

Abstract
The protein encoded by the wild-type p53 proto-oncogene has been shown to suppress transformation, whereas certain mutations that alter p53 become transformation competent. Fusion proteins between p53 and the GAL4 DNA binding domain were made to anchor p53 to a DNA target sequence and to allow measurement of transcriptional activation of a reporter plasmid. The wild-type p53 stimulated transcription in this assay, but two transforming mutations in p53 were unable to act as transcriptional activators. Therefore, p53 can activate transcription, and transformation-activating mutations result in a loss of function of the p53 protein. The inability of the p53 mutant proteins to activate transcription may enable them to be transformation competent.
The wild-type p53 protein functions to suppress transformation, and mutations have been reported that result not only in the loss of this function, but the gain of another function, the ability to actively transform a cell (1-6). In these tissue culture experiments, the expression of the normal p53 gene appears to inactivate the tumorigenic potential of the mutant p53 gene. Other data also suggest that the function of the wild-type p53 gene suppresses transformation. One of the most common genetic alterations that occurs during the development of colorectal tumors is a deletion of the short arm of human chromosome 17, the location of the p53 gene. In two colorectal tumors, both p53 alleles were shown to be altered (7), and the tumors did not make a normal p53 gene product. Additional studies with small cell lung cancer tumors and osteosarcoma samples have shown that these types of tumors also contain genomic rearrangements, deletions, or small mutations in the p53 gene in approximately half of the samples studied (8, 9).
Experiments with cells in culture suggest that p53 has a regulatory role, although the actual function has remained elusive. Both the p53 mRNA and protein have extremely short half lives, and a nuclear targeting signal directs p53 to the nucleus (10-13). In addition, p53 also appears to be involved in regulation of the cell cycle (10). The concentrations of p53 mRNA are decreased in primary cells in culture on contact inhibition and in F9 cells on differentiation with retinoic acid (14). Furthermore, a 10- to 20-fold increase in mRNA and protein is seen in Swiss 3T3 cells within 6 hours of serum stimulation immediately before the onset of DNA synthesis (10).
The amino acid sequence of p53 resembles that of transcription factors (15). The NH2-terminus of p53 is 20% acidic, similar to other acidic domains of transcription factors such as Fos, GAL4, and the glucocorticoid hormone receptor (16-18). This acidic domain is followed by a proline-rich stretch of amino acids analogous to the domain in the transcription factor CTF that is responsible for transactivation (19). Proline-rich sequences have also been found in other mammalian transcription factors including Fos, Jun, Oct-2, and SRF (20). The COOH-terminus of p53 encodes a basic domain that may bind DNA. Although the COOH-terminus of p53 proteins from Xenopus, mouse, and man are not well conserved, 17 of the basic residues are located in similar positions (21). DNA cellulose chromatography indicates that p53 can bind DNA, although a specific DNA binding sequence has not yet been identified (22).
To assay for the possibility that p53 functions as a transcription factor, we made p53-GAL4 fusion proteins with the GAL4 DNA binding domain (amino acids 1–147), which has no transactivation ability (23). Since the DNA sequence targeted by the GAL4 binding domain is known, we could thus anchor p53 to a reporter plasmid (CAT) via this binding domain (24). A similar approach has been used to examine the transcriptional activities of Fos, Myb, and ElA (16, 25, 26). We tested two p53-GAL4 fusion proteins containing p53 amino acids 1–343 or 1–330, followed by a short linker and the GAL4 DNA binding domain (amino acids 4–147) in-frame. The plasmids that encode these fusion proteins were cotransfected with the target plasmid CAT. Our CAT plasmids (derived from pAl0CAT2) contain 23 nucleotides of one SV40 72-bp enhancer, the SV40 21-bp repeats, TATA box, and CAT coding sequences (27). These plasmids have low amounts of CAT expression. At position –155 from the start of transcription, we inserted two or four GAL4 binding sites.
