

IMP dehydrogenase from C. neoformans and C. gattii has been crystallized in complex with the substrate IMP and the cofactor NAD+.
Keywords: inosine monophosphate dehydrogenase, Cryptococcus neoformans, Cryptococcus gattii
Abstract
Fungal human pathogens such as Cryptococcus neoformans are becoming an increasingly prevalent cause of human morbidity and mortality owing to the increasing numbers of susceptible individuals. The few antimycotics available to combat these pathogens usually target fungal-specific cell-wall or membrane-related components; however, the number of these targets is limited. In the search for new targets and lead compounds, C. neoformans has been found to be susceptible to mycophenolic acid through its target inosine monophosphate dehydrogenase (IMPDH); in contrast, a rare subtype of the related C. gattii is naturally resistant. Here, the expression, purification, crystallization and preliminary crystallographic analysis of IMPDH complexed with IMP and NAD+ is reported for both of these Cryptococcus species. The crystals of IMPDH from both sources had the symmetry of the tetragonal space group I422 and diffracted to a resolution of 2.5 Å for C. neoformans and 2.6 Å for C. gattii.
1. Introduction
Fungal infections of humans are highly refractive to pharmacological intervention because of the similarities in our shared eukaryotic physiology. Emerging drug resistance is further exacerbating this problem, as is the growing cohort of immunocompromised individuals susceptible to opportunistic infections. One such opportunist is Cryptococcus neoformans, a basidiomycete yeast that predominantly infects immunocompromised patients and causes fungal meningoencephalitis (Casadevall & Perfect, 1998 ▶). Recent figures from the US Centers for Disease Control and Prevention estimate an annual global incidence of cryptococcal meningitis of almost 958 000 cases, resulting in more than 624 000 deaths per year. The vast majority of cases involve AIDS patients in sub-Saharan Africa and mirror the prevalence of HIV in the region (Park et al., 2009 ▶). The development of more effective, readily available and cheaper antifungals is therefore a matter of urgency.
Rational drug design was pioneered in the purine metabolic pathway, which has continued to serve as a fertile source of drug targets and therapeutic agents for over fifty years (Elion, 1989 ▶). One of the key well studied enzymes from this pathway is inosine monophosphate dehydrogenase (IMPDH), which is the rate-limiting enzyme and catalyses the first committed step of de novo GTP biosynthesis: the conversion of inosine monophosphate (IMP) to xanthosine monophosphate (XMP). The mechanism involves two reactions: initially, both IMP and the cofactor NAD+ bind and IMP is oxidized, forming the covalent intermediate E–XMP*, while NAD+ is reduced to NADH. The E–XMP* complex undergoes a conformational change and is subsequently hydrolysed to form XMP (Jackson et al., 1977 ▶; Kohler et al., 2005 ▶; Guillen Schlippe & Hedstrom, 2005 ▶; Sintchak et al., 1996 ▶). XMP is then converted to guanosine monophosphate (GMP) via GMP synthase.
As a rate-limiting central metabolic enzyme, IMPDH is highly expressed in proliferating cells and has become a therapeutic target for antimicrobial, antiproliferative and immunosuppressive treatments (Kaur et al., 2005 ▶; Allison & Eugui, 2000 ▶; Chen & Pankiewicz, 2007 ▶). One such therapeutic agent is mycophenolic acid (MPA), an uncompetitive inhibitor of IMPDH that acts as an NAD+ analogue and traps the covalently bound E–XMP* enzyme intermediate.
IMPDH displays a variability in MPA sensitivity across different species; mammalian enzymes are largely sensitive and in humans MPA acts as a potent immunosuppressant via inhibition of B and T lymphocyte proliferation and is commonly prescribed following renal transplantation. Microorganisms, including bacteria, protozoans and fungi, range from extremely sensitive to highly resistant (Hedstrom, 2009 ▶; Zhang et al., 1999 ▶). Treatment with MPA dramatically reduces the intracellular GTP pool and evokes a range of phenotypes, including abnormal cell morphology, polarity and cell-cycle progression, as well as inhibition of translation and G-protein signalling, in the yeast Saccharomyces cerevisiae (Escobar-Henriques et al., 2001 ▶; Sagot et al., 2005 ▶).
MPA inhibits the growth of C. neoformans, yet we have found that a rare subtype of the closely related C. gattii is naturally resistant. Kinetic and structural studies have consistently revealed significant differences between microbial IMP dehydrogenases and their human counterparts. Subtle changes in the enzyme conformation, structure of the active site and structure of the cofactor-binding site can lead to substantial changes in IMPDH kinetic parameters and drug resistance, suggesting that highly species-specific inhibitors that do not affect a human host could be developed as novel therapeutics (McMillan et al., 2000 ▶; Prosise et al., 2002 ▶; Umejiego et al., 2004 ▶; Kohler et al., 2005 ▶; Riera et al., 2008 ▶).
