

The E. coli antitoxin MqsA (YgiT/b3021) has been cocrystallized with mqsRA promoter DNA. A native data set has been collected to a resolution of 2.1 Å.
Keywords: MqsA, Escherichia coli, DNA-binding proteins, antitoxins, protein–DNA complexes
Abstract
The Escherichia coli proteins MqsR and MqsA comprise a novel toxin–antitoxin (TA) system. MqsA, the antitoxin, defines a new family of antitoxins because unlike other antitoxins MqsA is structured throughout its entire sequence, binds zinc and coordinates DNA via its C-terminal and not its N-terminal domain. In order to understand how bacterial antitoxins, and MqsA in particular, regulate transcription, the MqsA protein was cocrystallized with a 26-mer duplex DNA corresponding to the palindromic region of the mqsRA promoter. The merohedrally twinned crystal belonged to space group P41, with unit-cell parameters a = 60.99, b = 60.99, c = 148.60 Å. A complete data set was collected to a resolution of 2.1 Å. The solvent content of the crystal was consistent with the presence of two MqsA molecules bound to the duplex DNA in the asymmetric unit.
1. Introduction
One well recognized but poorly understood mechanism used by bacteria to survive environmental stress is through the formation of persisters, a subpopulation of cells that survive prolonged exposure to antibiotics (Balaban et al., 2004 ▶; Bigger, 1944 ▶) and exhibit multidrug tolerance (Keren et al., 2004 ▶). The detailed molecular events that lead to the persistence phenotype are still elusive. Recent studies have demonstrated that chromosomal toxin–antitoxin (TA) genes are highly upregulated in persister cells (Keren et al., 2004 ▶). TA pairs are composed of two genes organized in an operon that encode an unstable antitoxin and a stable toxin (Magnuson, 2007 ▶). Under normal conditions, the toxin and antitoxin associate to form a tight nontoxic complex. However, under conditions of stress the antitoxins are rapidly degraded by proteases, leaving the stable toxins to exert their toxic effects, ultimately resulting in growth arrest or cell death (Christensen et al., 2003 ▶; Hiraga et al., 1986 ▶; Lehnherr & Yarmolinsky, 1995 ▶). TA complexes also regulate their own transcription by functioning as transcriptional repressor–corepressor complexes, with the antitoxin binding to the promoter DNA within the TA operon and the toxin enhancing DNA binding (Magnuson, 2007 ▶).
The gene most highly upregulated in Escherichia coli persister cells is mqsR (ygiU/b3022; Keren et al., 2004 ▶). Recently, we showed that the mqsRA operon defines a novel family of TA systems in E. coli (Brown et al., 2009 ▶), in which MqsR is the toxin and MqsA is the antitoxin. MqsA, in particular, is unique among all known antitoxins. Firstly, MqsA is the first antitoxin to be described that requires a metal, zinc, for structural stability (Brown et al., 2009 ▶). Secondly, all other canonical E. coli antitoxins whose structures are known have at least one intrinsically unstructured domain and all of them, with the exception of HipB, become ordered upon toxin binding (Schumacher et al., 2009 ▶; Takagi et al., 2005 ▶; Kamada et al., 2003 ▶; Kamada & Hanaoka, 2005 ▶; Kumar et al., 2008 ▶). In contrast, MqsA is well ordered throughout its entire sequence and its structure does not change when bound to MqsR (Brown et al., 2009 ▶). In addition, all other known antitoxins bind DNA via their N-terminal domains (Schumacher et al., 2009 ▶; Takagi et al., 2005 ▶; Kamada et al., 2003 ▶; Kamada & Hanaoka, 2005 ▶; Kumar et al., 2008 ▶). In contrast, MqsA is the first antitoxin shown to bind DNA via its C-terminal domain (Brown et al., 2009 ▶). Finally, in addition to binding MqsA’s own promoter, MqsA and the MqsR–MqsA complex also bind the promoters of genes that play important roles in E. coli physiology, including mcbR and spy (Brown et al., 2009 ▶).
We recently showed that the C-terminal domain of MqsA is both necessary and sufficient for binding DNA, as the MqsA C-terminal domain is capable of shifting DNA corresponding to the mqsRA promoter, PmqsRA, in EMSA assays, while the MqsA N-terminal domain is not (Brown et al., 2009 ▶). However, the structure of the MqsA dimer suggests that the N-terminal domains may also play a role in DNA binding (Brown et al., 2009 ▶). In order to understand the molecular basis of DNA binding and regulation by the MqsA antitoxin, we initiated an effort to crystallize and determine the structure of MqsA bound to PmqsRA DNA. Here, we report the crystallization and preliminary crystallographic analysis of MqsA bound to a 26-mer DNA element derived from the mqsRA promoter.