Both p53-GAL4 constructs were able to activate transcription of the CAT gene from 33- to 500-fold in HeLa cells, but only in the presence of the GAL4 DNA recognition sequence (Fig. 1). The CAT activity was increased by increasing the number of DNA binding sites. In addition, the GAL4 DNA binding domain (amino acids 4–147) could not by itself activate transcription in this assay. The entire GAL4 sequence activated transcription as expected, but to a lesser extent than the p53-GAL4 fusions. A p53 plasmid truncated at amino acid 343 had little or no transcriptional activity. These experiments indicated that wild-type p53 could specifically activate expression of CAT when anchored by a DNA binding domain.
Fig. 1.
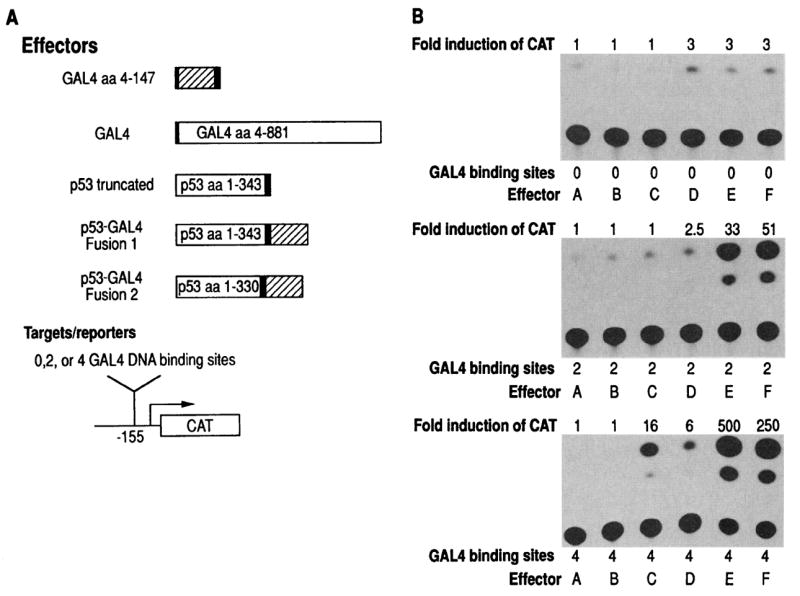
The p53-GAL4 fusion proteins specifically activate transcription of CAT. (A) A diagrammatic representation of the different plasmids used. The effector plasmids contain the NH2-terminal amino acids of p53 and the sequence coding for the GAL4 DNA binding domain; the reporter plasmid contains CAT coding sequences driven by the SV40 promoter. Two individual in-frame p53-GAL4 fusion proteins were made that contain the p53 transactivating domain and the GAL4 DNA binding domain. The p53-GAL4 plasmids consist of the Harvey murine sarcoma virus (H-MSV)–long terminal repeat, wild-type p53 coding sequences from amino acid 1–343 (fusion 1) or amino acids 1–330 (fusion 2) dispersed by p53 introns 2 and 3 and fused to GAL4 sequences (amino acids 4–147). The DNA construct labeled “p53 truncated” contains the H-MSV LTR and encodes only p53 amino acids 1–343. All p53 clones retain the nuclear localization signal of p53. GAL4 (amino acids 4–147) encodes only the GAL4 DNA binding domain. GAL4 (amino acids 4–881) encodes the entire GAL4 protein consisting of both the DNA binding domain and the transactivating domain (31). The GAL4 plasmids also use the H-MSV LTR enhancer-promoter. All regions of fusion were sequenced to ensure that the protein sequences remained in-frame. The CAT reporter plasmid used contains 23bp of the SV40 enhancer, the SV40 21-bp repeats, and the TATA box and CAT sequences in the bluescript vector (27). The oligonucleotide 5′-CTAGACGGAAGACTCTCCTCCGT-3′, which contains the GAL4 recognition sequence bounded by Xba I linkers, was inserted at the Xba I site ofthe CAT plasmid,155 bp upstream of the start of transcription (32). The reporter, CAT, contains either 0,2, or 4 DNA recognition sequences for the GAL4 binding domain. (B) The activator and reporter plasmids (10 μg of each) were cotransfected with a plasmid containing the β-galactosidase gene (5 μg) with calcium phosphate precipitation into HeLa cells essentially as described (33). A β-galactosidase plasmid was used to monitor and normalize for transfection efficiency. The activity ofCAT is measured by the conversion of [14C]chloramphenicol (lower dot) to acetyl and diacetyl chloramphenicol, respectively (34). Effector A, none; effector B, GAL4 (amino acids 4–147); effector C, GAL4; effector D, truncated p53; effector E, p53-GAL4 (fusion 1); and effector F, p53-GAL4 (fusion 2).
