

Abstract
The nicotinic acetylcholine receptors (nAChR) assembled from α4 and β2 subunits are the most densely expressed subtype in the brain. Concentration-effect curves for agonist activation of α4β2*-nAChR are biphasic. This biphasic agonist sensitivity is ascribed to differences in subunit stoichiometry. The studies described here evaluated desensitization elicited by low concentrations of epibatidine, nicotine, cytisine or methylcarbachol of brain α4β2-nAChR function measured with acetylcholine stimulated 86Rb+ efflux from mouse thalamic synaptosomes. Each agonist elicited concentration-dependent desensitization. The agonists differed in potency. However, IC50 values for each agonist for desensitization of 86Rb+ efflux both with high (EC50≈3 μM) and low (EC50≈ 150 μM) acetylcholine sensitivity were not significantly different. Concentrations required to elicit desensitization were higher that their respective KD values for receptor binding. Even though the two components of α4β2*-nAChR mediated 86Rb+ efflux from mouse brain differ markedly in EC50 values for agonist activation, they are equally sensitive to desensitization by exposure to low agonist concentrations. Mice were also chronically treated with nicotine by continuous infusion of 0, 0.5 or 4.0 mg/kg/hr and desensitization induced by nicotine was evaluated. Consistent with previous results, chronic nicotine treatment increased the density of epibatidine binding sites. Acute exposure to nicotine also elicited concentration-dependent desensitization of both high sensitivity and low sensitivity acetylcholine-stimulated 86Rb+ efflux from cortical and thalamic synaptosomes. Although chronic nicotine treatment reduced maximal 86Rb+ efflux from thalamus, IC50 values in both brain regions were unaffected by chronic nicotine treatment.
Keywords: nicotinic acetylcholine receptor, desensitization, nicotine, epibatidine, cytisine, methylcarbachol
1. Introduction
Prolonged or repeated exposure of nicotinic acetylcholine receptors (nAChR) to nicotine or other agonists elicits desensitization of receptor function (reviewed by Quick and Lester [1]). Desensitization is observed for many nAChR subtypes, but details of the desensitization differ among subtypes [2]. Desensitization can be elicited by exposure to agonist concentrations considerably lower than the concentrations that elicit receptor activation [1-5]. However, there is often a concentration range at which activation occurs before desensitization is complete leading to an overlap between the desensitization and activation concentration-effect curves resulting in a residual or persistent activity analogous to the “window current’ observed for voltage gated ion channels [3]. This overlapping concentration range is observed in the presence of continuous agonist exposure. The residual activity could be a particularly important contributor to the effects of nicotinic agonists encountered with the use of tobacco products or the administration of nicotinic agents therapeutically.
Desensitization of the most widely expressed subtype in the brain, α4β2*-nAChR, has been investigated in heterologous systems. Reversible time and concentration dependent desensitization following agonist exposure has been observed [2, 4, 6]. We have studied agonist-induced desensitization of nAChR mediated 86Rb+ efflux from mouse brain synaptosomes [7-8] and have also observed that treatment with agonist concentrations generally lower than those required to stimulate 86Rb+ efflux elicits desensitization. The range of concentrations over which the persistent activity was observed differed among the agonists with the range being significantly wider for partial agonists such as cytisine [8].
Agonist stimulation of α4β2-nAChR is biphasic. Studies using heterologous expression systems have demonstrated that the component of agonist stimulated responses activated at lower concentrations is mediated by α4β2-nAChR with a stoichiometry of (α4)2(β2)3, while responses activated at higher concentrations are mediated by receptors with a stoichiometry of (α4)3(β2)2 [9-12]. ACh-stimulated 86Rb+ efflux in mouse brain is also biphasic and in most brain regions, including cortex and thalamus, both components are eliminated by deletion of either the α4 [13] or β2 [14] subunits. Changes in the relative proportion of high sensitivity (HS) and low sensitivity (LS) components observed for mice heterozygotic for α4 or β2 gene expression are consistent with the hypothesis that these components reflect the activity of receptors with different α/β stoichiometries [15], although the α5 subunit contributes to the HS activity, particularly in thalamus [16]. Our previous studies of agonist-induced desensitization were conducted before the biphasic nature of agonist-stimulated 86Rb+ efflux was appreciated and used only relatively low agonist concentrations to activate the efflux. Consequently, desensitization of the LS component of the response was not investigated. Such an investigation would evaluate whether the concentration of agonists required to desensitize the HS and LS components is similar, reflecting affinity at the ligand binding sites, or different reflecting the affinities for receptor activation. The outcome of this investigation would also provide information about potential for persistent activity of the LS component.
Chronic nicotine treatment alters the expression and activity of nAChR in mouse brain [17], but the relative sensitivity to desensitization by nicotine of the α4β2*-nAChR component with high sensitivity (HS) and that with low sensitivity (LS) to agonist activation in mouse brain following chronic nicotine treatment has not been investigated. Chronic treatment does not alter the KD for nicotine and other ligands for binding to the high affinity agonist binding sites [17-19]. It is generally believed that high affinity agonist binding is to the desensitized state of the receptor and that functional desensitization of the receptors by exposure to subactivating concentrations of agonists also involves interaction with these high affinity sites resulting in a decrease in ground-state, activatable receptors. Consequently, we measured the activation by ACh and desensitization by nicotine of 86Rb+ efflux from cortical and thalamic synaptosomes of mice chronically treated with nicotine.
2. Materials and Methods
2.1 Materials
NaCl, KCl, MgSO4, CaCl2, glucose and sucrose were obtained from Fisher Scientific, Pittsburgh, PA. Nicotine free base, nicotine tartrate, methycarbachol chloride, acetylcholine iodide, cytisine, (±)-epibatidine, atropine sulfate, tetrodotoxin, bovine serum albumin, diiosopropylfluorophosphate (DFP) and polyethylenimine were obtained from Sigma Chemical Co., St. Louis, MO. HEPES and NaHEPES, products of BDH, were obtained from VWR International, West Chester, PA. Glass fiber filters (Type GB) were obtained from Micro Filtration Systems, Dublin, CA and (Type A/E) were obtained from Gelman Sciences, Ann Arbor, MI. [3H]Nicotine (78.4 Ci/mmol), [3H] cytisine (38.4 Ci/mmol), [3H] methylcarbachol (78.0 Ci/mmol), [3H] epibatidine (48 Ci/mmol), [125I] epibatidine (2200 Ci/mmol) and 86RbCl (average initial specific activity 15 Ci/mg) as well as Optiphase Supermix scintillation cocktail were purchased from Perkin-Elmer NEN, Boston, MA. BudgetSolve scintillation cocktail was obtained from Research Products International, Arlington Heights, IL.
2.2 Mice
C57BL/6J mice were bred at the Institute for Behavioral Genetics. Following weaning, five mice were housed in each cage and allowed free access to food (Teklad Lab Blox) and tap water. Animals were housed in a vivarium maintained at 23±2° C with lights on between 7 AM and 7 PM. Experimental procedures were evaluated and approved by the Animal Care and Utilization Committee of the University of Colorado, Boulder.
2.3 Surgery and Chronic Nicotine Treatment
Cannulas constructed of silastic tubing were inserted into the right jugular veins of female C57BL/6J mice. Briefly, mice were anesthetized by intraperitoneal injection of pentobarbital (55 mg/kg) and chloral hydrate (100 mg/kg). An incision was made and the right jugular vein was exposed. A small hole was made in the vein and a cannula filled with isotonic saline containing 0.3% citric acid was inserted 8 mm into the vein and secured to the underlying tissue with surgical thread. The cannula tubing was subsequently passed through the skin at the nape of the neck and secured with a stainless steel wound clip. A head mount constructed of polyacrylic dental cement was affixed to the skull. The mouse was gently warmed until fully awake and active.
Following recovery from surgery, each mouse was transferred to a 15 cm × 15 cm Plexiglas cage and its cannula was attached to tubing connected to a 1 ml glass syringe mounted on a Harvard Infusion Pump. Sterile saline was continuously infused at a rate of 35 μl/hr. Following two days of saline infusion, nicotine treatment was begun. Final nicotine treatment doses of 0, 0.5 or 4.0 mg/kg/hr (free base) were administered to three groups of mice for 8 days. The nicotine solutions were prepared from liquid nicotine dissolved in sterile saline and neutralized with HCl. Flow rates were maintained at 35 μl/hr.
2.4 Tissue Preparation
Following the 8-day treatment, each mouse was disconnected from the infusion pump, its cannula was checked for free flow, and following a 2-hr period (to allow metabolism of nicotine) it was killed by cervical dislocation. The brain was removed and placed on an ice-cold platform. Thalamus was dissected from naïve mice and thalamus and cerebral cortex were dissected from nicotine-treated mice.
Brain regions were placed in ice-cold 0.32 M sucrose buffered to pH 7.4 with HEPES and homogenized by hand in a glass-Teflon tissue grinder. This homogenate was centrifuged at 20000 × g for 20 min. The resulting pellet was resuspended in isotonic salt solution (NaCl, 140 mM; KCl, 1.5 mM; CaCl2, 2 mM; MgSO4, 1 mM; glucose, 25 mM; HEPES(½Na), 25 mM; pH=7.5). This suspension of crude synaptosomes was used for measuring 86Rb+ efflux.
Synaptosomes remaining that were not used for the functional assay were used for ligand binding. These synaptosomes were lysed by dilution in hypotonic buffered salt solution (NaCl, 14 mM; KCl, 0.15 mM; CaCl2, 0.2 mM; MgSO4, 0.1 mM; HEPES(½Na), 2.5 mM; pH=7.5) and centrifuged at 20000 × g for20 min. The resulting pellet was resuspended in hypotonic salt solution and centrifuged at 20000 × g for 20 min. Following three more cycles of resuspension and centrifugation, the pellets were stored frozen underneath fresh hypotonic buffer until assay.
