

A common biotransformation of arsenic is methylation to monomethylated, dimethylated and trimethylated species, which is catalyzed by the ArsM (or AS3MT) arsenic(III) S-adenosylmethionine methyltransferase. ArsM from the acidothermophilic alga Cyanidioschyzon sp. 5508 was expressed, purified and crystallized by the hanging-drop vapor-diffusion method and diffraction data were collected to 1.76 Å resolution.
Keywords: ArsM, arsenic(III) S-adenosylmethionine methyltransferases, methylation, Cyanidioschyzon sp. 5508
Abstract
Arsenic is the most ubiquitous environmental toxin and carcinogen and consequently ranks first on the Environmental Protection Agency’s Superfund Priority List of Hazardous Substances. It is introduced primarily from geochemical sources and is acted on biologically, creating an arsenic biogeocycle. A common biotransformation is methylation to monomethylated, dimethylated and trimethylated species. Methylation is catalyzed by the ArsM (or AS3MT) arsenic(III) S-adenosylmethionine methyltransferase, an enzyme (EC 2.1.1.137) that is found in members of every kingdom from bacteria to humans. ArsM from the thermophilic alga Cyanidioschyzon sp. 5508 was expressed, purified and crystallized. Crystals were obtained by the hanging-drop vapor-diffusion method. The crystals belonged to the monoclinic space group C2, with unit-cell parameters a = 84.85, b = 46.89, c = 100.35 Å, β = 114.25° and one molecule in the asymmetric unit. Diffraction data were collected at the Advanced Light Source and were processed to a resolution of 1.76 Å.
1. Introduction
Arsenic is the most ubiquitous environmental toxin and carcinogen and consequently ranks first on the Environmental Protection Agency’s Superfund Priority List of Hazardous Substances (http://www.atsdr.cdc.gov/cercla/07list.html). It is introduced primarily from geochemical sources and is acted on biologically, creating an arsenic biogeocycle. Members of every kingdom methylate arsenite in a series of alternating oxidative methylations and reductions, producing the pentavalent species methylarsenate (MAsV), dimethylarsenate (DMAsV) and trimethylarsine oxide (TMAsVO) and the trivalent species MAsIII, DMAsIII and TMAsIII (Challenger, 1951 ▶). Humans and other mammals methylate arsenite to species such as DMAsV, which is excreted in the urine (Thomas et al., 2004 ▶). The mammalian liver enzyme that catalyzes transfer of the methyl group of S-adenosylmethionine to AsIII has been called AS3MT (Styblo et al., 2002 ▶). It is not clear whether methylation in humans is a detoxification process. Because the more toxic trivalent intermediates MAsIII and DMAsIII are formed during liver biotransformation of inorganic arsenate and arsenite, methylation may activate inorganic arsenic to more carcinogenic species.
In contrast, it has been clearly demonstrated that microbial methylation detoxifies arsenic. Genes for orthologues of the human AS3MT are found in bacteria, archaea, fungi and lower plants. These genes have been termed arsM and their protein product has been termed ArsM (arsenite S-adenosylmethyltransferase; Qin et al., 2006 ▶). Since ArsMs are widespread in every kingdom, we have hypothesized that this biotransformation has a significant impact on the global arsenic cycle (Qin et al., 2009 ▶). ArsM (accession No. ACN39191) from the Yellowstone acidothermoacidophilic eukaryotic red alga Cyanidioschyzon sp. 5508 is a 400-residue enzyme [411 residues in the cloned gene after the addition of a His tag (AAALEHHHHHH) at the C-terminus] with a mass of 44 980 Da that methylates AsIII to a final product of volatile TMAsIII. When the algal arsM gene was expressed in an arsenic-hypersensitive strain of Escherichia coli, it conferred AsIII resistance in E. coli in the absence of any other arsenic-resistance genes, demonstrating that methylation is sufficient to detoxify arsenic. Purified ArsM is a thermostable enzyme with a temperature optimum of 333–343 K.