Since experiments in tissue culture and analysis of tumors suggested the existence of mutations in p53 that make it transformation competent, we analyzed the effects of these mutations on transcriptional activation. Two well-characterized p53 mutants known to be transformation competent were examined (28). Mutation 1 is a single base pair substitution that changes amino acid 135 from Ala to Val. This single, conservative amino acid substitution allows p53 to cooperate with activated Ras to transform primary rat embryo fibroblasts. The second mutation contains the insertion of a linker in-frame that alters a Val-Pro dipeptide to Pro-Ser-Leu-Ala at position 215 (29). This mutation also produces a p53 protein capable of transformation. Neither of these mutations alters the net negative charge associated with the p53 transactivation domain.
Both mutations were inserted into p53- GAL4 fusion plasmids containing p53 amino acids 1–343 and were cotransfected into HeLa cells with reporter plasmids (Fig. 2). The results of transfecting identical p53-GAL4 fusion plasmids that differ only in the single Ala to Val alteration are shown in Fig. 2A. This mutation completely inhibited the ability of p53 to transactivate expression of CAT. The results of mutation 2 on the ability of p53 to function in transactivation are shown in Fig. 2B. Again, this mutation completely eliminated the capability of the p53-GAL4 fusion protein to activate transcription. Since both mutations do not alter any negative amino acids of p53, these experiments argue that the acidic domain of p53 by itself when attached to a DNA binding domain was insufficient to activate transcription. All the p53 fusion plasmids and GAL4 control plasmids used in these assays have been transcribed and translated in vitro (Fig. 3). All DNA constructions made the appropriate in-frame protein. In addition, to confirm that both wild-type and mutant p53-GAL4 fusion proteins were present in the cells at comparable levels, we performed immunoprecipitation assays of cells transfected with plasmids encoding the p53-GAL4 fusion proteins. The two mutant p53-GAL4 fusion proteins that showed no transactivation activity were actually present at slightly higher concentrations than the wild-type p53-GAL4 (Fig. 4). Within the context of the p53 protein, both p53 mutations described here stabilized the protein and extended the half-life of p53 from 4 to 20 times higher (28).
Fig. 2.
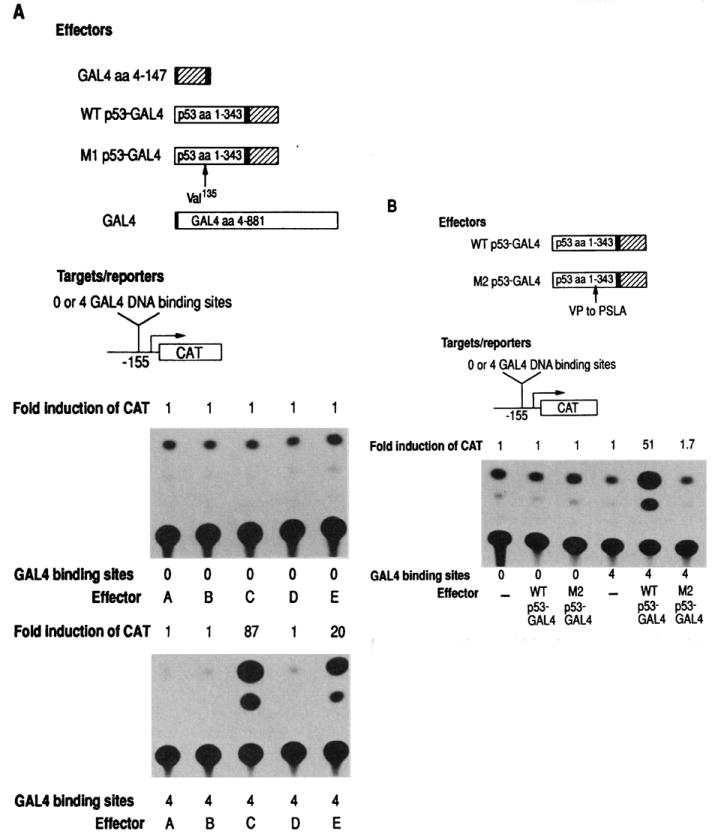
Mutations within the p53-activating domain of the p53-GAL4 fusion protein eliminate transcriptional activity. Mutant 1 (A)and mutant 2 (B)sequences were fused to GAL4 (amino acids 4–147) sequences as in Fig. 1, fusion 1. Plasmids were separately transfected into HeLa cells with the CAT target plasmid and β-galactosidase marker gene as in Fig. 1. In (A) effector A, none; effector B, GAL4 (amino acids 4–147); effector C, wild-type p53-GAL4; effector D, MI p53-GAL4;and effector E,GAL4.