2. Materials and methods
2.1. Cloning, expression and purification
MqsA (residues 1–131; full-length protein) was produced as described previously (Brown et al., 2009 ▶). Briefly, MqsA was PCR-amplified from genomic DNA and subcloned into a modified pET28a vector which contained an N-terminal His6 tag with a TEV cleavage site (Peti & Page, 2007 ▶). The MqsA protein was expressed in One Shot E. coli BL21 (DE3) cells (Invitrogen) at 291 K for 18 h following induction with 0.5 mM IPTG. The cells were lysed by high-pressure homogenization (Emulsiflex C3, Avestin) followed by centrifugation to remove cell debris. The clarified lysate was applied onto a HisTrap HP Ni2+-affinity column (GE Healthcare) pre-equilibrated with 50 mM Tris pH 8.0, 500 mM NaCl, 5 mM imidazole. The protein was eluted with a 5–350 mM imidazole gradient. The fractions containing MqsA were pooled and digested with TEV protease with overnight dialysis at 277 K against 50 mM Tris pH 7.0, 500 mM NaCl. Following dialysis, the cleaved affinity tag and TEV protease (which also contained a His6 tag) were removed from the MqsA protein using a second Ni2+-affinity purification (Ni–NTA agarose resin, Invitrogen). In a final step, the cleaved MqsA was purified using size-exclusion chromatography using a Superdex 75 26/60 column (GE Healthcare) pre-equilibrated in protein stabilization buffer (10 mM Tris pH 7.0, 50 mM NaCl, 0.5 mM TCEP). The final purified protein sample was concentrated to 7 mg ml−1 and either used immediately or flash-frozen in liquid nitrogen and stored at 193 K until needed.
2.2. Preparation of dsDNA
DNA oligonucleotides (Table 1 ▶) corresponding to the palindromic region of PmqsRA (Yamaguchi et al., 2009 ▶) were synthesized (Integrated DNA Technologies). All DNA oligonucleotides were dissolved in DNA stabilization buffer (10 mM Tris pH 7.5, 50 mM NaCl, 1 mM EDTA) to a final concentration of 1 mM. Various combinations of complementary oligonucleotides were mixed in equal ratios, annealed by heating to 368 K for 5 min and gradually cooled to room temperature (298 K) by reducing the temperature by 1 K min−1. The dsDNAs used for cocrystallization trials with MqsA are reported in Table 2 ▶.
Table 1. PmqsRA oligonucleotides used to generate dsDNA for cocrystallization with MqsA.
| Oligonucleotide† | Length | Sequence (5′→3′)‡ |
|---|---|---|
| F1 | 23 | TAATTAACCTTTTAGGTTATAAC |
| F2 | 26 | TGTAATTAACCTTTTAGGTTATAACT |
| F3 | 25 | GTAATTAACCTTTTAGGTTATAACT |
| F4 | 17 | TTAACCTTTTAGGTTAT |
| Fov | 24 | GCTAATTAACCTTTTAGGTTATAA |
| R1 | 23 | AGTTATAACCTAAAAGGTTAATT |
| R2 | 26 | AGTTATAACCTAAAAGGTTAATTACA |
| R3 | 25 | GTTATAACCTAAAAGGTTAATTACA |
| R4 | 23 | GTTATAACCTAAAAGGTTAATTA |
| R5 | 17 | ATAACCTAAAAGGTTAA |
| Rov | 24 | GCTTATAACCTAAAAGGTTAATTA |
F, forward; R, reverse; ov, the 5′-ends of the primers were engineered to include a GC overhang.
The palindromic sequence is highlighted in bold.
Table 2. dsDNA used in the MqsA–PmqsRA crystallization trials.
The italicized row indicates the dsDNA fragment cocrystal (MqsA–dsDNA-3) used for data collection.
