

Native FrpD43–271 and selenomethionine-derivative FrpD43–271 proteins were purified, crystallized and diffracted to resolutions of 2.25 and 2.00 Å, respectively.
Keywords: Fe-regulated protein D, iron-regulated proteins, outer membrane lipoproteins, Neisseria meningitidis
Abstract
Fe-regulated protein D (FrpD) is a Neisseria meningitidis outer membrane lipoprotein that may be involved in the anchoring of the secreted repeat in toxins (RTX) protein FrpC to the outer bacterial membrane. However, the function and biological roles of the FrpD and FrpC proteins remain unknown. Native and selenomethionine-substituted variants of recombinant FrpD43–271 protein were crystallized using the sitting-drop vapour-diffusion method. Diffraction data were collected to a resolution of 2.25 Å for native FrpD43–271 protein and to a resolution of 2.00 Å for selenomethionine-substituted FrpD43–271 (SeMet FrpD43–271) protein. The crystals of native FrpD43–271 protein belonged to the hexagonal space group P62 or P64, while the crystals of SeMet FrpD43–271 protein belonged to the primitive orthorhombic space group P212121.
1. Introduction
Neisseria meningitidis is a Gram-negative bacterium that colonizes the nasopharynx of about 10% of healthy humans. In most instances it colonizes a human host without causing disease; however, occasionally its invasion can lead to devastating invasive meningococcal diseases such as septicaemia and/or meningitis (Rosenstein et al., 2001 ▶). The molecular basis of meningococcal disease remains difficult to analyze because the available animal models do not adequately reproduce the natural route of infection and human pathology. However, several traits that are potentially required for the virulence of meningococci have been identified, including the production of a capsule conferring resistance to serum, the secretion of an IgA protease, the high antigenic variability of pili and of several nonfimbrial adhesins and the presence of several iron-acquisition systems (Tzeng & Stephens, 2000 ▶).
Under conditions of limited iron availability, N. meningitidis produces the iron-regulated proteins FrpC and FrpD, which are encoded consecutively in the iron-regulated frpDC operon controlled by a ferric uptake regulator protein (Fur; Grifantini et al., 2003 ▶). FrpC belongs to the family of type I secreted repeat in toxins (RTX) proteins that are characterized by the presence of a variable number of carboxy-terminal glycine- and aspartate-rich repetitions of the nonapeptide RTX consensus motif (L/I/F)XGGXG(D/N)DX (Thompson et al., 1993 ▶; Osicka et al., 2001 ▶, 2004 ▶). While the biological activity of the meningococcal FrpC protein remains unknown, a number of other RTX proteins have been shown to act as exotoxins that play important roles in the virulence of Gram-negative pathogens (Welch, 2001 ▶).
FrpD is a highly conserved lipoprotein of N. meningitidis that is anchored to the bacterial outer membrane (Prochazkova et al., 2005 ▶). The frpD gene sequence contains two translation-initiation sites, which give rise to the production of the full-length FrpD protein (FrpD1–271) harbouring an N-terminal signal peptide promoting FrpD export across the cytoplasmic membrane by Sec translocase; truncated FrpD protein (FrpD22–271) lacks the signal peptide and remains in the cytoplasm of the bacteria. The exported FrpD1–271 precursor is processed to its mature form on the periplasmic side of the cytoplasmic membrane, sequentially modified by a lipid molecule at the Cys25 residue and sorted as mature lipidated FrpD25–271 to the outer bacterial membrane. The biological function of FrpD appears to be linked to the FrpC protein, as FrpD was found to bind the N-terminal part of FrpC with very high affinity (K d = 0.2 nM; Prochazkova et al., 2005 ▶). This interaction probably occurs outside the cytosol of the bacterial cell. The mechanism of FrpD–FrpC interaction is unknown owing to the absence of any structural information on these proteins. Moreover, the primary amino-acid sequence of FrpD does not exhibit any similarity to known protein sequences of other organisms and therefore a new type of protein fold can be expected.