The available structures of eukaryotic IMPDHs include human type I and type II IMPDH and those from Cricetulus griseus (Chinese hamster) and a number of protozoan parasites including Tritrichomonas foetus and Cryptosporidium parvum. The Cryptococcus enzymes (both 544 residues) share ∼70% identity with human type I (563 residues) and type II (514 residues) IMPDH, but only ∼44% identity with the well characterized T. foetus enzyme (503 residues). Cryptococcus IMPDH is slightly longer than most comparable enzymes owing to a unique insertion in part of the active site. No IMPDH structures from the fungal kingdom are available, despite the extensive genetic and biochemical characterization of the enzymes from the model yeast Saccharomyces cerevisiae and the human pathogen Candida albicans (Shaw et al., 2001 ▶; Hyle et al., 2003 ▶; Kohler et al., 2005 ▶).
To investigate the mechanism of resistance and the potential of Cryptococcus IMPDH as an effective therapeutic target, here we describe the crystallization of an MPA-sensitive IMPDH from the clinically prevalent C. neoformans and of a rare naturally occurring MPA-resistant IMPDH from C. gattii.
2. Materials and methods
2.1. Cloning
Total RNA was isolated from C. neoformans var. grubii strain H99 and C. gattii strain MMRL2651 using TRIzol (Invitrogen, USA). Intron-free cDNA was subsequently synthesized using the Bioline cDNA Synthesis Kit (Bioline, UK) and the IMPDH-encoding IMD1 gene from both species was PCR-amplified using primers designed to introduce novel restriction sites. The resulting amplicons were digested and cloned in-frame into the pQE-30 expression vector (Qiagen, Netherlands) to introduce an N-terminal 6×His tag (MRGSHHHHHHGS).
The C. neoformans strain H99 IMPDH nucleotide sequence (locus designation CNAG_00441) is available from the Broad Institute unpublished genome (http://www.broadinstitute.org/annotation/genome/cryptococcus_neoformans/MultiHome.html), while the C. gattii strain MMRL2651 IMPDH sequence is available in GenBank (FJ418781).
2.2. Expression and purification
IMD1 plasmids were cotransformed with the lac repressor vector pREP4 into Escherichia coli strain BL21 (DE3) pLysS (Novagen, Japan). The transformed cells were grown in LB medium at 310 K with 100 µg ml−1 ampicillin and 35 µg ml−1 kanamycin to an OD600 of ∼1.0 and induced with 1 mM IPTG. The cells were subsequently grown for 5 h at 293 K before harvesting. The pellets were resuspended in lysis buffer (50 mM HEPES pH 8.0, 1 M KCl, 30 mM imidazole, 1 mM benzamidine–HCl and 1 mM PMSF) prior to disruption by sonication. The expressed protein was purified via Ni-immobilized metal-affinity chromatography using 5 ml HisTrap columns (GE Healthcare, USA). The supernatant was loaded onto the columns and eluted in a linear gradient of 30–500 mM imidazole. Fractions were analysed via SDS–PAGE; the peak fractions were concentrated and buffer-exchanged into gel-filtration buffer (50 mM Tris–HCl pH 8.0, 150 mM KCl, 2 mM EDTA, 2 mM DTT). Both proteins were loaded onto Superdex 200 size-exclusion columns and eluted at a rate of 2.5 ml min−1 using an ÄKTA FPLC system (GE Healthcare). The peak fractions were again identified via SDS–PAGE, pooled and concentrated to ∼5 mg ml−1 before buffer-exchange into crystallization buffer (50 mM Tris–HCl pH 8.0, 50 mM KCl, 2 mM EDTA, 1 mM DTT).
2.3. Crystallization
The optimal protein concentration for crystallization was 5 mg ml−1 as determined using the Hampton PCT screen (Hampton Research, USA). For the initial screening, only the IMPDH inhibitor MPA (dissolved in dimethylsulfoxide) was added to a concentration of 100 µM prior to crystallization. Crystallization screening for both proteins was performed using hanging-drop vapour diffusion at 277 K and commercial sparse-matrix screens (Qiagen; Hampton Research; Emerald Biosciences, USA). The plates were set up using a Mosquito Nanodrop crystallization robot (TTP LabTech, UK) by combining 200 nl protein solution and 200 nl reservoir solution and inverting the drops over 100 µl reservoir solution. The drops were monitored and imaged using the Rock Imager system (Formulatrix, USA). An initial crystal cluster was observed for C. gattii IMPDH with reservoir solution containing 1.8 M lithium sulfate, 6.8%(v/v) 2-methyl-2,4-pentanediol and 0.09 M imidazole–HCl pH 6.5. The crystals were further optimized using factorial grid screening around this condition using sitting-drop vapour diffusion, varying the protein and salt concentration and the pH. In addition, we used an Additive Screen kit (Hampton Research). 1 µl protein solution was mixed with 1 µl reservoir solution and equilibrated against 100 µl reservoir solution. Small crystals obtained in 1.6–1.9 M lithium sulfate pH 6.4–6.6 were further optimized by using larger drop sizes and microseeding.