2.5 86Rb+ Efflux
86Rb+ efflux was measured essentially as described previously [14, 17]. Samples were loaded with 86Rb+ by incubating a 25μl aliquot of the crude synaptosomal suspension with 10 μl of buffer containing approximately 4 μCi of carrier-free 86Rb+ for 25 min at 22°C. Following this incubation, DFP was added to a final concentration of 10 μM and the incubation was continued for 5 more min to irreversibly inhibit AChE. Subsequently samples were filtered onto an 8-mm glass fiber filter disk and washed once with 500 μl of buffer. The filter paper containing the loaded synaptosomes was transferred to a platform and perfusion buffer (NaCl, 135 mM; CsCl, 5 mM; KCl, 1.5 mM; CaCl2, 2 mM; MgSO4, 1 mM; glucose, 25 mM; tetrodotoxin, 50 nM; atropine, 1 μM; bovine serum albumin, 0.1%; HEPES(½Na), 25 mM; pH=7.5) was applied to the top of the filter at a flow rate of 2.5 ml/min with a peristaltic pump. Buffer was actively removed from the bottom of the filter with a second peristaltic pump running at a flow rate of 3.5 ml/min. Samples were superfused with buffer for 7 min before 86Rb+ efflux was monitored by passing the effluent through a 200 μl Cherenkov cell mounted in an IN/US β-ram radioactivity detector. The efflux was monitored for 3 min with data points collected every 3 sec. nAChR mediated efflux was elicited by exposing the samples to ACh for 5 sec. Each sample was stimulated only once with one of the following ACh concentrations: 0.1, 0.3, 1.0, 3.0, and 10. 30, 100, 300, 1000 or 3000 μM. In order to evaluate the effects of prior exposure to low concentrations of the agonists nicotine, epibatidine, methylcarbachol or cytisine samples were exposed to a single concentration of the agonist during the prewash and the entire sample collection period (an 8-min treatment) before stimulation by ACh.
Time courses for the desensitization produced by exposure to each of the four agonists were determined by exposing samples to one of two different concentrations of each agonist for various times up to 8 min. Responses were measured following stimulation with either 30 μM or 1000 μM ACh.
2.6 [125I]Epibatidine Binding
The binding of [125I]epibatidine was measured using a modification of previously published procedures [17, 20]. Final incubation volume was 30 μL and the incubations were conducted in 96-well polystyrene plates with buffer of the same composition as that used to measure 86Rb+ efflux. The specific activity of the [125I]epibatidine was adjusted from 2200 Ci/mmol to 220 Ci/mmol by dilution with unlabeled I-epibatidine (a gift from Kenneth Kellar, Georgetown University, Washington, D.C.). Samples were incubated for 3 hr at 22°C. Blanks were determined by including 100 μM nicotine in some incubations. The binding reaction was terminated by filtration of the samples onto glass fiber filters that had been soaked in 0.5% polyethylenimine using an Inotech Cell Harvester. Two filters were used to collect the samples: The top filter was type MFB and the bottom filter was type A/E. Filters were then washed five times with an isotonic salt solution buffered to pH=7.5 with HEPES. Radioactivity was measured at 80% efficiency with a Packard Cobra Autogamma counter.
Inhibition of [3H]epibatidine binding by cytisine, epibatidine, methylcarbachol or nicotine was determined by including one of several concentrations of the potential inhibitor in the incubation. Inhibition studies were done using 200 pM [3H]epibatidine to determine ligand affinity at higher affinity [125I]epibatidine binding sites.
2.7 Time Courses for [3H]Epibatidine, [3H]Nicotine, [3H]Cytisine and [3H]Methylcarbachol Binding
Particulate fractions were prepared from homogenates of whole C57BL/6 mouse brains. All samples were arranged in a 96 well format. Final incubation volumes of 100 μL were used for [3H]nicotine, [3H]cytisine and [3H]methylcarbachol and a final incubation volume of 500 μL was used for [3H]epibatidine. Incubations were conducted at room temperature (22°C) in an isotonic salt solution (NaCl, 140 mM; KCl, 1.5 mM; CaCl2, 2 mM; MgSO4, 1 mM; HEPES(½Na), 25 mM; pH=7.5) and were initiated by the addition of homogenate. Incubation times were: 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3,, 4, 5, 10, and 20 min. The binding reaction was terminated by filtration using the Inotech harvester as described above. Samples were washed five times with ice-cold binding buffer. Nonspecific binding was determined by including 10 μM nicotine in the incubation. Filters were placed in 4 ml vials and radioactivity was measured at 45% efficiency using a Packard 1600 liquid scintillation spectrometer after the addition of Budget Solve scintillation cocktail. Association kinetics was measured using two concentrations of each ligand, one near the KD and a second higher concentration. [3H]Nicotine was purified as described previously [21].
2.8 Data Calculations
The algorithms in Sigma Plot were used for all curve fitting.
ACh-stimulated 86Rb+ efflux was expressed as the increase in signal above basal efflux. A non-linear least squares curve fit to a first order equation (Ct = C0*e-kt), where Ct is the basal efflux counts at time, t, C0 is the estimated efflux counts at t = 0 sec, and k is the first order decay constant) was used to estimate basal efflux for each sample. Counts in fractions preceding and following the peak were used for curve fitting. ACh-stimulated efflux was calculated by summing the counts in the fractions exceeding basal efflux during ACh exposure and dividing by the corresponding basal efflux counts. This value represents total peak relative to baseline. Concentration effect curves for ACh-stimulated 86Rb+ efflux were calculated for all data sets individually using a two-component model:
where, EACh is the total 86Rb+ efflux measured at each concentration of ACh, EHS and ELS are the maximal rates of 86Rb+ efflux with higher and lower sensitivity to stimulation by ACh (ECHS and ECLS, respectively).
EHS and ELS values were calculated for each concentration of nicotine, cytisine, epibatidine and methycarbachol to which the samples were exposed for 8 min (approximately equilibrium) and IC50 values were calculated using the following equation:
where EC is the 86Rb+ efflux measured for either the higher or lower sensitivity component after preexposure to an agonist concentration, C, and Emax is the maximal efflux measured when C = 0.
The complete data set at equilibrium was also subjected to curve fitting using the following equation:
where EACh is the amount of 86Rb+ efflux measured at a given concentration of ACh and prexposing agonist, C (other parameters as defined above). These curve fits were conducted using each individual datum point.
When results for chronically treated mice were analyzed, data from each brain region and nicotine treatment dose were initially calculated independently, followed by constraints on the variables within and between the treatment groups.
Inhibition of [125I]epibatidine binding by cytisine, nicotine, epibatidine and methycarbachol was measured using an[125I]epibatidine concentration of 400 pM Since agonist inhibition of at high affinity [125I]epibatidine binding is heterogeneous owing to multiple binding sites, the data were fit using the following equation:
Where BI is binding at inhibitor concentration, I, and BH is the density of high affinity and BL is the density of low affinity sites in the absence of inhibitor and IC50-H and IC50-L are the concentration yielding 50% inhibition, respectively. Apparent KI values were subsequently calculated from the IC50 values using the Cheng-Prussoff equation.
Time courses for association of [3H]nicotine, [3H]cytisine, [3H]methylcarbachol and [3H]epibatidine were fit to a two component model as described previously [22] [7-8] [23]) using the equation:
where binding at any time, t (Bt) is described by the sum of binding occurring rapidly (Bf) with an apparent rate constant, k1 and binding occurring slowly (Bs) with an apparent rate constant, k2.
Time courses for the onset of desensitization elicited by exposure to nicotine, cytisine, methycarbachol and epibatidine were fit to the following equation:
Where Et is the 86Rb+ efflux measured at time t, Ed is the total amount of efflux desensitized at the test agonist concentration occurring with a rate constant, k, and Er is the residual efflux not desensitized under those conditions.
SPSS was used for statistical analyses.
3. Results
3.1 Working Model For Evaluation of Desensitization of HS and LS ACh-Stimulated 86Rb+ Efflux
The schemes in Figure 1 illustrate the approach used to investigate the desensitization of ACh-stimulated 86Rb+ efflux in mouse brain synaptosomes. The biphasic concentration-effect curve for ACh-stimulated 86Rb+ efflux is shown in Figure 1A. The concentration effect curve can be resolved into two components with different sensitivity to ACh stimulation. In this illustration an EC50 value of 2.2±0.5 μM was calculated for the high-sensitivity (HS) component and an EC50 value of 120±35 μM was calculated for the low sensitivity (LS) component. The investigation of the desensitization of the HS and LS components was based on a Katz and Thesleff model [24] for both the HS and LS components as illustrated by the cyclical schemes in Figure 1B. The results to follow focus on the desensitization elicited by exposure to relatively low agonist concentrations at steady-state. It should be noted that equilibrium constants are shown rather than rate constants since most of the studies were conducted at or near equilibrium.
Figure 1. Concentration-Effect Curve for ACh Simulated 86Rb+ Efflux and Katz-Thesleff Models for HS and LS Activity.
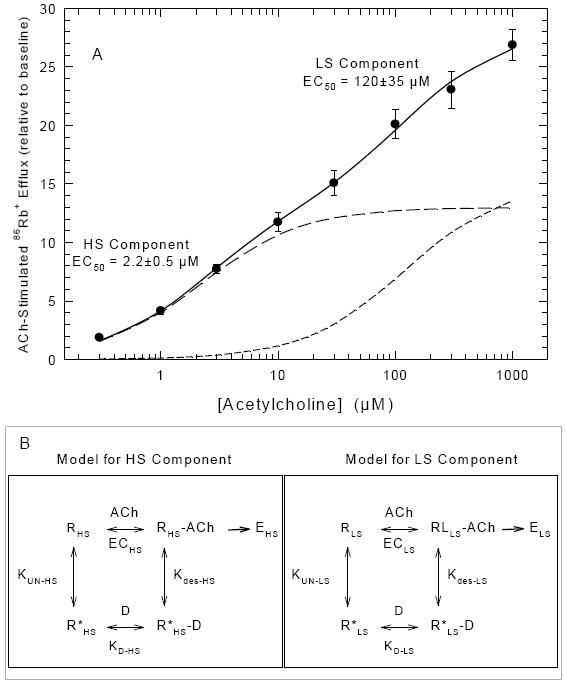
The upper panel of this figure shows the ACh concentration effect curve for stimulation of 86Rb+ efflux. Each data point represents the mean ± SEM for six independent studies. The solid line is the fit of the data to a two component model as described in the methods. The two dashed lines are the individual theoretical curves for the HS and LS components. The cyclical Katz-Thesleff diagrams in the boxes below the main figure illustrate the model used to evaluate the desensitization data. HS and LS 86Rb+ efflux observed following stimulation at each [ACh] are designated EHS and ELS, respectively. The ACh concentrations that elicit 50% maximal HS and LS 86Rb+ efflux are designated, ECHS and ECLS, respectively. The apparent KD for binding of the ligand (D) to the unliganded desensitized state of the receptor subtypes (R*HS and R*LS) are designated KD-HS and KD-LS. KUN-HS and KUN-LS are the equilibrium constants between the unliganded ground state (RHS and RLS) and unliganded desensitized state (R*HS and R*LS) and Kdes-HS and Kdes-LS are the equilibrium constants between the liganded active receptors (RHS-ACh and RLS-ACh) and the liganded desensitized receptors.