In the present work, we report progress in the structural study of the FrpD protein, describing the purification, crystallization and diffraction data analysis of native FrpD43–271 and selenomethionine-subsituted (SeMet) FrpD43–271 proteins.
2. Materials and methods
2.1. Plasmid construction
The expression vector (pET28frpD 250) for the production of C-terminally His-tagged FrpD22–271 protein with a truncated N-terminal signal peptide (residues 1–21) has been described previously (Prochazkova et al., 2005 ▶). For the production of the FrpD43–271 protein, the frpD 43–271 open reading frame was amplified by polymerase chain reaction (PCR) with the following primers: the upstream primer 5′-AAACCATGGCTAAAGAACAAACCAGTTTCAAC-3′ containing an NcoI site (italicized) and the downstream primer 5′-AAACTCGAG GCCCTGGAAGTACAGGTTTTCTTTTTTATTTGAATTGTTTACAAAA-3′ containing an XhoI site (italicized) and the nucleotide sequence encoding the tobacco-etch virus (TEV) protease recognition site (ENLYFQG; bold). The purified PCR product was cut with NcoI and XhoI (New England Biolabs, Ipswich, USA) and cloned in pET28b (Novagene, Merck KGaA, Darmstadt, Germany), yielding pET28bfrpD 229. The construct was confirmed by DNA-sequence analysis with an ABI Prism 3130XL analyzer (Applied Biosystems, Foster City, USA) using a Big Dye Terminator Cycle Sequencing Kit.
2.2. Protein expression and purification
For the production of the native FrpD22–271 protein, a culture of Escherichia coli BL21 (λDE3) cells carrying pET28frpD 250 was grown at 310 K in MDO medium supplemented with 60 µg ml−1 kanamycin to an optical density (OD) of 0.6 at 600 nm. The overexpression of FrpD protein was induced by the addition of 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG).
SeMet FrpD43–271 protein was expressed using a modification of a previously described protocol (Van Duyne et al., 1993 ▶). Briefly, E. coli BL21 (λDE3) cells carrying the pET28bfrpD 229 plasmid were grown at 310 K in M9 minimal medium supplemented with 60 µg ml−1 kanamycin to an OD600 of 0.6. At this point, 50 mg selenomethionine (Molecular Dimensions, Suffolk, England) together with 100 mg lysine, 100 mg threonine, 100 mg phenylalanine, 50 mg leucine, 50 mg isoleucine and 50 mg valine were added as solids per litre of culture medium. After 15 min, overexpression of SeMet FrpD43–271 protein was induced by the addition of 1 mM IPTG.
After induction, the bacterial cultures were grown for 4 h and then harvested by centrifugation. The cells were washed in PBS buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.76 mM KH2PO4 pH 7.4), disrupted by sonication (45 W, Misonix Sonicator 3000, Misonix, Farmingdale, USA) on ice and the homogenate was centrifuged at 20 000g at 277 K for 30 min.
To purify the native FrpD22–271 protein, the supernatant after cell disruption was loaded onto an Ni-Sepharose 6 Fast Flow column (GE Healthcare, Chalfont St Giles, England) and washed with 50 mM imidazole in PBS buffer; the protein was eluted by 200 mM imidazole in PBS buffer. The collected fractions were pooled and dialyzed at 277 K overnight against a buffer consisting of 20 mM Tris–HCl pH 7.4 and 50 mM NaCl. The dialyzed sample was further loaded onto a Q-Sepharose Fast Flow column (GE Healthcare, Chalfont St Giles, England) and washed with dialysis buffer; the protein was eluted by 100 mM NaCl in 20 mM Tris–HCl pH 7.4. The eluate was concentrated with an Amicon YM10 ultrafiltration membrane and loaded onto an Ultrapack TSK G-2000SWG gel-permeation column equilibrated with a buffer consisting of 10 mM Tris–HCl pH 7.4, 150 mM NaCl and 0.01% NaN3. The collected fractions were pooled, concentrated to 10 mg ml−1 with an Amicon YM10 ultrafiltration membrane and stored at 277 K.
