatory and profibrogenic responses in the murine liver induced by hepatotoxic chemicals. Hepatology. 2001;34:288–297. doi: 10.1053/jhep.2001.26518. [DOI] [PubMed] [Google Scholar]
- 39.Leclercq IA, Farrell GC, Schriemer PJ, Robertson GR. Leptin is required for the development of hepatic fibrosis [Abstract] Hepatology. 2000;32(Suppl):302A. [Google Scholar]
- 40.Gariano RF, Nath AK, D’Amico DJ, Lee T, Sierra-Honigmann MR. Elevation of vitreous leptin in diabetic retinopathy and retinal detachment. Invest Ophthalmol Vis Sci. 2000;11:3576–3581. [PubMed] [Google Scholar]
- 41.Bartoli M, Gu X, Tsai NT, Venema RC, Brooks SE, Marrero MB, Caldwell RB. Vascular endothelial growth factor activates STAT proteins in aortic endothelial cells. J Biol Chem. 2000;275:33189–33192. doi: 10.1074/jbc.C000318200. [DOI] [PubMed] [Google Scholar]
- 42.Takahashi Y, Okimura Y, Mizuno I, Iida K, Takahashi T, Kaji H, Abe H, et al. Leptin induces mitogen-activated protein kinase-dependent proliferation of C3H10T1/2 cells. J Biol Chem. 1997;272:12897–12900. doi: 10.1074/jbc.272.20.12897. [DOI] [PubMed] [Google Scholar]
- 43.Bouloumie A, Marumo T, Lafontan M, Busse R. Leptin induces oxidative stress in human endothelial cells. FASEB J. 1999;13:1231–1238. [PubMed] [Google Scholar]
- 44.Cortez-Pinto H, Camilo ME, Baptista A, De Oliveira AG, De Moura MC. Non-alcoholic fatty liver: another feature of the metabolic syndrome? Clin Nutr. 1999;18:353–358. doi: 10.1016/s0261-5614(99)80015-6. [DOI] [PubMed] [Google Scholar]
- 45.Nakamura T, Nagasaka S, Ishikawa S, Kusaka I, Hayashi H, Saito T, Higashiyama M, et al. Clinical implication of serial leptin measurement in subjects with type 2 diabetes mellitus. Endocr J. 2001;48:87–94. doi: 10.1507/endocrj.48.87. [DOI] [PubMed] [Google Scholar]