3.2 Time courses for the binding of nicotine, cytisine, methylcarbachol and epibatidine
In order to determine whether binding of the ligands to the desensitized conformation (R’+D↔R’D) of the receptor is more rapid than the transition between unliganded ground state and unliganded desensitized state (R↔R*), the rates of binding of the four ligands were measured. Previously published studies have shown that the time courses for the association of [3H]-nicotine to brain preparations are biphasic, with an initial fast phase postulated to correspond to binding to the high affinity desensitized conformation and a slower phase corresponding to isomerization from the ground state to the desensitized state [8, 22-23]. This route corresponds to the lower left corners of the Katz-Thesleff schemes in Figure 1B. Desensitization elicited by exposure to low concentrations of nicotinic agonists is postulated to be dependent on the binding of the agonist to the desensitized state of the receptor followed by a conformational change from unliganded ground state to unliganded desensitized state. Thus, time courses for association of the four agonists to whole brain particulate samples were measured at two concentrations of each ligand to determine the rates of fast (binding) and slow (isomerization) processes (Figure 2). Biphasic association curves were clearly observed for nicotine (2A), cytisine (2B) and epibatidine (2C) but to a lesser extent with methylcarbachol (2D). Apparent rate constants for fast and slow phase binding are compiled in Table 1. The rate constants measured for fast phase binding for the four ligands differ by less than a factor of 2. With the exception of epibatidine, these constants tended to be larger at the higher ligand concentrations, as expected. These rate constants indicate that the t½ for fast phase binding ranged from 8 to 16 sec, and is at least 10 times greater than the rate constants measured for slow phase binding.
Figure 2. Time Courses for the Binding of [3H]-Epibatidine, [3H]-Cytisine, [3H]-Nicotine and [3H]-Methylcarbachol to Mouse Brain Samples.
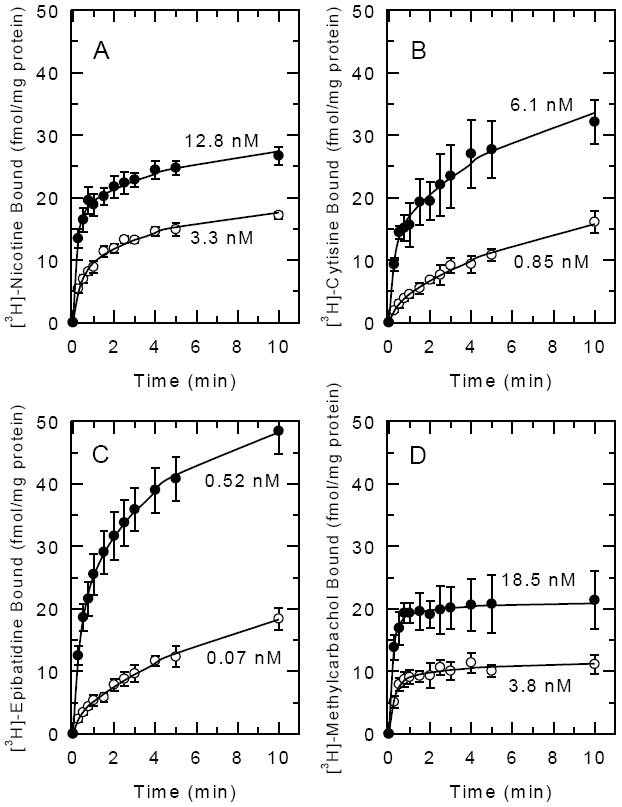
Whole mouse brain particulate fractions were incubated for the times shown with two concentrations of the indicated ligand as indicated. Binding at the lower concentration is illustrated with open circles (○) and binding at higher concentrations is illustrated with closed circles (●). Each point represents the mean±SEM of three separate experiments. Curves were determined by nonlinear least squares fitting of the data as a two component process as described in the Methods.
Table 1.
Estimated Rate Constants for Ligand Binding and Desensitization
| Binding | Time Courses For Agonist Binding | Time Courses For Functional Desensitization | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Agonist | Ki (nM) |
Conc (nM) |
kfast (min-1)t |
kslow | Conc | kfast (min-1) |
kslow (min-1)- |
Conc (nM) |
kdes 30 μM (min-1) |
% des | Kdes 1 mM (min-1) |
% des |
Conc (nM) |
Kdes 30 μM (min-1) |
% des |
Kdes 1 mM (min-1) |
% des |
| epibatidine | .024 ± .008 | 0.07 | 2.9 ± 1.36 | 0.11 ± .01 | 0.52 | 2.85 ± .29 | 0.23 ± .03 | 0.3 | .29 ± .06 | 69.9 ± 6.1 | .41 ± .17 | 64.5 ± 11.7 | 3 | 5.4 ± 1.9 | 96.7 ± 8.9 | 3.1 ± 0.7 | 93.8 ± 7.4 |
| cytisine | .32 ± .02 | 0.85 | 2.61 ± 1.03 | 0.13 ± .01 | 6.13 | 3.96 ± .82 | 0.16 ± .01 | 20 | .65 ± .17 | 66.0 ± 10.6 | .53 ± .16 | 65.5 ± 7.1 | 200 | 2.9 ± 0.5 | 85.2 ± 15.2 | .98 ± .22 | 78.4 ± 14.7 |
| nicotine | 2.0 ± 0.2 | 3.3 | 2.92 ± .69 | .23 ± .03 | 12.8 | 4.99 ± .60 | .20 ± .02 | 10 | .30 ± .34 | 20.1 ± 9.9 | .30 ± .27 | 41.4 ± 14.7 | 200 | .81 ± .10 | 98.6 ± 5.0 | .84 ± .21 | 95.1 ± 8.8 |
| Methyl-carbachol | 7.9 ± 1.7 | 3.8 | 3.68 ± .77 | .29 ± .13 | 18.5 | 5.22 ± .51 | .39 ± .15 | 100 | .41 ± .13 | 50.9 ± 8.8 | .36 ± .14 | 56.0 ± 8.1 | 500 | .77 ± .16 | 94.3 ± 7.3 | .82 ± .16 | 88.7 ± 6.4 |
Binding and kinetic parameters for epibatidine, cytisine nicotine and methylcarbachol are summarized. Binding affinities were determined by inhibition of [3H]epibatidine binding. Rate constants for agonist binding at the indicated agonist concentrations were determined from data shown in Figure 2. Rate constants and the extent of desensitization at the indicated concentrations were calculated from the data shown in Figure 3. All values are mean±SEM.
3.3 Time Courses for Desensitization of 86Rb+ efflux ACh-Stimulated by Low Agonist Concentrations
Since the experiments described below are intended to measure the extent of desensitization at equilibrium, the desensitization rates observed following exposure to each of the four agonists were measured. The effect of time of exposure to the nicotinic agonists on subsequent stimulation of 86Rb+ efflux by either 30 μM or 1000 μM ACh was measured to evaluate whether the rate and/or extent of desensitization varied as a function of stimulating ACh concentration. These two ACh concentrations were chosen to provide estimates of the 86Rb+ efflux mediated by nAChR sensitive to stimulation by low ACh (high sensitivity=HS responses) and the 86Rb+ efflux mediated by both nAChR HS and LS (responses requiring higher ACh concentrations, that is low sensitivity=LS). These studies also were used to select an appropriate exposure time for the construction of the dose-response curves described in the following section. The effect of exposure time on subsequent ACh-stimulated 86Rb+ efflux is illustrated in Figure 3 for nicotine (3A), cytisine (3B), epibatidine (3C) and methylcarbachol (3D). A time-dependent decrease in 86Rb+ efflux stimulated by either 30 μM or 1000 μM ACh was observed. Rate constants and extents of decrease in ACh-stimulated 86Rb+ efflux calculated from the data in Figure 3 are summarized in Table 1. No significant differences in either the rate constants for decrease in activity or the extent of the decrease were noted for responses stimulated by 30 μM and 1000 μM ACh following treatment with the lower concentration of any agonist indicating that HS and LS components desensitized at similar rates. It should be noted, however, that the rate constants for cytisine were higher than those for the other three agonists. While the extent of desensitization observed following treatment with the higher concentrations of any of the agonists were not significantly different, faster desensitization rates were measured for responses stimulated with 30 μM ACh than for responses stimulated with 1000 μM ACh following treatment with either 200 μM cytisine or 3 μM epibatidine. The lowest desensitization rate measured in these time course studies was 0.29 min-1, which corresponds to a t½ of 2.37 min and results in 90% desensitization following an 8-min exposure, and 95% desensitization following a 10 min exposure.
Figure 3. Time Courses for the Inhibition of ACh-Stimulated 86Rb+ Efflux Elicited by Exposure to Epibatidine, Cytisine, Nicotine or Methylcarbachol.
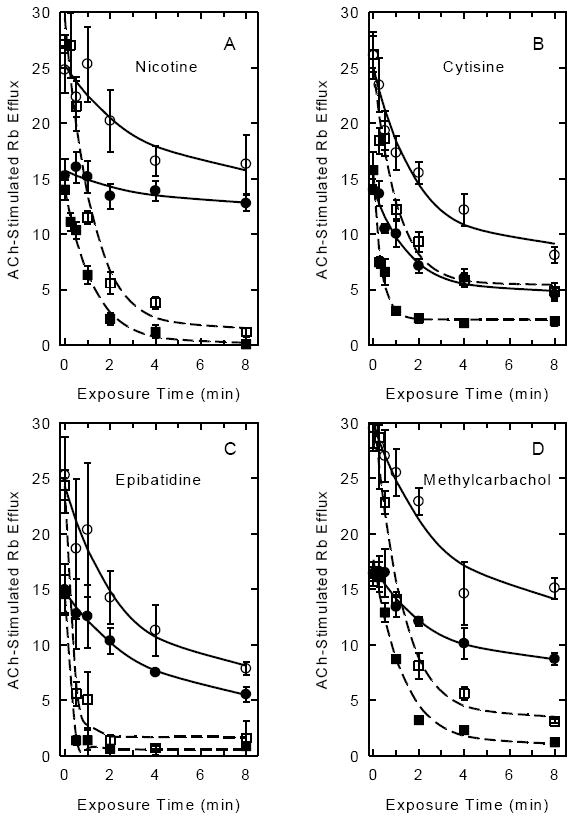
Mouse thalamic synaptosomes loaded with 86Rb+ were exposed to two concentrations of the each agonist for the indicated times (0 to 8 min) and then stimulated by exposure to either 30 μM ACh (filled symbols: ●lower agonist concentration; ■ higher agonist concentration) or 1000 μM ACh (open symbols: ○lower agonist concentration; □ higher agonist concentration). The agonist concentrations used for exposure prior to stimulation were: nicotine; 10 nM and 200 nM; cytisine; 20 nM and 200 nM; epibatidine 0.3 nM and 3 nM and methylcarbachol; 100 nM and 500 nM. Each data point represents the mean±SEM of 3-5 separate experiments. Curves were determined by nonlinear least squares fitting of the data as an exponential decay with residual activity as described in the methods.