The SeMet FrpD43–271 protein was initially purified on an Ni-Sepharose column following the same protocol as used for the native protein, with the exception that 5 mM β-mercaptoethanol was added to all buffers. The collected fractions were pooled, mixed with purified recombinant TEV protease [1:30(w:w)] and dialyzed at 277 K overnight against a buffer consisting of 20 mM Tris–HCl pH 7.4, 100 mM NaCl and 5 mM β-mercaptoethanol. A second Ni-Sepharose purification was performed and unbound SeMet FrpD43–271 protein was collected as the flowthrough fraction in 20 mM Tris–HCl pH 7.4, 150 mM NaCl and 5 mM β-mercaptoethanol. The protein was concentrated with an Amicon YM10 ultrafiltration membrane and loaded onto an Ultrapack TSK G-2000SWG gel-permeation column equilibrated with a buffer consisting of 10 mM Tris–HCl pH 7.4, 150 mM NaCl, 5 mM β-mercaptoethanol and 0.01% NaN3. The collected fractions were pooled, concentrated to 12 mg ml−1 with an Amicon YM10 ultrafiltration membrane and stored at 277 K.
Protein concentration was determined by the Bradford assay (Bio-Rad, Hercules, USA) using bovine serum albumin as a standard.
2.3. Protein analyses
In order to control the purity and behaviour of the native FrpD22–271 protein, a comparison of the first batch used for crystallization trials and new freshly isolated batches of the FrpD22–271 protein was performed. SDS–PAGE with a total acrylamide content of 12% in the separating gel was performed. The gels were stained with Coomassie Brilliant Blue R-250 dye (Fluka, Buchs, Switzerland). As shown in Fig. 1 ▶(a), SDS–PAGE analysis revealed a reduction in the molecular weight of the first batch of protein.
Figure 1.

(a) SDS–PAGE of native FrpD protein. Lane 1, molecular-weight markers (kDa); lanes 2 and 3, freshly purified FrpD22–271 and FrpD22–271 truncated to FrpD43–271 after storage at 277 K, respectively. (b) SDS–PAGE of purified SeMet FrpD43–271. Lane 1, molecular-weight markers (kDa); lane 2, purified SeMet FrpD43–271.
The identity of the protein was determined by matrix-assisted laser desorption/ionization time-of-flight (MALDI–TOF/TOF) mass spectrometry using a Bruker Ultraflex III (Bruker Daltonics, Bremen, Germany). Samples were desalted using Michrom MicroTrap cartridges (Unichrom, Minsk, Belarus). The intact molecular mass was measured using sinapinic acid as a matrix. MALDI–TOF analysis of the native FrpD22–271 protein used in crystallization trials revealed that the intact molecular weight of His-tagged FrpD22–271 was about 26 750 Da. This molecular weight is lower than the predicted molecular weight of the His-tagged FrpD22–271 protein (30 068 Da).
N-terminal amino-acid sequencing of the proteins was performed on a Procise 491 protein sequencer (Applied Biosystems, Foster City, USA) according to a standard protocol using methylpiperazine as the coupling amine during eight cycles of automated Edman degradation. N-terminal sequence analysis of this band led to a sequence of 13 amino acids (KEQTSFNNPEPMT) that was identical to a position between amino-acid residues 43 and 55 of the FrpD protein. These data clearly demonstrated that the specific cleavage of 21 amino-acid residues at the N-terminus and six amino-acid residues representing a His6 affinity tag at the C-terminus of the FrpD22–271 protein took place after protein purification, yielding FrpD43–271 protein.
Binding experiments of FrpD and FrpC were performed using an ELISA-based binding assay as described previously (Prochazkova et al., 2005 ▶).