Here, we report the crystallization of two active derivatives of the 400-residue ArsM7 from Cyanidioschyzon sp. 5508, one lacking the C-terminal 28 residues (ArsM7A) and the other lacking these residues plus the N-terminal 31 residues (ArsM7B). ArsM7A crystallized in the monoclinic space group C2, with unit-cell parameters a = 84.85, b = 46.89, c = 100.35 Å, β = 114.25° and one molecule in the asymmetric unit. Diffraction data were collected at the Advanced Light Source and processed to a resolution of 1.76 Å.
2. Materials and methods
2.1. Construction of arsM genes, ArsM expression and purification
Attempts to crystallize full-length ArsM were unsuccessful. From alignment of putative ArsM orthologues, it appears that the algal ArsM has N-termini and C-termini that are not conserved in other members of the group. For this reason, the genes for two catalytically active derivatives lacking the C-terminal 28 residues or both the C-terminal 28 residues and the N-terminal 31 residues were constructed from the sequence of the full-length arsM7 gene (Qin et al., 2009 ▶).
For construction of a plasmid with the arsM7A deletion, a PCR fragment was amplified from the full-length arsM7 gene using the forward primer 5′-GATATACCATGGCGTGCAGCTGTGCGTCTGG-3′ (NcoI site in bold) and the reverse primer 5′-TGCGGCCGCTTCACAGACAAGCTGC-3′ (NotI site in bold) and then cloned into vector plasmid pET28a(+) (Novagen) as an NcoI/NotI digest, generating plasmid pET28-arsM7A, in which the arsM7A gene is under the control of the T7 promoter. This construct encodes an arsM7 gene lacking the last 28 residues but with the last two residues (Cys-Glu) changed to Ser-Gly; the sequence for the same histidine tag was added at the 3′-end. The 383-residue ArsM7A protein has a molecular mass of 42 228 Da.
While the location of the C-terminal truncation was decided by inspection of a multiple alignment, the decision for truncation from the N-terminus was made following limited trypsin digestion of full-length ArsM7. Limited trypsin digestion was performed at room temperature in 50 mM KHPO4 pH 7.4 buffer and 2 mg ml−1 protein. The ArsM7:trypsin ratio was 500:1(w:w). Digestion was initiated by the addition of N-p-tosyl-l-phenylalanine chloromethyl ketone-treated trypsin (Sigma–Aldrich, St Louis, Missouri, USA). Proteolysis was terminated at times between 10 and 300 min by the addition of 0.2% trifluoroacetic acid and was analyzed by 12% SDS–PAGE (Laemmli, 1970 ▶) with Coomassie Blue staining. During this entire period, a fragment of approximately 38 kDa remained resistant to trypsin digestion. Gel slices were excised from the trypsin-resistant band, cut into 1 mm3 cubes and transferred into Eppendorf tubes. The bands were subjected to in-gel trypsin digestion and the peptides were analyzed by mass spectrometry by the Proteomics Facility Core of the Institute of Environmental Health Sciences at Wayne State University, Detroit, Michigan, USA. From this analysis, the sites of limited trypsin digestion were determined to be Lys31 at the N-terminal end and Lys375 at the C-terminal end, indicating a trypsin-resistant core of 344 residues with a mass of 38 185 Da. Since ArsM7A was already truncated at Val370, the gene for an N-terminally truncated derivative was constructed from the arsM7A gene. In this construct, the first 31 residues of the sequence were deleted and the codons for two residues (Met-Ala) were added to the 5′-end, encoding a protein of 354 residues with a mass of 39 223 Da. Plasmid pET28-arsM7A was used as a template for PCR amplification using the forward primer 5′-GATATACCATGGAAACGCTTCAGTCAAG-3′ (NcoI site in bold) and the reverse primer 5′-TGCGGCCGCTTCACAGACAAGCTGC-3′ (NotI site in bold). The PCR product was cloned into pET28a(+) as an NcoI/NotI digest, generating plasmid pET28-arsM7B encoding the doubly N- and C-terminally truncated ArsM. All genes were verified by DNA sequencing. All ArsM proteins were purified as described in Qin et al. (2009 ▶).