The rate of desensitization is significantly slower than the rate of fast phase binding of the agonists, but is similar to that for slow phase binding. This result is consistent with the predictions of the Katz-Thesleff model [24] (Figure 1) that postulates that the decrease of the unliganded desensitized receptor (R*) by binding of agonist (forming R*D) disrupts the equilibrium between R and R* leading to the relatively slow conformational transition from R to R*. This decrease in R lowers ground state receptor available for activation [1, 3, 25].
3.4 Concentration Dependence for Desensitization of ACh-Stimulated 86Rb+ Efflux by Exposure to Low Concentrations of Nicotinic Agonists
The concentration-response curve for ACh-stimulated 86Rb+ efflux from mouse brain synaptosomes is biphasic [13-15] (and Figure 1A). ACh-stimulated 86Rb+ efflux can be resolved into two components differing in apparent EC50 for ACh: A higher-sensitivity component (HS, EC50 ≈ 2 μM) and a lower-sensitivity component (LS, EC50 ≈ 120 μM). The time course experiments presented in Figure 3 and summarized in Table 1 indicate that treatment with a low concentration of agonist for 8 min elicits over 90% steady-state desensitization. Thus, a 10 min exposure to agonist was used to investigate the concentration dependence of the agonist-induced desensitization. This exposure time elicits extensive desensitization, but the extent of desensitization may not be complete, especially when low agonist concentrations are used. For illustration, the effects of acute exposure to relatively low concentrations of nicotine on 86Rb+ efflux stimulated by various concentrations of ACh are presented in Figure 4A. ACh elicits a concentration dependent increase in 86Rb+ efflux. However, exposure to nicotine for 10 min decreases the response. The percentage decrease elicited by nicotine treatment is similar at each ACh concentration as illustrated for the ACh concentration-effect curve for samples exposed to 20 nM nicotine in Figure 4B. The curve for samples exposed to 20 nM nicotine is also biphasic. The EC50 values for ACh for the two components (3.4 μM and 134 μM) are similar to those of the samples that were not exposed to nicotine.
Figure 4. ACh-Stimulated 86Rb+ Efflux After Acute Exposure to Nicotine.
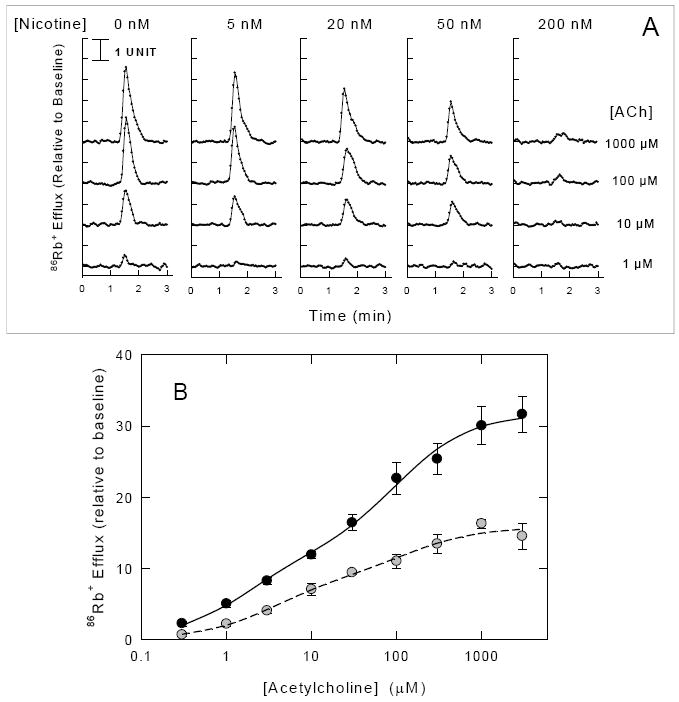
Panel A. Mouse thalamic synaptosomes loaded with 86Rb+ were superfused with buffer containing the concentrations of nicotine indicated at the top of each panel (0 nM, 5 nM, 20 nM, 50 nM or 200 nM) for 10 min before stimulation with ACh. Each 3-sec datum point represents 86Rb+ efflux (normalized to baseline) stimulated by a 5 sec exposure to the ACh concentrations indicated at the right of the figure (1 μM, 10 μM, 100 μM or 1000 μM). Panel B. Concentration-effect curves for ACh-stimulated 86Rb+ efflux measured after exposure to 0 nM (●) or 20 nM (○) nicotine are shown. Each point represents the mean ±SEM of 4-6 individual experiments. Curves were obtained by nonlinear least squares fitting of the data to a two component process as described in the Methods.
The families of ACh-concentration effect curves shown in Figure 5 show that desensitization of ACh-stimulated 86Rb+ efflux was elicited by exposure to relatively low concentrations of nicotine (5A), cytisine (5B), epibatidine (5C) and methylcarbachol (5D). Exposure to each of these agonists produces a concentration-dependent decrease in 86Rb+ efflux, but the concentrations that elicit desensitization varied among the compounds.
Figure 5. Concentration-Effect Curves for ACh-Stimulated 86Rb+ Efflux After Acute Exposure to Cytisine, Epibatidine, Methylcarbachol or Nicotine.
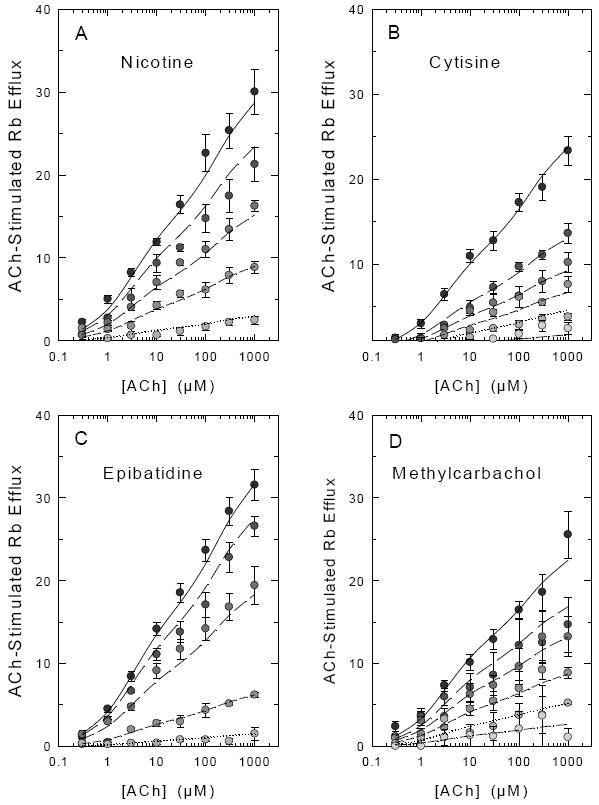
Mouse thalamic synaptosomes loaded with 86Rb+ were superfused with buffer containing nicotine (0 nM, 5 nM, 20 nM, 50 nM or 200 nM), cytisine (0 nM, 2 nM, 5 nM, 10 nM, 20 nM or 100 nM), epibatidine (0 nM, 0.1 nM, 3 nM, 10 nM or 30 nM), or methylcarbachol (0 nM, 10 nM, 30 nM, 100 nM, 300 nM or 1000 nM) for 10 min prior to stimulation with the indicated concentration of ACh (0.1 μM to 1000 μM) for 5 sec. Each point represents the mean ± SEM of 6- separate experiments. Symbol shading becomes less intense with increasing agonist concentration. Curves represent the fit of the entire data set to the two component activation curves (high ACh sensitivity and low ACh sensitivity) with the simplifying assumption that the IC50 for each agonist is the same for the high and low ACh sensitive components as described in the Methods and Results.
In order to provide estimates of the inhibitory concentrations for each agonist, the maximal efflux for the HS and LS components of 86Rb+ efflux was calculated by non-linear least-squares fitting of the full ACh concentration-effect curves at each concentration of nicotine, epibatidine, cytisine and methylcarbachol. Exposure to the agonists did not significantly affect the EC50 values for ACh at either the high sensitivity (average EC50 ≈ 3.3 μM) or the low sensitivity (average EC50 ≈ 250 μM) component at any concentration of the desensitizing agonist. Concentration-effect curves for the desensitization of the high-sensitivity and low-sensitivity components by the four agonists generated by calculating maximal efflux for the two components at each concentration are shown in Figure 6. Although the effective concentrations of the four agonists that induce desensitization differ, the IC50 values for desensitization of the HS and LS components are similar for each agonist (nicotine (Figure 6A): 20.6±6.0 nM and 18.5±3.4 nM; cytisine (Figure 6B): 2.7±0.6 and 5.4±1.4 nM; epibatidine (Figure 6C): 0.33±0.09 nM and 0.38±0.09 nM; and methylcarbachol(Figure 6D): 78.6±17.8 and 45.2±7.6 nM, for the components with high and low ACh sensitivity, respectively).
Figure 6. Inhibition Curves for Acute Desensitization of ACh-Stimulated 86Rb+ Efflux by Nicotine, Epibatidine, Cytisine or Methylcarbachol.