2.4. Crystallization
A sample of FrpD43–271 at a concentration of 10 mg ml−1 in 10 mM Tris–HCl pH 7.4, 150 mM NaCl and 0.01% NaN3 buffer (stored at 277 K) was used for crystallization experiments. Initial crystallization trials were performed using the sitting-drop vapour-diffusion technique (Ducruix & Giegé, 1999 ▶) in CombiClover crystallization plates (Emerald BioSystems, Bainbridge Island, USA) and in CrystalClear P strips (Douglas Instruments, Berkshire, England) at both 277 and 293 K. The commercial screeening kits Crystal Screen and Crystal Screen 2 (Hampton Research, Aliso Viejo, USA), MemStart and MemSys HT-96 (Molecular Dimensions, Suffolk, England), JBScreen Classic 1-10 (Jena Bioscience, Jena, Germany) and Precipitant Synergy Primary 64 (Emerald BioSystems, Bainbridge Island, USA), as well as in-house solutions, were tested to determine initial crystallization conditions. Crystallization drops consisting of 0.3 µl protein solution and 0.3 µl precipitant solution were equilibrated against 50 µl reservoir solution.
Crystallization screening of the SeMet FrpD43–271 protein was performed by the sitting-drop vapour-diffusion technique in Cryschem plates (Hampton Research) at 293 K using protein at a concentration of 7–9 mg ml−1 in buffer consisting of 10 mM Tris–HCl pH 7.4, 150 mM NaCl, 0.01% NaN3 and 5 mM β-mercaptoethanol. Drops consisting of 1 µl protein solution and 1 µl reservoir solution were equilibrated over 700 µl reservoir solution.
2.5. Data collection and processing
Diffraction data were collected on the BESSY beamline MX 14.1 for macromolecular crystallography (Berlin, Germany) equipped with a MAR Mosaic 225 mm detector. The crystals were mounted in nylon loops and flash-cooled in a 100 K liquid-nitrogen stream without any additional cryoprotection. Diffraction data for the native FrpD43–271 crystals were collected to a resolution of 2.25 Å using synchrotron radiation of wavelength 0.918 Å. A set of 120 images was recorded with a 1° oscillation angle, an exposure time of 6 s per image and a crystal-to-detector distance of 260 mm. Single-wavelength anomalous diffraction (SAD) data for the SeMet FrpD43–271 crystals were collected to a resolution of 2.00 Å using synchrotron radiation of wavelength 0.979 Å. A set of 320 images was collected with a 0.5° oscillation angle, an exposure time of 1.5 s per image and a crystal-to-detector distance of 225 mm. The diffraction data were processed using the HKL-3000 program package (Minor et al., 2006 ▶). Crystal parameters and data-collection statistics are summarized in Table 1 ▶.
Table 1. Data-collection statistics for the native FrpD43–271 and SeMet FrpD43–271 crystals.
Values in parentheses are for the highest resolution shell.
| Native FrpD43–271 | SeMet FrpD43–271 | |
|---|---|---|
| Wavelength (Å) | 0.918 | 0.979 |
| Resolution range (Å) | 50–2.25 (2.33–2.25) | 50–2.00 (2.07–2.00) |
| Unit-cell parameters (Å, °) | a = b = 115.4, c = 38.8, α = β = 90, γ = 120 | a = 38.1, b = 38.7, c = 165.5α = β = γ = 90 |
| Space group | P62 or P64 | P212121 |
| Multiplicity | 6.8 (4.4) | 5.9 (4.3) |
| Observed reflections† | 95502 | 100435 |
| Measured unique reflections | 14134 (1277) | 17108 (1487) |
| Completeness (%) | 98.6 (90.6) | 98.2 (88.1) |
| Rmerge‡ (%) | 6.6 (41.1) | 5.5 (11.9) |
| Mean I/σ(I) | 23.0 (2.3) | 77.4 (20.0) |
The criterion used for observed reflections was I/σ(I) > 0.