| dsDNA | F/R | DNA ends | MqsA–dsDNA crystals | Resolution (Å) |
|---|---|---|---|---|
| 1 | F1/R1 | Overhang | Y | 3.5 |
| 2 | F1/R4 | Blunt | Y | 3.8 |
| 3 | F2/R2 | Blunt | Y | 2.1 |
| 4 | F2/R3 | Overhang | N | — |
| 5 | F3/R3 | Overhang | Y | ∼15 |
| 6 | F3/R2 | Overhang | N | — |
| 7 | F1/R2 | Overhang | N | — |
| 8 | F1/R3 | Overhang | Y | 7.5 |
| 9 | F2/R1 | Overhang | N | — |
| 10 | F2/R4 | Overhang | N | — |
| 11 | F3/R1 | Overhang | N | — |
| 12 | F3/R4 | Overhang | N | — |
| 13 | F4/R5 | Blunt | Y | 4.5 |
| 14 | Fov/Rov | Overhang | N | — |
2.3. Crystallization and DNA-binding assay
The MqsA–PmqsRA dsDNA complex (hereafter referred to as MqsA–dsDNA) was prepared by incubating 24 µM MqsA with 30 µM PmqsRA dsDNA in protein stabilization buffer. After incubation at room temperature for 30 min, the complex was concentrated tenfold using 3000 Da molecular-weight cutoff centrifugal filters (Millipore) and then immediately subjected to crystallization trials. Multiple cocrystallization trials were performed with MqsA complexed with 14 distinct PmqsRA dsDNA duplexes with varying lengths (17–26 base pairs) and Watson–Crick base-pairing ends (Table 2 ▶). All initial crystallization trials were performed at 277 and 295 K by the sitting-drop vapour-diffusion method using three-well Intelli-Plates and the Phoenix liquid-handling system (Art Robbins). A total of eight sparse-matrix screens and three coarse-grid screens were used for initial crystallization trials, including Wizard I, Wizard II, Cryo I, Cryo II (Emerald BioSystems), PEG/Ion, PEG/Ion 2, Natrix 1 and Natrix 2 (Hampton Research).
Initial crystals of the MqsA–dsDNA-2 complex (dsDNA-2 corresponds to dsDNA entry 2 in Table 2 ▶) formed after 2 d at 277 K in 0.1 M imidazole pH 8.0, 1.0 M sodium citrate. To verify that these crystals contained the MqsA–dsDNA-2 complex, the crystals were harvested, washed twice with reservoir solution and then once with Milli-Q water and finally transferred to an Eppendorf tube containing 5 µl SDS loading buffer. The sample was heated and then run on a 4–12% SDS–PAGE gel together with the appropriate controls (the MqsA–dsDNA-2 protein–DNA complex sample used for the crystallization trials, MqsA alone and the dsDNA-2 duplex alone). In order to visualize the protein and the DNA, the gel was placed in Coomassie stain or in TAE buffer supplemented with ethidium bromide, respectively.
The MqsA–dsDNA-2 crystals initially diffracted to between 4 and 8 Å resolution. Multiple strategies were used to improve the diffraction quality of these crystals. Firstly, both additive screening (ADDit Screen, Emerald BioSystems) and fine screening around the pH and precipitant concentration of five distinct crystallization conditions was performed. In parallel, microseeding (Seed Bead, Hampton Research) into conditions that produced initial crystals, as well as conditions which previously did not lead to crystal formation, was also tested. In addition, because initial crystals were obtained in drops with either 1:1, 1:2 or 2:1(v:v) protein:condition ratios, 3:1 ratios were also screened. Crystal dehydration, in which the crystals were transferred into conditions with increasing concentrations of precipitant prior to harvesting, was also tested. Finally, trials using batch crystallization under oil were also performed. Despite this extensive optimization (consisting of more than 3000 independent trials), the MqsA–dsDNA-2 crystals never diffracted beyond 3.5 Å resolution. Similar results were obtained for the other MqsA–dsDNA crystals that formed (MqsA–dsDNA-1, MqsA–dsDNA-5, MqsA–dsDNA-8 and MqsA–dsDNA-13; see Table 2 ▶). A total of more than 23 000 trials were carried out on the various MqsA–dsDNA complexes. Fortuitously, after 14 d a single merohedrally twinned crystal of the MqsA–dsDNA-3 complex formed in 0.2 M ammonium chloride, 20% PEG 3350 at 277 K in a drop containing 0.2 µl protein and 0.4 µl crystallization condition. In spite of repeated trials, no other crystals formed in this condition and thus we were not able to analyze this crystal by gel electrophoresis prior to diffraction screening. The single crystal obtained diffracted to 2.1 Å resolution and was used for data collection.
2.4. X-ray diffraction data collection and processing
The MqsA–dsDNA-3 crystal was cryoprotected by a short soak in mother liquor supplemented with 20% glycerol and then frozen by direct transfer into liquid nitrogen. X-ray diffraction data were collected on the NSLS X25 microfocus beamline at a wavelength of 0.9788 Å using an ADSC Q315 CCD detector. Data were indexed, integrated and scaled with DENZO and SCALEPACK from the HKL-2000 program package (Otwinowski & Minor, 1997 ▶). The crystal parameters and data-processing statistics are summarized in Table 3 ▶.