Fig. 3.
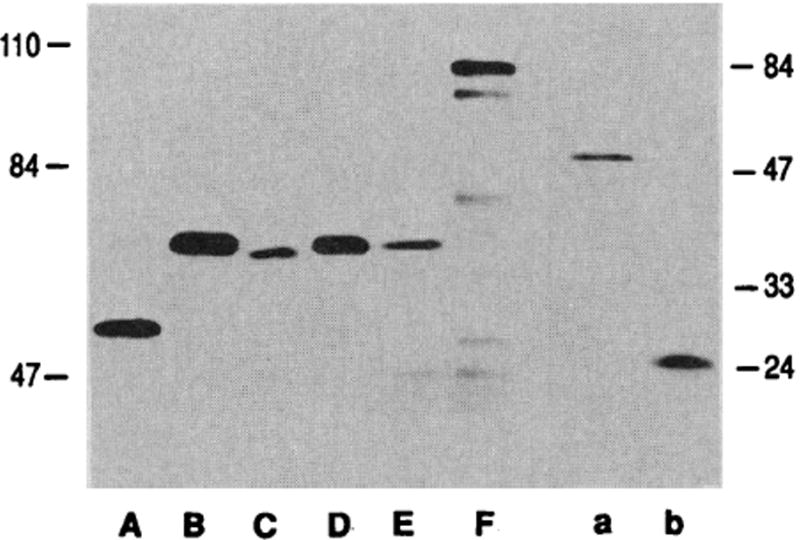
In vitro translation of p53-GAL4 fusion proteins and GAL4 proteins. To ensure that the appropriate proteins were produced, we made RNA in vitro and translated it into protein with rabbit reticulocyte lysate and a mixture of 35S-labeled Cys and 35S-labeled Met according to manufacturer’s instructions. On the left is a 7.5% acrylamide gel with the higher molecular mass proteins. On the right is a 12% acrylamide gel with the wild-type p53 protein product and the small GAL4 DNA binding domain protein. Size markers are shown in kilodaltons on both sides of the figure. Lane A, wild-type p53; lane B, p53-GAL4 (fusion 1); lane C, p53-GAL4 (fusion 2); lane D, p53-GAL4 (Ml); lane E, p53-GAL4 (M2); and lane F, GAL4. Lane a, wild-type p53; and lane b, GAL4 (amino acids 4–147).
Fig. 4.
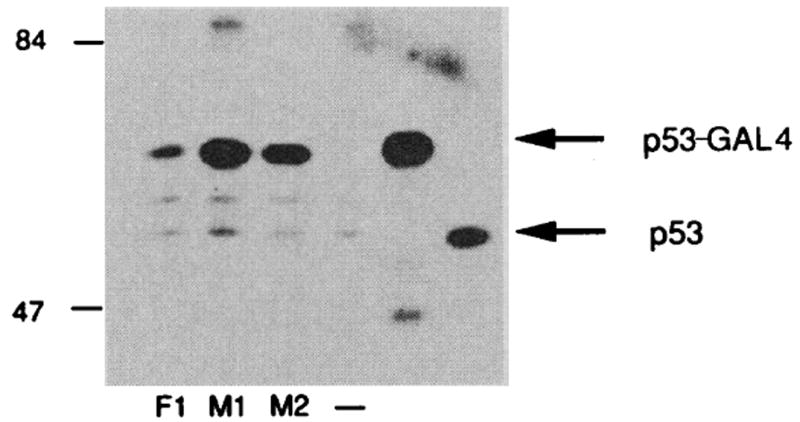
Immunoprecipitation of p53-GAL4 fusion proteins. Plasmids encoding wild-type p53-GAL4 (Fl), p53-GAL4 mutant 1 (Ml), p53-GAL4 mutant 2 (M2), or vector alone (–) were cotransfected with a plasmid containing the β-galactosidase gene into COS cells with DEAE dextran. The β-galactosidase plasmid was used to monitor transfection efficiency. Transfected cells were labeled with a mixture of 35S-labeled Cys and 35S-labeled Met. Immunoprecipitations were performed as described with p53 monoclonal antibody PAb248 and equal amounts of trichloroacetic acid–precipitable counts (28). Also shown are in vitro–translated wild-type p53- GAL4 fusion 1 (lane 5) and p53 mutant 1 immunoprecipitated from cells that overexpress this protein (lane 6). Molecular weight markers are shown in kilodaltons.