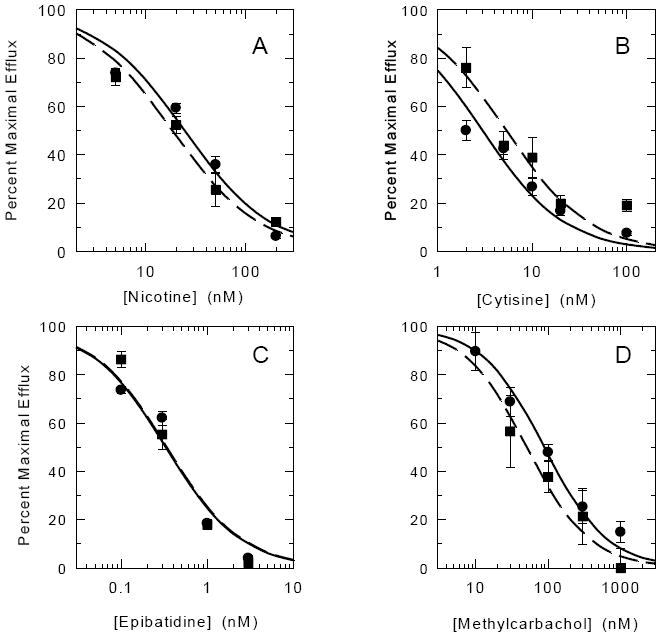
Individual ACh concentration-effect curves obtained in the presence of the indicated concentrations of agonists were fit to the two-site model and HS and LS components of the curves were calculated. IC50 values for inhibition of maximal HS and LS responses determined following exposure to agonists were then calculated as described in the Methods. Each point is the mean ± SEM calculated from the fits of the ACh concentration effect curves at each agonist concentration [HS (●), LS (■)]. Lines are theoretical inhibition curves for HS (solid) or LS (dashed).
The complete data set for ACh-stimulated 86Rb+ efflux assayed after exposure to low concentrations of each agonist was fit as described in the methods based on the following equation:
This equation formally treats the steady-state desensitization occurring following exposure to low agonist concentrations as a noncompetitive process, that is, the effects restricted to the maximal efflux rates, EHS and ELS. The most general form of this equation includes eight possible independent variables: EHS, ECHS, IC50-HS, nHS, ELS, EC50-LS, IC50-LS, and nLS. As mentioned above, pretreatment with any concentration of the four agonists did not significantly affect either EC50-HS (average 3.3 μM) or EC50-LS (average 250 μM). Consequently, these two variables were fixed in all calculations. The simplest model assumes that IC50-1 = IC50-2 and that nHS = nLS = 1 (agonists desensitize HS and LS 86Rb+ efflux equally and noncooperatively), while the most complex model allows that IC50-HS ≠ IC50-LS and that nHS ≠ nLS≠1. The more complex models did not provide significant improvements in the curve fits. Indeed, the IC50 values for the HS and LS components of 86Rb+ efflux calculated by fitting each individual curve do not differ significantly indicates that the simpler model is sufficient. Therefore, results obtained from the simpler model are presented as the lines on the concentration-effect curves in Figure 5. Using this model, the following IC50 values were obtained: Nicotine: 21.5±1.7 nM; cytisine: 2.7±0.2 nM; epibatidine: 0.37±0.02 nM; and methylcarbachol: 50.7±8.3 nM. These values are similar to those obtained from the inhibition curves in Figure 5.
In order to provide a comparison between the agonist concentrations that elicit desensitization and the affinities of the four ligands for their binding sites, inhibition of [3H]epibatidine binding was measured. Apparent KI values are summarized in Table 2. The rank order for the potency of these four agonists as inhibitors of [3H]epibatidine binding and desensitizers of 86Rb+ efflux was the same (epibatidine>cytisine>nicotine>methylcarbachol). However, the apparent affinities for each of the agonists were significantly higher (approximately 10-fold) as inhibitors of [3H]epibatidine binding sites than for desensitization of ACh-stimulated 86Rb+ efflux.
Table 2.
Summary of Desensitization and Binding Properties of Four Nicotinic Agonists.
| Agonist | IC50 Desensitization (nM) | KI, High Affinity Epibatidine Binding (nM) | Ratio KI High Affinity Binding/Desensitization IC50 | Ratio: HS EC50/Desensitization IC50 | Ratio: LS EC50/Desensitization IC50 |
|---|---|---|---|---|---|
| Epibatidine | 0.37±0.02 | 0.024.008 | 0.065±0.022 | 154±44 | 6000±870 |
| Cytisine | 2.7±0.2 | 0.32±0.02 | 0.119±0.011 | 4.8±4.1 | 56000±23000 |
| Nicotine | 21.5±1.7 | 2.0±0.2 | 0.093±0.012 | 65±20 | 6000±930 |
| Methylcarbachol | 50.7±8.3 | 7.9±1.7 | 0.156±0.042 | 380±120 | 14400±2800 |
IC50 for desensitization for each agonist was determined by fitting all data to the equation given in the Methods and Results. KI were determined by inhibition of [3H]epibatidine binding and converting the resulting IC50 values to KI. Ratios between the indicated values and IC50 values for desensitization were calculated and errors estimated using Taylor’s expansion. EC50 values for HS and LS ACh-stimulated 86Rb+ efflux were obtained from Marks et al., 1999.
3.5 86Rb+ Efflux and Desensitization by Nicotine Following Chronic Nicotine Treatment
C57BL/6 mice were chronically treated with 0, 0.5 or 4.0 mg/kg/hr nicotine by continuous intravenous infusion for 10 days. The mice were removed from the infusion chamber and their cannulas checked for patency. Two hours later the mice were sacrificed and crude synaptosomes were prepared from cortex and thalamus. Subsequently, ACh concentration-effect curves were constructed for both brain regions and each nicotine treatment. Samples were superfused with buffer containing one of the following nicotine concentrations of 0 nM, 10 nM, 20 nM, 50 nM or 200 nM before stimulation with ACh. The curves are shown in Figure 7. Results from all experiments at each chronic nicotine treatment dose were analyzed. The EC50 values for the HS and LS components of the ACh concentration effect curves did not differ among the treatment groups (average 0.55 μM and 135 μM for cortex and 2.1 μM and 180 μM for thalamus for the high and low sensitivity EC50 values, respectively). Therefore, these EC50 values were fixed for the subsequent analyses.
Figure 7. Concentration-Effect Curves for ACh Stimulated 86Rb+ Efflux After Chronic Nicotine Treatment and Effect of Acute Nicotine Desensitization.
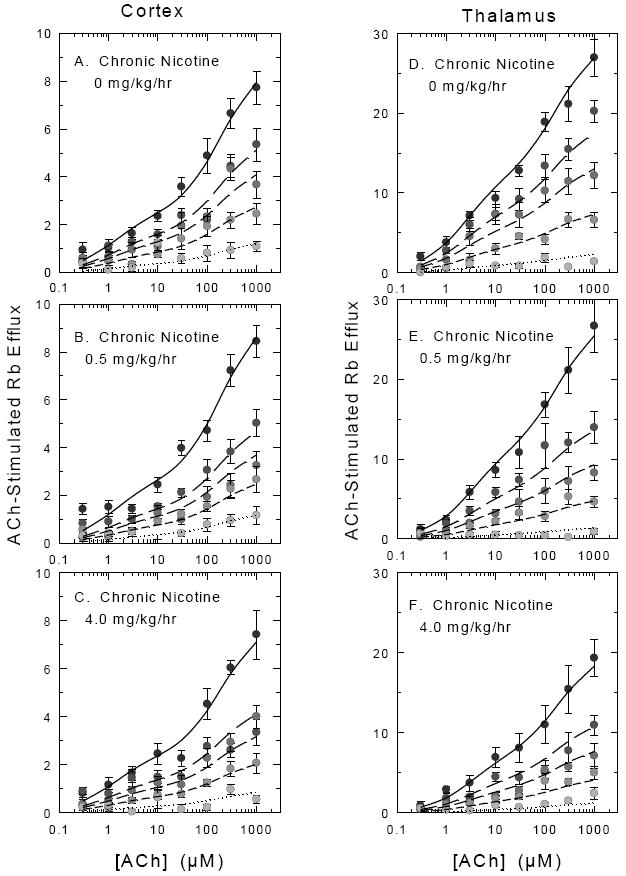
C57BL/6 mice were chronically treated with saline, 0.5 mg/kg/hr nicotine or 4 mg/kg/hr nicotine. Mice were withdrawn from treatment for 2 hr and crude synaptosomes were then prepared from cortex and thalamus and loaded with 86Rb+. Samples were subsequently treated with various concentrations of nicotine (0 nM, 5 nM, 20 nM, 50 nM or 200 nM) for 10 min before a 5 sec stimulation with the indicated concentrations of nicotine. Each point represents the mean ± SEM for 9-11 separate experiments for cortex and 7 separate experiments for thalamus. Symbol shading becomes less intense with increasing nicotine concentration. Curves represent the fit of the entire data set for each region and each treatment dose to the two component activation curves (high ACh sensitivity and low ACh sensitivity) with the simplifying assumption that the IC50 for each agonist is the same for the high and low ACh sensitive components as described in the Methods and Results. The EC50 values for ACh were fixed while the IC50 value for nicotine were allowed to float.
ACh activation and nicotine desensitization of 86Rb+ efflux from cortical synaptosomes of mice chronically treated with nicotine treatment are shown in the panels on the left of Figure 7. The IC50 values for nicotine calculated from these curves from mice treated with saline (Figure 7A). 0.5 mg/kg/hr (Figure 7B) and 4.0 mg/kg/hr (Figure 7C) were not significantly different, although the IC50 for the saline mice tended to be somewhat higher than that for the nicotine-treated mice (23.7±2.4 nM, 15.0±1.3 nM and 16.4±1.8 nM, respectively). No significant differences in EC50 values for either the high sensitivity or low sensitivity components were observed (average EC50 values were 0.55 μM and 135 μM). Values summarized in Table 3 represent the averages of the high- and low-sensitivity components obtained each individual experiment with fixed kinetic constants. Chronic nicotine treatment did not significantly affect either HS or LS components as determined by one-way ANOVA (F2,27=0.27 and F2,27=1.12, p>0.05 HS and LS components, respectively).
Table 3.
Maximal HS and LS ACh-Stimulated 86Rb+ Efflux and Cytisine-Sensitive [125I]Epibatidine Binding in Cortex and Thalamus of Mice Chronically Treated with Nicotine
| Nicotine Infusion Dose | 0.0 mg/kg/hr | 0.5 mg/kg/hr | 4.0 mg/kg/hr |
|---|---|---|---|
| CORTEX | |||
| High Sensitivity 86Rb+ Efflux | 2.11±0.11 | 2.06±0.19 | 1.88±0.11 |
| Low Sensitivity 86Rb+ Efflux | 6.81±0.59 | 6.80±0.50 | 5.63±0.80 |
| [125I]-Epibatidine Bound | 88.2±0.7 | 127.9±1.1 | 139.0±1.2 |
| THALAMUS | |||
| High Sensitivity 86Rb+ Efflux | 13.07±0.89 | 10.19±0.99 | 6.76±1.19 |
| Low Sensitivity 86Rb+ Efflux | 16.26±1.27 | 13.84±2.48 | 12.52±1.96 |
| [125I]-Epibatidine Bound | 198.4±2.7 | 240.7±2.5 | 256.8±2.9 |
HS and LS ACh-stimulated 86Rb+ efflux were calculated as described in the Methods. Values represent mean ± SEM for 9-11 individual experiments for Cortex and 7 individual experiments for Thalamus. 86Rb+ efflux data are presented as total efflux normalized to baseline. [125I]Epibatidine binding is given as fmol/mg protein.