Table 3. Data-collection, processing and scaling statistics and twinning analysis.
| Space group | P41 |
| Unit-cell parameters (Å) | a = b = 60.99, c = 148.60 |
| Resolution range (Å) | 50.0–2.10 (2.14–2.10) |
| No. of observations | 211416 |
| No. of unique reflections | 31421 |
| Completeness (%) | 99.5 (93.5) |
| Rmerge† | 0.075 (0.515) |
| Mean I/σ(I) | 29.13 (2.39) |
| Average redundancy | 6.7 (3.5) |
| Estimated twinning fraction (%) | 37.2 |
| Mean |L|‡ | 0.40 |
| Mean |L2|‡ | 0.224 |
R
merge = 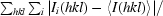
 , where I
i(hkl) is the ith observation of a symmetry-equivalent reflection hkl.
, where I
i(hkl) is the ith observation of a symmetry-equivalent reflection hkl.
Mean |L| = 0.5 and mean |L 2| = 0.333 for untwinned data; mean |L| = 0.375 and mean |L 2| = 0.2 for twinned data.
3. Results and discussion
The crystallization of protein–DNA complexes is highly dependent on the DNA construct, with small changes in nucleotide sequence (i.e. the addition or removal of one nucleotide or the engineering of a GC overhang) making the difference between production of diffraction-quality crystals versus crystals that diffract poorly or simply do not form at all (Tahirov et al., 2001 ▶). We designed 11 forward and reverse oligonucleotides corresponding to the palindromic sequence of PmqsRA (5′-TAACCTTTTAGGTTA-3′; Table 1 ▶). These oligonucleotides allowed us to generate a diverse set of dsDNAs that were used for cocrystallization with MqsA. Fourteen different MqsA–dsDNA complexes were screened for crystal formation against an array of commercially available sparse-matrix conditions (Table 2 ▶).
Crystals suitable for diffraction screening were obtained for six of the 14 complexes (Figs. 1 ▶ a and 2 ▶ a; Table 2 ▶). Prior to diffraction screening and optimization, we verified that the crystals contained the protein–DNA complex as opposed to just DNA or just protein alone. Specifically, the crystals were harvested, extensively washed and then analyzed using SDS–PAGE electrophoresis. As can be seen in Fig. 1 ▶, the crystals contained both dsDNA (Fig. 1 ▶ b) and MqsA protein (Fig. 1 ▶ c), confirming that the crystals did indeed contain the MqsA–dsDNA complex and that the MqsA–dsDNA interaction is sufficiently stable for crystallization. Unfortunately, despite extensive optimization using fine screening, microseeding, additive screening, microbatch crystallization, crystal dehydration and varying the protein:condition ratio, the crystals obtained with dsDNA fragments 1, 2, 5, 8 and 13 never diffracted beyond 3.5 Å resolution.
Figure 1.

Crystals of the the MqsA–dsDNA complex contain both DNA and protein. (a) Crystals of the MqsA–dsDNA-2 complex. (b) Crystals from (a) were washed sequentially in two drops of fresh mother liquor and then one drop of water and were finally transferred to 5 µl SDS–PAGE loading buffer. The sample was heated, run on an SDS–PAGE gel and the gel was stained in TAE buffer supplemented with ethidium bromide to stain for DNA. Lane 1, washed crystals; lane 2, the MqsA–dsDNA-2 protein–DNA complex sample used for crystallization trials; lane 3, dsDNA-2 alone. (c) Crystals treated identically as in (b) except that the gel was transferred to Coomassie stain in order to stain for protein. Lane 1, washed crystals; lane 2, MqsA–dsDNA-2 protein–DNA complex sample used for crystallization trials; lane 3, MqsA protein alone.
Figure 2.

Crystal and diffraction pattern of the MqsA–dsDNA complex used for data collection. (a) The MqsA–dsDNA-3 crystal used for data collection was grown in 0.2 M ammonium chloride, 20% PEG 3350 at 277 K in a drop containing 0.2 µl protein solution and 0.4 µl crystallization solution. (b) A representative diffraction image from the MqsA–dsDNA-3 crystal.