We have shown that a direct correlation exists between transcriptional activation by p53 and its ability to suppress transformation. The wild-type p53 protein, a tumor suppressor, activates transcription. Mutations within the p53 protein that result in loss of the suppression phenotype and gain of transformation ability show no transactivation, which correlates well with the fact that p53 is inactive in the transformed state. Deletion analysis and site-specific mutagenesis of the p53 transactivation domain will help pinpoint the precise sequence involved in transactivation. The evidence that p53 is a tumor suppressor and that it activates transcription also implies that the gene or genes being transactivated should function as suppressors of transformation.
Our data can be modeled in two ways. Wild-type p53 could bind directly to DNA and activate transcription. This model is supported by the fact that p53 contains a basic COOH-terminus and that p53 is known to bind DNA cellulose. An alternative model is one in which p53 does not bind DNA directly but interacts with other proteins that bind DNA. In either case, the two mutations analyzed may have major effects on the conformation of p53, since both of these mutations are no longer recognized by the conformation-dependent p53 monoclonal antibody PAb246 (28). These conformational changes might not allow p53 to dimerize or cooperate with other factors to transactivate expression of other genes.
The observation that p53 is involved in the transcriptional machinery is not totally unexpected. Other proto-oncogene products have been shown to be transcriptional activators. The fos and jun oncogenes form dimers and together activate transcription (30). The myb oncogene, which has similarities to p53, has also been shown to activate transcription (25). In both of these examples, the proto-oncogenes c-fos and c-myb activated transcription as well as their transforming counterparts v-fos and v-myb (16, 25). Thus p53 is a paradigm for the model whereby a tumor-suppressing gene is inactivated by loss of its ability to function as a transcriptional activator.
REFERENCES AND NOTES
- 1.Eliyahu D, Raz A, Gruss P, Givol D, Oren M. Nature. 1984;312:646. doi: 10.1038/312646a0. [DOI] [PubMed] [Google Scholar]
- 2.Parada LF, Land H, Weinberg RA, Wolf D, Rotter V. ibid. :649. doi: 10.1038/312649a0. [DOI] [PubMed] [Google Scholar]
- 3.Jenkins JR, Rudge K, Currie GA. ibid. :651. doi: 10.1038/312651a0. [DOI] [PubMed] [Google Scholar]
- 4.Hinds P, Finlay C, Levine AJ. J Virol. 1989;63:739. doi: 10.1128/jvi.63.2.739-746.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finlay CA, Hinds PW, Levine AJ. Cell. 1989;57:1083. doi: 10.1016/0092-8674(89)90045-7. [DOI] [PubMed] [Google Scholar]
- 6.Eliyahu D, Michaelovitz D, Eliyahu S, Pinhasi-Kimhi O, Oren M. Proc Natl Acad Sci USA. 1989;86:8763. doi: 10.1073/pnas.86.22.8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker SJ, et al. Science. 1989;244:217. doi: 10.1126/science.2649981. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi T, et al. ibid. 1989;246:491. [Google Scholar]
- 9.Masuda H, Miller C, Koeffler HP, Battifora H, Cline MJ. Proc Natl Acad Sci U S A. 1987;84:7716. doi: 10.1073/pnas.84.21.7716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reich NC, Levine AJ. Nature. 1984;308:199. doi: 10.1038/308199a0. [DOI] [PubMed] [Google Scholar]