ACh activation and nicotine desensitization of 86Rb+ efflux from thalamic synaptosomes of mice chronically treated with nicotine are shown in the panels on the right of Figure 7. As was the case with cortex, the IC50 values for nicotine calculated from these curves in thalamus from mice treated with saline (figure 7D), 0.5 mg/kg/hr (Figure 7E) and 4.0 mg/kg/hr (Figure 7F) were not significantly different, although the IC50 for the saline mice tended to be somewhat higher than that for the nicotine-treated mice (19.5 ±1.8 nM, 12.3 ± 1.2 nM, and 14.5±1.9 nM, respectively). No significant differences in EC50 values for either the high sensitivity or low sensitivity components were observed (average EC50 values were 2.1 μM and 190 μM). Values summarized in Table 3 represent the averages of the high- and low-sensitivity components obtained each individual experiment calculated by fixing the two EC50 values and the IC50 value at 2.1 μM, 190 μM and 15 nM, respectively. A chronic nicotine dose-dependent decrease in maximal HS 86Rb+ efflux was noted (one-way AVOVA: F2,18=9.58, P=0.001) and the activity measured following chronic treatment with 4.0 mg/kg/hr nicotine was approximately one-half that for the saline group. Although there was a tendency for response of the LS component to decrease, this was not statistically significant (F2,18=0.93, P>0.05).
3.6 Epibatidine Binding following Chronic Nicotine Treatment
In order to compare the effects of chronic nicotine treatment on function to changes in nAChR density, binding sites were measured using [3H]epibatidine binding to particulate samples prepared from synaptosomes used to measure 86Rb+ efflux. Bmax values for both cortex and thalamus are shown in Table 3. Chronic nicotine treatment increased maximal [3H]epibatidine binding in cortex (F2,16=21.56, P<0.001) and Bmax values following treatment with 0.5 or 4.0 mg/kg/hr nicotine were significantly higher than Bmax for saline-treated mice. Apparent KD values were unaffected by chronic nicotine treatment (F2,16=0.67, P>0.05). While Bmax values in thalamus tended to be higher following chronic nicotine treatment, these apparent changes were not statistically significant (F2,13=2.71, P>0.05). Chronic nicotine treatment did not significantly affect apparent KD values in thalamus (F2,11=0.61, P>0.05).
4. Discussion
The current study indicates that exposure of mouse brain synaptosomes to nicotine, epibatidine, cytisine or methylcarbachol at concentrations that elicit little if any activation of α4β2*nAChR desensitize ACh-stimulated 86Rb+ efflux. Although the four agonists differ in potency, the concentrations of each agonist that elicited desensitization of HS and LS α4β2*nAChR mediated 86Rb+ efflux were not significantly different. The concentrations required to desensitize the functional responses were approximately 10-fold higher than the Ki values for inhibiting high affinity [3H]epibatidine binding. Furthermore, the concentration dependence for desensitization by nicotine of ACh-stimulated 86Rb+ efflux was not significantly affected by chronic nicotine treatment, indicating that exposure to nicotine in vivo did not alter steady-state desensitization of α4β2*-nAChR in either cortical or thalamic synaptosomes.
The fact that we measured an approximate 10-fold difference in the affinity for the binding site and the concentration required to desensitize the functional response is somewhat unexpected since induction of desenstitization likely occurs through binding to the high affinity desensitized state of the receptor with a subsequent depletion of ground state receptors. Indeed a close correspondence between these variables has been noted in heterologously expressed α4β2-nAChR [6]. Our study of the desensitization of HS responses noted a smaller difference of about 3-fold between binding site affinity and desensitizing concentrations that were highly correlated [8]. In that study we postulated that these small differences could occur as a result of the cyclical nature of the Katz-Thesleff desensitization model that is influenced by the initial equilibrium between activatable and desensitized conformations of the receptor. Another contributor to the difference could be strictly procedural. If desensitization had not completely reached equilibrium the extent of desensitization elicited especially at low agonist concentrations may not yet be complete. Consequently, the concentration-effect curve would be shifted to the right resulting in apparently higher IC50 values for desensitization. Comparison of concentration-effect curves following different times of exposure to low agonist concentrations will address this question. It should be noted that the IC50 values measured for nicotine in the current study are similar to those determined with transfected cells (≈10 nM) [4] and dopaminergic cells in the ventral tegmental areal (≈20 nM) [5], which also desensitized at a very similar rate to that reported here.
The EC50 values for ACh activation of the HS and LS responses mediated by α4β2-nAChR differ by a factor of approximately 100 both in the heterologous systems [9-12, 26-28] and in mouse brain synaptosomes [13-15]. Differences in EC50 values for stimulation of HS and LS responses in mouse brain synaptosomes also have been observed for nicotine (94-fold), epibatidine (39-fold), cytisine (11,500-fold) and methylcarbachol (38-fold) [14], the agonists investigated for their desensitization properties in the current study. Both the HS and LS components of nAChR-mediated 86Rb+ efflux in as well as high affinity epibatidine binding in thalamus and cortex of mouse brain are mediated by the α4β2*-nAChR subtype as demonstrated by elimination of ACh-stimulated 86Rb+ efflux and epibatidine (or nicotine) binding following deletion of either the α4 or the β2 subunit in knockout mice [13-14, 29-31] Considerable evidence using heterologous expression systems supports the proposal that the HS and LS α4β2-nAChR responses are mediated by receptors differing in subunit stoichiometry (HS component (α4)2(β2)3 and LS component (α4)3(β2)2) [9-12, 26-28]. Functional and immunochemical evidence with cortical and thalamic tissue is generally consistent with the contention that stoichiometric differences also underlie the heterogeneous agonist stimulation of 86Rb+ efflux mediated by α4β2*-nAChR in mouse brain [15]. However, a significant reduction of HS function in thalamus and a smaller fraction in cortex occurs in α5 knockout mice, indicating the role of the α5 subunit in nAChR mediated HS 86Rb+ efflux in these regions that is thought to be mediated by an receptor with an implied stoichiometry of (α4)2(β2)2(α5) [16]. The gene dose dependent reduction in the expression of α5 subunit protein upon deletion of the both the α4 and β2 genes supports the contention that the α5 subunit protein is expressed in a subset of α4β2*-nAChR [15]. Inclusion of the α5 subunit could possibly modify the pharmacology of the HS component. Deletion of the α5 subunit did reduce the amount of HS 86Rb+ efflux, but had no significant effect on the EC50 for agonists [16]. This result is similar to the observation made after coassembly of the α5 subunit with α4 and β2 subunits in an heterologous expression system in which only HS responses with EC50 values similar to or slightly higher than those for HS receptors assembled with only α4 and β2 subunits [32]. Immunochemical experiments have also demonstrated that α5 subunit protein in rat cortex and thalamus as well as measureable amounts of α3 and β4 contribute to epibatidine binding and therefore receptors composed of this subunits are likely to contribute functional nAChR [33]. The fact that no detectable 86Rb+ efflux persisted in either α4 or β2 null mutants indicates that the contribution of non-α4β2*-nAChR to ACh-mediated 86Rb+ efflux in the mouse cortical and thalamic synaptosomes evaluated here is unlikely. However, 86Rb+ efflux mediated by α3β4-nAChR has been measured in other brain regions [34].
It has been known for over 50 years that prolonged or repeated exposure of nAChR to agonists desensitizes the receptors [24]. Exposure to nicotine also effectively desensitizes α4β2-nAChR [1-2, 4]. We have reported that desensitization of the HS component of 86Rb+ efflux by nicotine and several other agonists occurs at concentrations significantly below those required for activation by these agonists [8]. We have reported here that desensitizing concentrations do not differ between HS and LS, while activating concentrations differ markedly [14]. This result is consistent with the proposal that agonist binding to the desensitized state of both HS and LS stoichiometries is the same [35], which would be expected given that the α/β interface at which ligand binding occurs is likely to be very similar if not identical for the receptors with alternate α4/β2 subunit stoichiometries or containing an accessory subunit such as α5. The observation that desensitization by prolonged exposure to low agonist concentrations does not differ between HS and LS forms of α4β2*-nAChR also has implications for the degree of residual activity observed in the presence of low agonist concentrations. The ratios of EC50 for HS stimulation of 86Rb+ efflux to IC50 for desensitization range from 4.8 for cytisine to 380 for methylcarbachol, while the ratios of EC50 for LS stimulation of 86Rb+ efflux to IC50 for desensitization are much higher and range from 6000 for epibatidine and nicotine to 56,000 for methylcarbachol. If the relative potency for these and other nicotinic agonists are similar in vivo, little or no residual LS activity for a given agonist would persist, while some HS activity may persist.
The wide range of ratios between concentrations leading to desensitization and those eliciting functional activity for the HS sites suggests that the activity persisting with prolonged exposure would differ markedly among agonists. The small ratio observed for cytisine, a partial α4β2-nAChR agonist, is comparable to that reported for ABT-089, which is also a partial agonist [36]. It remains to be determined if the high degree of persistent activity is a common property of partial agonists and, if so, whether this explains, in part, the therapeutic efficacy of compounds such as varenicline [37-39].