R
merge = 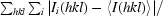
 , where I
i(hkl) is an individual intensity of the ith observation of reflection hkl and 〈I(hkl)〉 is the average intensity of reflection hkl with summation over all data.
, where I
i(hkl) is an individual intensity of the ith observation of reflection hkl and 〈I(hkl)〉 is the average intensity of reflection hkl with summation over all data.
3. Results and discussion
For structural studies, a construct encoding the FrpD22–271 protein was selected. The N-terminal truncation encompasses the 21-amino-acid secretion signal, which is cleaved off during protein transport to the outer membrane of the bacterial cells in vivo. Recombinant expression of this construct in E. coli thus results in a cytosolic product that is not modified by a lipid molecule at Cys25 and that is not anchored to the bacterial membrane. The binding affinity of the FrpD22–271 protein towards FrpC was not impaired by this truncation (Prochazkova et al., 2005 ▶).
Using complex protein analysis (see §2.3), it was found that a specific spontaneous cleavage of 21 amino-acid residues at the N-terminus and six amino-acid residues representing a His6 affinity tag at the C-terminus of the FrpD22–271 protein occurred after protein purification and storage, yielding FrpD43–271 protein. This recombinant iron-regulated FrpD43–271 protein from N. meningitidis was used for crystallization experiments (Fig. 1 ▶ a). Colourless hexagonal-shaped crystals of FrpD43–271 protein grew within 2 d during transport to the synchrotron in a reservoir solution consisting of 0.1 M Tris–HCl buffer pH 8.5 and 2 M ammonium sulfate at 293 K. A diffraction data set was collected to a resolution of 2.25 Å (Fig. 2 ▶ a). The FrpD43–271 protein crystallized in the hexagonal space group P62 or its enantiomorph P64, with unit-cell parameters a = b = 115.4, c = 38.8 Å, α = β = 90, γ = 120°. The calculated Matthews coefficient of 2.62 Å3 Da−1 (Matthews, 1968 ▶) corresponded to a solvent content of 54.3%, indicating the presence of one molecule in the asymmetric unit. Merohedral twinning was excluded by analysis of the cumulative intensity distribution using the Padilla–Yeates algorithm (Padilla & Yeates, 2003 ▶). The complete data-collection statistics are summarized in Table 1 ▶.
Figure 2.

Diffraction images from (a) native FrpD43–271 and (b) SeMet FrpD43–271 crystals. Resolution rings are labelled in Å.
As found previously, crystallization of the FrpD protein might be hampered by the presence of the N-terminal 21 residues of the FrpD22–271 protein; therefore, a new plasmid construct for production of the truncated protein FrpD43–271 was prepared. In this construct, the C-terminal His6 tag was preceded by a tobacco etch virus (TEV) protease recognition site, which was used for the specific removal of the affinity tag during purification of the protein. The ability of the truncated FrpD43–271 protein to bind to the FrpC protein was confirmed using an ELISA-based binding assay. The apparent dissociation constant K d of the FrpD43–271–FrpC complex was found to be about 0.35 nM, closely resembling the values of between 0.16 and 0.24 nM previously observed for the FrpD22–271–FrpC complex (Prochazkova et al., 2005 ▶). This suggested that the N-terminal part of FrpD does not interact with the FrpC protein.