For the final round of crystallization, the IMPDH proteins from C. neoformans and C. gattii were mixed with a tenfold molar excess of the substrate IMP and the cofactor NAD+. Large crystals were obtained using wells containing 1.9 M lithium sulfate and 0.09 M imidazole–HCl pH 6.5 plus either 3.0–12.0%(v/v) 2-methyl-2,4-pentanediol, 3.0–12.0% pentaerythritol ethoxylate (15/04 EO/OH) or no additive.
2.4. Data collection and processing
Crystals were mounted in nylon loops and immersed in the cryoprotectant Paratone-N (Hampton Research) before flash-cooling in liquid nitrogen. Diffraction data were collected from cryocooled single crystals on the MX2 undulator beamline of the Australian Synchrotron (Clayton, Australia). Reflections were indexed and integrated using the program XDS (Kabsch, 2010 ▶) and then scaled using SCALA as implemented within the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶).
3. Results and discussion
Both C. neoformans and C. gattii IMPDH were expressed heterologously in soluble form in E. coli after 5 h induction at 293 K. Both proteins appeared as a single band of approximately 58 kDa on reducing SDS–PAGE gels, matching the estimated size of His-tagged IMPDH monomers, and their purity was estimated to be >99%.
Following initial screening, three further rounds of optimization led to large diffraction-quality crystals in 1.9 M lithium sulfate and 0.09 M imidazole–HCl pH 6.5 plus either 3–12% 2-methyl-2,4-pentanediol, 3–12% pentaerythritol ethoxylate (15/04 EO/OH) or no further additive. Crystals of both proteins (Fig. 1 ▶) appeared after 3 d at 277 K.
Figure 1.

Crystals of cryptococcal IMP dehydrogenases in complex with IMP and NAD+, space group I422. (a) C. neoformans IMP dehydrogenase, (b) C. gattii IMP dehydrogenase.
The crystals of IMPDH from the two species visually had different morphologies, with the largest IMPDH crystals growing to maximum dimensions of 100 × 150 × 200 µm. However, the crystals of C. neoformans and C. gattii IMPDH both had the body-centred tetragonal symmetry of space group I422. The unit-cell parameters for the C. neoformans and C. gattii IMPDH crystals were a = b = 148.8, c = 106.4 Å and a = b = 148.4, c = 106.2 Å, respectively. The resolution of the best diffracting crystals was 2.5 Å for C. neoformans IMPDH and 2.6 Å for C. gattii IMPDH. Data-collection and refinement statistics are summarized in Table 1 ▶.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the outer shell.
| C. neoformans IMPDH | C. gattii IMPDH | |
|---|---|---|
| No. of crystals | 1 | 1 |
| Beamline | Australian Synchrotron MX2 | Australian Synchrotron MX2 |
| X-ray wavelength (Å) | 0.95 | 0.95 |
| Detector | ADSC Quantum 315r | ADSC Quantum 315r |
| Crystal-to-detector distance (mm) | 350 | 320 |
| Rotation range per image (°) | 1 | 1 |
| Total rotation range (°) | 180 | 180 |
| Exposure time per image (s) | 2 | 2 |
| Data-collection temperature (K) | 100 | 100 |
| Space group | I422 | I422 |
| Unit-cell parameters (Å, °) | a = b = 148.8, c = 106.4, α = β = γ = 90 | a = b = 148.8, c = 106.2, α = β = γ = 90 |
| Resolution range (Å) | 20–2.5 (2.5–2.42) | 20–2.6 (2.74–2.6) |
| Unique reflections | 20908 | 18484 |
| Total observations | 305209 | 491039 |
| 〈I/σ(I)〉 | 23.6 (4.9) | 22.5 (6.1) |
| Rmerge† (%) | 11.7 (78.1) | 13.5 (79.6) |
| Rmeas = Rr.i.m.‡ (%) | 12.1 (80.9) | 13.8 (81.3) |
| Wilson B factor (Å2) | 46.6 | 52.7 |
| Completeness (%) | 99.8 (100) | 99.8 (100) |
| Mosaicity (°) | 0.40 | 0.41 |
| Multiplicity | 14.6 | 26.6 |
| Monomer molecular weight (kDa) | 57.9 | 57.9 |
R
merge = 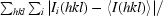
 , where I
i(hkl) is the intensity of an individual measurement of the reflection with Miller indices hkl and 〈I(hkl)〉 is the mean intensity of that reflection. Calculated for I > −3σ(I).