The observation that prolonged exposure to nicotinic agonists induces desensitization for the HS and LS forms of α4β2*-nAChR could also have important consequences for receptor responses to nicotine following tobacco use. Chronic exposure to nicotine increases the numbers of α4β2*-nAChR in brain of rodents [17-19, 40-43] humans [44-46] and cell lines [47-49]. The increases observed in cell lines is generally greater than those observed in brain, but the extent of the increase of α4β2*-nAChR binding sites also varies among brain areas [17, 19, 42, 50-53]. It has been proposed that chronic receptor desensitization is an important factor underlying the nicotine-induced increases in receptor expression [6, 19, 54], perhaps by inducing a conformational change or receptor state that decreases the turnover of the receptors [47]. Consequently, the fact that nicotine is equally effective in desensitizing both HS and LS responses implies that the α4β2*-nAChR subtypes mediating these responses will have similar extents of desensitization during chronic nicotine exposure and potentially respond similarly to chronic treatment. However, chronic exposure to nicotine elicited differential changes in the expression of HS and LS forms of heterologously expressed α4β2-nAChR [27, 55]. Furthermore, the α4β2α5-nAChR that displays HS pharmacology is resistant to upregulation in vivo [33] but perhaps not for heterologously expressed receptors [32]. These results suggest that the simple desensitization hypothesis may not adequately account for all the data. While the concentration of nicotine required to desensitize and upregulate α4β2-nAChR expressed in Xenopus oocytes is virtually identical [6], considerably higher agonist concentrations have been reported to be required to elicit upregulation of this subtype in transfected cells [35, 49]. We have compared the plasma levels of nicotine observed with chronic treatment to the extent of upregulation of α4β2*-nAChR binding sites in mice treated chronically with different nicotine doses and observed that the average dose for half-maximal upregulation was 0.44 mg/kg/hr, which gave an estimated plasma concentration of 22 ng/ml (0.13 μM) [17]. Half-maximal upregulation of high affinity binding sites in rats occurred at a very similar plasma concentration [40]. The plasma concentration eliciting half-maximal upregulation is approximately three times higher than the nicotine concentration inducing 50% desensitization. This difference could occur because there is a temperature-dependent affinity difference between 21°C (functional measurements) and 37°C (body temperature) as noted for [3H]nicotine binding [56] or a higher nicotine concentration in brain compared to plasma [40]. Perhaps a fundamentally different mechanism operates such as nicotine serving as a molecular chaperone with somewhat lower affinity for less mature receptors [35, 57]. Nevertheless, interaction with the agonist binding site seems to be an important component of up-regulation [58].
Data presented in the current study indicate that chronic nicotine treatment did not significantly change the ability of nicotine (IC50 values) to elicit desensitization nor did chronic treatment significantly affect the EC50 values for ACh stimulation of either HS or LS forms of the receptor. However, consistent with our previous studies [17, 59] a dose-dependent decrease in maximal response was noted for the HS component in thalamus. Although activity tended to decrease with nicotine treatment no statistically significant changes in HS in cortex or LS in either cortex or thalamus were observed. When total functional responses are normalized to the density of α4β2*-nAChR binding sites a decrease of approximately 50% is noted for both regions investigated here, suggesting that the upregulated receptors are not fully functional. Although this could occur as a result of continued desensitization by residual nicotine, the two-hour withdrawal period from nicotine treatment before sacrifice and the extensive washing of the synaptosomes before stimulation makes this unlikely. However, the continued persistence of deeply desensitized receptor states cannot be discounted. There is also evidence that upregulated receptors may be retained intracellularly where they are measurable with ligand binding, but are not functional owing to their absence from the cell surface [35].
Differing results have been obtained about the extent of receptor function following chronic nicotine treatment. Similar to our results, decreases in function per unit binding site have been reported; [6, 48, 60]. Gopalakrishnan et al. [48] noted decreased function per binding site following treatment of a cell line stably expressing α4β2-nAChR. Kuryatov et al. [35] reported a small decrease in the function/binding ratio for surface receptors. In contrast, absolute increases in function after chronic nicotine treatment have also been observed [10, 35, 48, 55, 61]. For example, Nguyen et al. [61] reported that function increased in proportion to the number of binding sites following chronic nicotine treatment in rats. Thus, the consequences of chronic nicotine treatment on receptor function are unresolved. Differences in the results could occur because of differences in functional assays used, differences in species or subtle differences in receptor composition, such as incorporation of the α5 subunit.
In summary, desensitization properties of nAChR subtypes are known to differ so that acute or chronic agonist exposure alters the normal balance among receptor subtypes with different desensitization properties such as α4B2*-nAChR and α7-nAChR that subsequently alter physiological and behavioral effects of nicotinic drugs [5, 42, 62-64]. We reported here that86Rb+ efflux mediated by α4β2*-nAChR that differ in sensitivity to activation by ACh and other nicotinic agonists, perhaps because of differences in subunit stoichiometry, show very similar desensitization upon exposure to low concentrations of each of four agonists. Consequently, the subset of receptors activated at lower agonist concentrations display significantly greater residual activity than the subset activated at higher agonist concentrations. Since both activation and desensitization are likely to be important in mediating the physiological and behavioral responses to nicotinic drugs in vivo [65-67], this difference in persistent activity of α4β2*-nAChR subtypes, in addition to differences between other nAChR subtypes, could have significant consequences when the receptors are exposed to nicotinic agonists for prolonged periods of time, either from smoking or therapeutically.
Acknowledgments
This work was supported by grants R01 DA003194 and P30 DA015663 from the National Institutes on Drug Abuse, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Michael J. Marks, Email: marksm@colorado.edu.
Natalie M. Meinerz, Email: natalie.meinerz@colorado.edu.
Robert W. B. Brown, Email: robwbbrown@googlemail.com.
Allan C. Collins, Email: al.collins@colorado.edu.
References
- 1.Quick MW, Lester RA. Desensitization of neuronal nicotinic receptors. J Neurobiol. 2002;53:457–78. doi: 10.1002/neu.10109. [DOI] [PubMed] [Google Scholar]
- 2.Fenster CP, Rains MF, Noerager B, Quick MW, Lester RA. Influence of subunit composition on desensitization of neuronal acetylcholine receptors at low concentrations of nicotine. J Neurosci. 1997;17:5747–59. doi: 10.1523/JNEUROSCI.17-15-05747.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lester RA. Activation and desensitization of heteromeric neuronal nicotinic receptors: implications for non-synaptic transmission. Bioorg Med Chem Lett. 2004;14:1897–900. doi: 10.1016/j.bmcl.2004.02.081. [DOI] [PubMed] [Google Scholar]
- 4.Paradiso KG, Steinbach JH. Nicotine is highly effective at producing desensitization of rat alpha4beta2 neuronal nicotinic receptors. J Physiol. 2003;553:857–71. doi: 10.1113/jphysiol.2003.053447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wooltorton JR, Pidoplichko VI, Broide RS, Dani JA. Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J Neurosci. 2003;23:3176–85. doi: 10.1523/JNEUROSCI.23-08-03176.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fenster CP, Whitworth TL, Sheffield EB, Quick MW, Lester RA. Upregulation of surface alpha4beta2 nicotinic receptors is initiated by receptor desensitization after chronic exposure to nicotine. J Neurosci. 1999;19:4804–14. doi: 10.1523/JNEUROSCI.19-12-04804.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marks MJ, Grady SR, Yang JM, Lippiello PM, Collins AC. Desensitization of nicotine-stimulated 86Rb+ efflux from mouse brain synaptosomes. J Neurochem. 1994;63:2125–35. doi: 10.1046/j.1471-4159.1994.63062125.x. [DOI] [PubMed] [Google Scholar]
- 8.Marks MJ, Robinson SF, Collins AC. Nicotinic agonists differ in activation and desensitization of 86Rb+ efflux from mouse thalamic synaptosomes. J Pharmacol Exp Ther. 1996;277:1383–96. [PubMed] [Google Scholar]
- 9.Zwart R, Vijverberg HP. Four pharmacologically distinct subtypes of alpha4beta2 nicotinic acetylcholine receptor expressed in Xenopus laevis oocytes. Mol Pharmacol. 1998;54:1124–31. [PubMed] [Google Scholar]
- 10.Nelson ME, Kuryatov A, Choi CH, Zhou Y, Lindstrom J. Alternate stoichiometries of alpha4beta2 nicotinic acetylcholine receptors. Mol Pharmacol. 2003;63:332–41. doi: 10.1124/mol.63.2.332. [DOI] [PubMed] [Google Scholar]
- 11.Zhou Y, Nelson ME, Kuryatov A, Choi C, Cooper J, Lindstrom J. Human alpha4beta2 acetylcholine receptors formed from linked subunits. J Neurosci. 2003;23:9004–15. doi: 10.1523/JNEUROSCI.23-27-09004.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moroni M, Bermudez I. Stoichiometry and pharmacology of two human alpha4beta2 nicotinic receptor types. J Mol Neurosci. 2006;30:95–6. doi: 10.1385/JMN:30:1:95. [DOI] [PubMed] [Google Scholar]
- 13.Marks MJ, Meinerz NM, Drago J, Collins AC. Gene targeting demonstrates that alpha4 nicotinic acetylcholine receptor subunits contribute to expression of diverse [3H]epibatidine binding sites and components of biphasic 86Rb+ efflux with high and low sensitivity to stimulation by acetylcholine. Neuropharmacology. 2007;53:390–405. doi: 10.1016/j.neuropharm.2007.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marks MJ, Whiteaker P, Calcaterra J, Stitzel JA, Bullock AE, Grady SR, et al. Two pharmacologically distinct components of nicotinic receptor-mediated rubidium efflux in mouse brain require the beta2 subunit. J Pharmacol Exp Ther. 1999;289:1090–103. [PubMed] [Google Scholar]