- 11.Reich NC, Oren M, Levine AJ. Mol Cell Biol. 1983;3:2143. doi: 10.1128/mcb.3.12.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rotter V, Abutbul H, Ben-Ze’ev A. EMBO J. 1983;2:1041. doi: 10.1002/j.1460-2075.1983.tb01543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dang CV, Lee WMF. J Biol Chem. 1989;264:18019. [PubMed] [Google Scholar]
- 14.Oren M, Reich NC, Levine AJ. Mol Cell Biol. 1982;2:443. doi: 10.1128/mcb.2.4.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pennica D, et al. Virology. 1984;134:477. doi: 10.1016/0042-6822(84)90316-7. [DOI] [PubMed] [Google Scholar]
- 16.Lech K, Anderson K, Brent R. Cell. 1988;52:179. doi: 10.1016/0092-8674(88)90506-5. [DOI] [PubMed] [Google Scholar]
- 17.Ma J, Ptashne M. ibid. 1987;48:847. doi: 10.1016/0092-8674(87)90081-x. [DOI] [PubMed] [Google Scholar]
- 18.Hollenberg SM, Evans RM. ibid. 1988;55:899. doi: 10.1016/0092-8674(88)90145-6. [DOI] [PubMed] [Google Scholar]
- 19.Mermod N, O’Neill EA, Kelly TJ, Tjian R. ibid. 1989;58:741. doi: 10.1016/0092-8674(89)90108-6. [DOI] [PubMed] [Google Scholar]
- 20.Mitchell PJ, Tjian R. Science. 1989;245:371. doi: 10.1126/science.2667136. [DOI] [PubMed] [Google Scholar]
- 21.Soussi T, Caron de Fromentel C, Mechali M, May P, Kress M. Oncogene. 1987;1:71. [PubMed] [Google Scholar]
- 22.Steinmeyer K, Deppert W. ibid. 1988;3:501. [PubMed] [Google Scholar]
- 23.Keegan L, Gill G, Ptashne M. Science. 1986;231:699. doi: 10.1126/science.3080805. [DOI] [PubMed] [Google Scholar]
- 24.Kakidani H, Ptashne M. Cell. 1988;52:161. doi: 10.1016/0092-8674(88)90504-1. [DOI] [PubMed] [Google Scholar]
- 25.Weston K, Bishop JM. ibid. 1989;58:85. doi: 10.1016/0092-8674(89)90405-4. [DOI] [PubMed] [Google Scholar]
- 26.Lillie JW, Green MR. Nature. 1989;338:39. doi: 10.1038/338039a0. [DOI] [PubMed] [Google Scholar]
- 27.Laimins LA, Gruss P, Pozzatti R, Khoury G. J Virol. 1984;49:183. doi: 10.1128/jvi.49.1.183-189.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finlay CA, et al. Mol Cell Biol. 1988;8:531. doi: 10.1128/mcb.8.2.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan T-H, Wallis J, Levine AJ. J Virol. 1986;59:574. doi: 10.1128/jvi.59.3.574-583.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rauscher FJ, Sambucetti LC, Curran T, Distel RJ, Spiegelman BM. Cell. 1988;52:471. doi: 10.1016/s0092-8674(88)80039-4. [DOI] [PubMed] [Google Scholar]
- 31.Laughon A, Gesteland RF. Mol Cell Biol. 1984;4:260. doi: 10.1128/mcb.4.2.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giniger E, Vamum SM, Ptashne M. Cell. 1985;40:767. doi: 10.1016/0092-8674(85)90336-8. [DOI] [PubMed] [Google Scholar]
- 33.Chen C, Okayama H. Mol Cell Biol. 1987;7:2745. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gorman CM, Moffat LF, Howard BH. ibid. 1982;2:1044. doi: 10.1128/mcb.2.9.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.We thank A. J. Levine for availability of p53 plasmids before publication and P. Silver for GAL4 plasmids. We also thank B. de Crombrugghe and G. Karsenty for helpful discussions. Support was provided by NIH grants CA47296 and CA16672, and March of Dimes grant 5–740.