2.2. Crystallization
Proteins were exchanged into a buffer consisting of 50 mM MOPS pH 7.0 containing 0.5 M NaCl and 10 mM DTT. Initially, sitting-drop crystallization experiments were set up with 3.0 µl 15 mg ml−1 protein solution in 96-well plates (Corning 3785) at 293 K using a variety of crystal screens from Hampton Research (Aliso Viejo, California, USA), Emerald BioSystems Inc. (Bainbridge Island, Washington, USA) and Jena Bioscience GmbH (Jena, Germany). Drops consisting of 1.5 µl protein solution mixed with 1.5 µl reservoir solution were equilibrated against 100 µl reservoir solution. Crystals were obtained under condition No. 18 of Wizard II from Emerald BioSystems [20.0%(w/v) polyethylene glycol 3000, 0.1 M Tris–HCl pH 7.0 containing 0.2 M calcium acetate]. Optimization of the initial crystal conditions were performed by microseeding for ArsM7A. Initial crystals of ArsM7A were transferred to an Eppendorf tube containing Seed Beads (Hampton Research) and 50 µl reservoir solution. The sample was vortexed for 90 s and three serial dilutions (1:10, 1:100 and 1:1000) were made before seeding. Drops were prepared by mixing 2.5 µl 15 mg ml−1 ArsM7A solution with 2.5 µl seed stock consisting of 20% polyethylene glycol 3000, 0.2 M calcium acetate, 0.1 M Tris–HCl pH 7.0. Diffraction-quality crystals were obtained from 1:100 dilution drops. The ArsM7B construct produced suitable crystals without microseeding by mixing 2.5 µl 15 mg ml−1 ArsM7B solution with 2.5 µl reservoir solution consisting of 18% polyethylene glycol 3350, 0.2 M calcium acetate and 0.1 M Tris–HCl pH 7.0. The hanging drops were equilibrated against 0.5 ml well solution and crystals were obtained at 293 K using Linbro 24-well plates (HR3-110) from Hampton Research.
2.3. Data collection
Crystals were transferred to a cryoprotectant solution (25% polyethylene glycol 3350, 0.2 M calcium acetate, 0.1 M Tris–HCl pH 7.0 and 10% glycerol) and flash-cooled in liquid nitrogen. A native data set was recorded on beamline 5.02 of the Lawrence Berkeley National Laboratory Advanced Light Source. Data were collected using an ADSC Quantum 315r (3 × 3 CCD array) detector at 100 K under a liquid-nitrogen stream. Data obtained from 143 images using a crystal-to-detector distance of 250 mm, 1° oscillation per image and an exposure time per image of 2 s were indexed, integrated and scaled using HKL-2000 (Otwinowski & Minor, 1997 ▶).
3. Results and discussion
Crystallization trials were attempted with native ArsM7, but this protein did not yield crystals. The active derivatives ArsM7A and ArsM7B were purified (Fig. 1 ▶) and diffraction-quality crystals were obtained with both. Initial crystals of ArsM7A diffracted to only 4 Å resolution, but microseeding using small crushed crystals of ArsM7A as seeds was more successful. Crystals appeared within 1 d and reached maximum dimensions of 0.4 × 0.3 × 0.2 mm within 2–3 d (Fig. 2 ▶). Crystals kept for longer than 3 d began to disintegrate in the drop, so newly obtained crystals were used for diffraction. Complete data sets were collected to resolutions of 1.76 and 1.78 Å using single crystals of ArsM7A (Fig. 3 ▶) and ArsM7B, respectively. The data-collection statistics for ArsM7A and ArsM7B are shown in Table 1 ▶. The asymmetric unit contains a single molecule of ArsM7A; the crystal volume per unit molecular weight, V M, was calculated to be 2.22 Å3 Da−1, corresponding to a solvent content of 44.7% (Matthews, 1968 ▶). No structure of a homologous protein is available for molecular-replacement phasing, so crystals with heavy metals or of selenomethionine-labeled protein will be obtained for structure determination.