In order to determine the FrpD structure by experimental phasing, SeMet FrpD43–271 protein was prepared (Fig. 1 ▶ b). MALDI–TOF analysis revealed that about 4.5 of the five methionine residues present in FrpD43–271 were substituted by selenomethionine. Single crystals of SeMet FrpD43–271 protein with dimensions of about 0.03 × 0.15 × 0.35 mm grew within 2 d from precipitant solution consisting of 0.1 M Tris–HCl buffer pH 8.5, 20%(w/v) PEG 8000, 20%(v/v) PEG 400 and 0.1 M MgCl2 at 293 K (Fig. 3 ▶). Diffraction data were collected to 2.0 Å resolution (Fig. 2 ▶ b). The SeMet FrpD43–271 protein crystallized in the primitive orthorhombic space group P212121. The crystal had the potential to diffract to higher resolution; however, the detector position was optimized to ensure proper separation of the peaks in order to determine the anomalous signal accurately. Evaluation of the crystal-packing parameters indicated the presence of one molecule in the asymmetric unit, with a solvent content of 44.1% (V M = 2.2 Å3 Da−1). Crystal parameters and data-collection statistics are summarized in Table 1 ▶.
Figure 3.

Crystals of SeMet FrpD43–271 protein.
Structure determination of the SeMet FrpD43–271 protein is currently in progress using the SAD method. The calculated structure will be used as a molecular-replacement search model to determine the structure of the native FrpD43–271 protein.
Acknowledgments
We would like to thank Alexey Bondar, Sona Charvatova, Hana Kubinova and Helena Hessova for excellent technical help. We also thank Dr Uwe Müller and Dr Karthik Paithankar for their assistance with data collection at the BESSY MX 14.1 beamline and Dr Jiri Brynda for his help with data processing. The BESSY II ELISA programme (226716) for access to the BESSY MX 14.1 beamline in Berlin is gratefully acknowledged. This work was supported by the Ministry of Education of the Czech Republic (LC06010 and MSM6007665808) and by the Academy of Sciences of the Czech Republic (AV0Z60870520, AV0Z50520514 and AV0Z40550506). Additionally, ES was supported by the University of South Bohemia (grant GAJU 170/2010/P) and KP was supported by the Grant Agency of the Czech Republic (310/06/P150).
References
- Ducruix, A. & Giegé, R. (1999). Crystallization of Nucleic Acids and Proteins: A Practical Approach, 2nd ed. Oxford University Press.
- Grifantini, R., Sebastian, S., Frigimelica, E., Draghi, M., Bartolini, E., Muzzi, A., Rappuoli, R., Grandi, G. & Genco, C. A. (2003). Proc. Natl Acad. Sci. USA, 100, 9542–9547. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. (2006). Acta Cryst. D62, 859–866. [DOI] [PubMed]
- Osicka, R., Kalmusova, J., Krizova, P. & Sebo, P. (2001). Infect. Immun.69, 5509–5519. [DOI] [PMC free article] [PubMed]
- Osicka, R., Prochazkova, K., Sulc, M., Linhartova, I., Havlicek, V. & Sebo, P. (2004). J. Biol. Chem.279, 24944–24956. [DOI] [PubMed]
- Padilla, J. E. & Yeates, T. O. (2003). Acta Cryst. D59, 1124–1130. [DOI] [PubMed]
- Prochazkova, K., Osicka, R., Linhartova, I., Halada, P., Sulc, M. & Sebo, P. (2005). J. Biol. Chem.280, 3251–3258. [DOI] [PubMed]
- Rosenstein, N. E., Perkins, B. A., Stephens, D. S., Popovic, T. & Hughes, J. M. (2001). N. Engl. J. Med.344, 1378–1388. [DOI] [PubMed]
- Thompson, S. A., Wang, L. L., West, A. & Sparling, P. F. (1993). J. Bacteriol.175, 811–818. [DOI] [PMC free article] [PubMed]
- Tzeng, Y.-L. & Stephens, D. S. (2000). Microbes Infect.2, 687–700. [DOI] [PubMed]
- Van Duyne, G. D., Standaert, R. F., Karplus, P. A., Schreiber, S. L. & Clardy, J. (1993). J. Mol. Biol.229, 105–124. [DOI] [PubMed]
- Welch, R. A. (2001). Curr. Top. Microbiol. Immunol.257, 85–111. [DOI] [PubMed]