Fortunately, one crystal of MqsA bound to dsDNA-3 (Table 2 ▶; Fig. 2 ▶ a) was obtained which diffracted to 2.1 Å resolution (Fig. 2 ▶ b). The data were initially indexed and scaled in P4, with unit-cell parameters a = b = 60.99, c = 148.60 Å. The program POINTLESS (Evans, 2006 ▶) suggested space group P4122 or P4322. However, when the data were scaled in either P4122 or P4322 more than 11% of the reflections were rejected and the final χ2 values were moderately high (above 3) in the low-resolution shells. Moreover, the presence of one MqsA–dsDNA-3 complex per asymmetric unit in P4122 or P4322 gave a crystal volume per protein weight (V M) of 1.81 Å3 Da−1, with a correspondingly low solvent content of 31.9% (Kantardjieff & Rupp, 2003 ▶; Matthews, 1968 ▶). These observations collectively suggested that the apparent space group was incorrect. The data were then analyzed using phenix.xtriage (Adams et al., 2010 ▶) and the intensity statistics suggested merohedral twinning with an actual space group of P41 or P43 with the following twin law: h, −k, −l. In addition, the presence of one MqsA–dsDNA-3 complex per asymmetric unit in either of these space groups (P41 or P43) gives a V M of 3.6 Å3 Da−1, with a corresponding solvent content of 65.9%, values that are much more realistic for a protein–DNA complex. The data were reprocessed and scaled in P41 with a reported (phenix.xtriage) mean |L| of 0.4 and a mean |H| of 0.136, which were closely related to the values expected for twinned data (0.375 and 0.0, respectively) versus untwinned data (0.5 and 0.5, respectively). The estimated twin fraction was 37.2%. Because of the significant degree of twinning, the data were not detwinned. Rather, the twinned data were used directly for phasing by molecular replacement. A molecular-replacement solution was found in space group P41. Details of structure determination and refinement will be reported elsewhere.
Acknowledgments
This work was supported by an NSF-CAREER award to RP (MCB-0952550) and an NIH F31 NRSA pre-doctoral award to BLB (F31 NS062630). The data for this study were measured on beamline X25 of the National Synchrotron Light Source. Financial support comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US Department of Energy and from the National Center for Research Resources of the National Institutes of Health.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Balaban, N. Q., Merrin, J., Chait, R., Kowalik, L. & Leibler, S. (2004). Science, 305, 1622–1625. [DOI] [PubMed]
- Bigger, J. W. (1944). Lancet, 244, 497–500.
- Brown, B. L., Grigoriu, S., Kim, Y., Arruda, J. M., Davenport, A., Wood, T. K., Peti, W. & Page, R. (2009). PLoS Pathog.5, e1000706. [DOI] [PMC free article] [PubMed]
- Christensen, S. K., Pedersen, K., Hansen, F. G. & Gerdes, K. (2003). J. Mol. Biol.332, 809–819. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Hiraga, S., Jaffe, A., Ogura, T., Mori, H. & Takahashi, H. (1986). J. Bacteriol.166, 100–104. [DOI] [PMC free article] [PubMed]
- Kamada, K. & Hanaoka, F. (2005). Mol. Cell, 19, 497–509. [DOI] [PubMed]
- Kamada, K., Hanaoka, F. & Burley, S. K. (2003). Mol. Cell, 11, 875–884. [DOI] [PubMed]
- Kantardjieff, K. A. & Rupp, B. (2003). Protein Sci.12, 1865–1871. [DOI] [PMC free article] [PubMed]
- Keren, I., Shah, D., Spoering, A., Kaldalu, N. & Lewis, K. (2004). J. Bacteriol.186, 8172–8180. [DOI] [PMC free article] [PubMed]
- Kumar, P., Issac, B., Dodson, E. J., Turkenburg, J. P. & Mande, S. C. (2008). J. Mol. Biol.383, 482–493. [DOI] [PubMed]
- Lehnherr, H. & Yarmolinsky, M. B. (1995). Proc. Natl Acad. Sci. USA, 92, 3274–3277. [DOI] [PMC free article] [PubMed]
- Magnuson, R. D. (2007). J. Bacteriol.189, 6089–6092. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Peti, W. & Page, R. (2007). Protein Expr. Purif.51, 1–10. [DOI] [PubMed]
- Schumacher, M. A., Piro, K. M., Xu, W., Hansen, S., Lewis, K. & Brennan, R. G. (2009). Science, 323, 396–401. [DOI] [PMC free article] [PubMed]
- Tahirov, T. H., Inoue-Bungo, T., Sasaki, M., Fujikawa, A., Kimura, K., Sato, K., Adachi, S., Kamiya, N. & Ogata, K. (2001). Acta Cryst. D57, 854–856. [DOI] [PubMed]
- Takagi, H., Kakuta, Y., Okada, T., Yao, M., Tanaka, I. & Kimura, M. (2005). Nature Struct. Mol. Biol.12, 327–331. [DOI] [PubMed]
- Yamaguchi, Y., Park, J. H. & Inouye, M. (2009). J. Biol. Chem.284, 28746–28753. [DOI] [PMC free article] [PubMed]