, where I
i(hkl) is the intensity of an individual measurement of the reflection with Miller indices hkl and 〈I(hkl)〉 is the mean intensity of that reflection. Calculated for I > −3σ(I).
R
meas = R
r.i.m. = 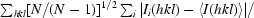 , where I
i(hkl) is the intensity of the ith individual measurement of reflection hkl and 〈I(hkl)〉 is the weighted average intensity of all measurements i of reflection hkl. N is the multiplicity.
, where I
i(hkl) is the intensity of the ith individual measurement of reflection hkl and 〈I(hkl)〉 is the weighted average intensity of all measurements i of reflection hkl. N is the multiplicity.
Current work is focused on refining the structure of IMPDH for both species and obtaining data sets for alternate ligand-bound forms, including inhibitor-bound MPA-resistant and MPA-sensitive IMPDH.
Acknowledgments
The authors would like to acknowledge the UQ ROCX facility (The University of Queensland) and the Australian Synchrotron. This work was supported by grants from the National Health and Medical Research Council (NHMRC) and the Australian Research Council (ARC). CAM is supported by an ANZ Trustees PhD Scholarship in Medical Research. JAF is the recipient of an NHMRC Career Development Award. BK is an ARC Federation Fellow and an NHMRC Honorary Research Fellow.
References
- Allison, A. C. & Eugui, E. M. (2000). Immunopharmacology, 47, 85–118. [DOI] [PubMed]
- Casadevall, A. & Perfect, J. R. (1998). Cryptococcus neoformans. Washington: ASM Press.
- Chen, L. & Pankiewicz, K. W. (2007). Curr. Opin. Drug Discov. Devel.10, 403–412. [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Elion, G. B. (1989). Science, 244, 41–47. [DOI] [PubMed]
- Escobar-Henriques, M., Balguerie, A., Monribot, C., Boucherie, H. & Daignan-Fornier, B. (2001). J. Biol. Chem.276, 46237–46242. [DOI] [PubMed]
- Guillen Schlippe, Y. V. & Hedstrom, L. (2005). Biochemistry, 44, 16695–16700. [DOI] [PubMed]
- Hedstrom, L. (2009). Chem. Rev.109, 2903–2928. [DOI] [PMC free article] [PubMed]
- Hyle, J. W., Shaw, R. J. & Reines, D. (2003). J. Biol. Chem.278, 28470–28478. [DOI] [PMC free article] [PubMed]
- Jackson, R. C., Morris, H. P. & Weber, G. (1977). Biochem. J.166, 1–10. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kaur, R., Klichko, V. & Margolis, D. (2005). AIDS Res. Hum. Retroviruses, 21, 116–124. [DOI] [PubMed]
- Kohler, G. A., Gong, X., Bentink, S., Theiss, S., Pagani, G. M., Agabian, N. & Hedstrom, L. (2005). J. Biol. Chem.280, 11295–11302. [DOI] [PubMed]
- McMillan, F. M., Cahoon, M., White, A., Hedstrom, L., Petsko, G. A. & Ringe, D. (2000). Biochemistry, 39, 4533–4542. [DOI] [PubMed]
- Park, B. J., Wannemuehler, K. A., Marston, B. J., Govender, N., Pappas, P. G. & Chiller, T. M. (2009). AIDS, 23, 525–530. [DOI] [PubMed]
- Prosise, G. L., Wu, J. Z. & Luecke, H. (2002). J. Biol. Chem.277, 50654–50659. [DOI] [PubMed]
- Riera, T. V., Wang, W., Josephine, H. R. & Hedstrom, L. (2008). Biochemistry, 47, 8689–8696. [DOI] [PMC free article] [PubMed]
- Sagot, I., Schaeffer, J. & Daignan-Fornier, B. (2005). BMC Cell Biol.6, 24. [DOI] [PMC free article] [PubMed]
- Shaw, R. J., Wilson, J. L., Smith, K. T. & Reines, D. (2001). J. Biol. Chem.276, 32905–32916. [DOI] [PMC free article] [PubMed]
- Sintchak, M. D., Fleming, M. A., Futer, O., Raybuck, S. A., Chambers, S. P., Caron, P. R., Murcko, M. A. & Wilson, K. P. (1996). Cell, 85, 921–930. [DOI] [PubMed]
- Umejiego, N. N., Li, C., Riera, T., Hedstrom, L. & Striepen, B. (2004). J. Biol. Chem.279, 40320–40327. [DOI] [PubMed]
- Zhang, R., Evans, G., Rotella, F., Westbrook, E., Huberman, E., Joachimiak, A. & Collart, F. R. (1999). Curr. Med. Chem.6, 537–543. [PubMed]