Figure 1.

ArsM7A was purified by Ni–NTA chromatography as described in Qin et al. (2009 ▶). The SDS–PAGE profile of ArsM7A is shown. Lane 1 contains molecular-weight markers (labeled in kDa). Lanes 2–5 are sequential fractions eluted with an imidazole gradient. Fractions 4 and 5 were used for crystallization trials. The arrow indicates the position of ArsM7A.
Figure 2.

Crystals of ArsM7A were grown in hanging drops as described in §2. The crystal indicated by the arrow has approximate dimensions of 0.4 × 0.3 × 0.2 mm.
Figure 3.

A 1° oscillation image collected from an ArsM7A crystal at the Lawrence Berkeley National Laboratory Advanced Light Source at 100 K. The edge of the oscillation image corresponds to 1.76 Å resolution.
Table 1. Data-collection and processing statistics.
Values in parentheses are for the highest resolution shell.
| ArsM7A | ArsM7B | |
|---|---|---|
| Wavelength (Å) | 0.9795 | 0.9795 |
| Space group | C2 | C2 |
| Unit-cell parameters (Å, °) | a = 84.85, b = 46.89, c = 100.35, β = 114.25 | a = 84.75, b = 47.20, c = 101.68, β = 115.67 |
| Resolution range (Å) | 45.78–1.76 (1.82–1.76) | 50.78–1.78 (1.84–1.78) |
| No. of observations | 99708 | 153634 |
| No. of unique reflections | 34898 (3092) | 34951 (3450) |
| Completeness (%) | 96.7 (86.2) | 99.9 (99.5) |
| Redundancy | 2.9 (2.7) | 4.4 (3.5) |
| 〈I〉/〈σ(I)〉 | 17.51 (2.47) | 37.06 (5.83) |
| Rmerge† (%) | 5.3 (35.2) | 3.4 (19.1) |
R
merge = 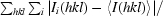
 , where Ii(hkl) is the intensity of the ith measurement of the reflection with Miller indices hkl and 〈I(hkl)〉 is the mean intensity of that reflection.
, where Ii(hkl) is the intensity of the ith measurement of the reflection with Miller indices hkl and 〈I(hkl)〉 is the mean intensity of that reflection.
Acknowledgments
This study was supported in part by US National Institutes of Health Grant GM55425. The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, National Institute of General Medical Sciences and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences of the US Department of Energy under Contract No. DE-AC02-05CH11231.
References
- Challenger, F. (1951). Adv. Enzymol. Relat. Subj. Biochem.12, 429–491. [DOI] [PubMed]
- Laemmli, U. K. (1970). Nature (London), 227, 680–685. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Qin, J., Lehr, C. R., Yuan, C., Le, X. C., McDermott, T. R. & Rosen, B. P. (2009). Proc. Natl Acad. Sci. USA, 106, 5213–5217. [DOI] [PMC free article] [PubMed]
- Qin, J., Rosen, B. P., Zhang, Y., Wang, G., Franke, S. & Rensing, C. (2006). Proc. Natl Acad. Sci. USA, 103, 2075–2080. [DOI] [PMC free article] [PubMed]
- Styblo, M., Drobna, Z., Jaspers, I., Lin, S. & Thomas, D. J. (2002). Environ. Health Perspect.110, Suppl. 5, 767–771. [DOI] [PMC free article] [PubMed]
- Thomas, D. J., Waters, S. B. & Styblo, M. (2004). Toxicol. Appl. Pharmacol.198, 319–326. [DOI] [PubMed]