- 15.Gotti C, Moretti M, Meinerz NM, Clementi F, Gaimarri A, Collins AC, et al. Partial deletion of the nicotinic cholinergic receptor alpha 4 or beta 2 subunit genes changes the acetylcholine sensitivity of receptor-mediated 86Rb+ efflux in cortex and thalamus and alters relative expression of alpha 4 and beta 2 subunits. Mol Pharmacol. 2008;73:1796–807. doi: 10.1124/mol.108.045203. [DOI] [PubMed] [Google Scholar]
- 16.Brown RW, Collins AC, Lindstrom JM, Whiteaker P. Nicotinic alpha5 subunit deletion locally reduces high-affinity agonist activation without altering nicotinic receptor numbers. J Neurochem. 2007;103:204–15. doi: 10.1111/j.1471-4159.2007.04700.x. [DOI] [PubMed] [Google Scholar]
- 17.Marks MJ, Rowell PP, Cao JZ, Grady SR, McCallum SE, Collins AC. Subsets of acetylcholine-stimulated 86Rb+ efflux and [125I]-epibatidine binding sites in C57BL/6 mouse brain are differentially affected by chronic nicotine treatment. Neuropharmacology. 2004;46:1141–57. doi: 10.1016/j.neuropharm.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz RD, Kellar KJ. Nicotinic cholinergic receptor binding sites in the brain: regulation in vivo. Science. 1983;220:214–6. doi: 10.1126/science.6828889. [DOI] [PubMed] [Google Scholar]
- 19.Marks MJ, Burch JB, Collins AC. Effects of chronic nicotine infusion on tolerance development and nicotinic receptors. J Pharmacol Exp Ther. 1983;226:817–25. [PubMed] [Google Scholar]
- 20.Whiteaker P, Marks MJ, Grady SR, Lu Y, Picciotto MR, Changeux JP, et al. Pharmacological and null mutation approaches reveal nicotinic receptor diversity. Eur J Pharmacol. 2000;393:123–35. doi: 10.1016/s0014-2999(00)00052-2. [DOI] [PubMed] [Google Scholar]
- 21.Romm E, Lippiello PM, Marks MJ, Collins AC. Purification of L-[3H]nicotine eliminates low affinity binding. Life Sci. 1990;46:935–43. doi: 10.1016/0024-3205(90)90095-9. [DOI] [PubMed] [Google Scholar]
- 22.Lippiello PM, Sears SB, Fernandes KG. Kinetics and mechanism of L-[3H]nicotine binding to putative high affinity receptor sites in rat brain. Mol Pharmacol. 1987;31:392–400. [PubMed] [Google Scholar]
- 23.Bhat RV, Marks MJ, Collins AC. Effects of chronic nicotine infusion on kinetics of high-affinity nicotine binding. J Neurochem. 1994;62:574–81. doi: 10.1046/j.1471-4159.1994.62020574.x. [DOI] [PubMed] [Google Scholar]
- 24.Katz B, Thesleff S. A study of the desensitization produced by acetylcholine at the motor end-plate. J Physiol. 1957;138:63–80. doi: 10.1113/jphysiol.1957.sp005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lester RA, Dani JA. Acetylcholine receptor desensitization induced by nicotine in rat medial habenula neurons. J Neurophysiol. 1995;74:195–206. doi: 10.1152/jn.1995.74.1.195. [DOI] [PubMed] [Google Scholar]
- 26.Khiroug SS, Khiroug L, Yakel JL. Rat nicotinic acetylcholine receptor alpha2beta2 channels: comparison of functional properties with alpha4beta2 channels in Xenopus oocytes. Neuroscience. 2004;124:817–22. doi: 10.1016/j.neuroscience.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 27.Moroni M, Zwart R, Sher E, Cassels BK, Bermudez I. alpha4beta2 nicotinic receptors with high and low acetylcholine sensitivity: pharmacology, stoichiometry, and sensitivity to long-term exposure to nicotine. Mol Pharmacol. 2006;70:755–68. doi: 10.1124/mol.106.023044. [DOI] [PubMed] [Google Scholar]
- 28.Carbone AL, Moroni M, Groot-Kormelink PJ, Bermudez I. Pentameric concatenated (alpha4)(2)(beta2)(3) and (alpha4)(3)(beta2)(2) nicotinic acetylcholine receptors: subunit arrangement determines functional expression. Br J Pharmacol. 2009;156:970–81. doi: 10.1111/j.1476-5381.2008.00104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marubio LM, del Mar Arroyo-Jimenez M, Cordero-Erausquin M, Lena C, Le Novere N, de Kerchove d’Exaerde A, et al. Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature. 1999;398:805–10. doi: 10.1038/19756. [DOI] [PubMed] [Google Scholar]
- 30.Picciotto MR, Zoli M, Lena C, Bessis A, Lallemand Y, Le Novere N, et al. Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature. 1995;374:65–7. doi: 10.1038/374065a0. [DOI] [PubMed] [Google Scholar]
- 31.Ross SA, Wong JY, Clifford JJ, Kinsella A, Massalas JS, Horne MK, et al. Phenotypic characterization of an alpha 4 neuronal nicotinic acetylcholine receptor subunit knock-out mouse. J Neurosci. 2000;20:6431–41. doi: 10.1523/JNEUROSCI.20-17-06431.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuryatov A, Onksen J, Lindstrom J. Roles of accessory subunits in alpha4beta2(*) nicotinic receptors. Mol Pharmacol. 2008;74:132–43. doi: 10.1124/mol.108.046789. [DOI] [PubMed] [Google Scholar]
- 33.Mao D, Perry DC, Yasuda RP, Wolfe BB, Kellar KJ. The alpha4beta2alpha5 nicotinic cholinergic receptor in rat brain is resistant to up-regulation by nicotine in vivo. J Neurochem. 2008;104:446–56. doi: 10.1111/j.1471-4159.2007.05011.x. [DOI] [PubMed] [Google Scholar]
- 34.Marks MJ, Whiteaker P, Grady SR, Picciotto MR, McIntosh JM, Collins AC. Characterization of [(125) I]epibatidine binding and nicotinic agonist-mediated (86) Rb(+) efflux in interpeduncular nucleus and inferior colliculus of beta2 null mutant mice. J Neurochem. 2002;81:1102–15. doi: 10.1046/j.1471-4159.2002.00910.x. [DOI] [PubMed] [Google Scholar]
- 35.Kuryatov A, Luo J, Cooper J, Lindstrom J. Nicotine acts as a pharmacological chaperone to up-regulate human alpha4beta2 acetylcholine receptors. Mol Pharmacol. 2005;68:1839–51. doi: 10.1124/mol.105.012419. [DOI] [PubMed] [Google Scholar]
- 36.Marks MJ, Wageman CR, Grady SR, Gopalakrishnan M, Briggs CA. Selectivity of ABT-089 for alpha4beta2* and alpha6beta2* nicotinic acetylcholine receptors in brain. Biochem Pharmacol. 2009;78:795–802. doi: 10.1016/j.bcp.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rollema H, Coe JW, Chambers LK, Hurst RS, Stahl SM, Williams KE. Rationale, pharmacology and clinical efficacy of partial agonists of alpha4beta2 nACh receptors for smoking cessation. Trends Pharmacol Sci. 2007;28:316–25. doi: 10.1016/j.tips.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 38.Tonstad S, Rollema H. Varenicline in smoking cessation. Expert Rev Respir Med. 2010;4:291–9. doi: 10.1586/ers.10.27. [DOI] [PubMed] [Google Scholar]
- 39.Keating GM, Lyseng-Williamson KA. Varenicline: a pharmacoeconomic review of its use as an aid to smoking cessation. Pharmacoeconomics. 2010;28:231–54. doi: 10.2165/11204380-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 40.Rowell PP, Li M. Dose-response relationship for nicotine-induced up-regulation of rat brain nicotinic receptors. J Neurochem. 1997;68:1982–9. doi: 10.1046/j.1471-4159.1997.68051982.x. [DOI] [PubMed] [Google Scholar]
- 41.Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ. A subtype of nicotinic cholinergic receptor in rat brain is composed of alpha 4 and beta 2 subunits and is up-regulated by chronic nicotine treatment. Mol Pharmacol. 1992;41:31–7. [PubMed] [Google Scholar]
- 42.Nashmi R, Xiao C, Deshpande P, McKinney S, Grady SR, Whiteaker P, et al. Chronic nicotine cell specifically upregulates functional alpha 4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J Neurosci. 2007;27:8202–18. doi: 10.1523/JNEUROSCI.2199-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgmeyer I, Heinemann SF, et al. Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. J Neurosci. 1992;12:2765–84. doi: 10.1523/JNEUROSCI.12-07-02765.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benwell ME, Balfour DJ, Anderson JM. Evidence that tobacco smoking increases the density of (-)-[3H]nicotine binding sites in human brain. J Neurochem. 1988;50:1243–7. doi: 10.1111/j.1471-4159.1988.tb10600.x. [DOI] [PubMed] [Google Scholar]
- 45.Breese CR, Marks MJ, Logel J, Adams CE, Sullivan B, Collins AC, et al. Effect of smoking history on [3H]nicotine binding in human postmortem brain. J Pharmacol Exp Ther. 1997;282:7–13. [PubMed] [Google Scholar]
- 46.Perry DC, Davila-Garcia MI, Stockmeier CA, Kellar KJ. Increased nicotinic receptors in brains from smokers: membrane binding and autoradiography studies. J Pharmacol Exp Ther. 1999;289:1545–52. [PubMed] [Google Scholar]
- 47.Peng X, Gerzanich V, Anand R, Whiting PJ, Lindstrom J. Nicotine-induced increase in neuronal nicotinic receptors results from a decrease in the rate of receptor turnover. Mol Pharmacol. 1994;46:523–30. [PubMed] [Google Scholar]
- 48.Gopalakrishnan M, Monteggia LM, Anderson DJ, Molinari EJ, Piattoni-Kaplan M, Donnelly-Roberts D, et al. Stable expression, pharmacologic properties and regulation of the human neuronal nicotinic acetylcholine alpha 4 beta 2 receptor. J Pharmacol Exp Ther. 1996;276:289–97. [PubMed] [Google Scholar]
- 49.Whiteaker P, Sharples CG, Wonnacott S. Agonist-induced up-regulation of alpha4beta2 nicotinic acetylcholine receptors in M10 cells: pharmacological and spatial definition. Mol Pharmacol. 1998;53:950–62. [PubMed] [Google Scholar]
- 50.Pauly JR, Marks MJ, Gross SD, Collins AC. An autoradiographic analysis of cholinergic receptors in mouse brain after chronic nicotine treatment. J Pharmacol Exp Ther. 1991;258:1127–36. [PubMed] [Google Scholar]
- 51.Sanderson EM, Drasdo AL, McCrea K, Wonnacott S. Upregulation of nicotinic receptors following continuous infusion of nicotine is brain-region-specific. Brain Res. 1993;617:349–52. doi: 10.1016/0006-8993(93)91104-z. [DOI] [PubMed] [Google Scholar]
- 52.Sparks JA, Pauly JR. Effects of continuous oral nicotine administration on brain nicotinic receptors and responsiveness to nicotine in C57Bl/6 mice. Psychopharmacology (Berl) 1999;141:145–53. doi: 10.1007/s002130050818. [DOI] [PubMed] [Google Scholar]
- 53.Nguyen HN, Rasmussen BA, Perry DC. Subtype-selective up-regulation by chronic nicotine of high-affinity nicotinic receptors in rat brain demonstrated by receptor autoradiography. J Pharmacol Exp Ther. 2003;307:1090–7. doi: 10.1124/jpet.103.056408. [DOI] [PubMed] [Google Scholar]
- 54.Schwartz RD, Kellar KJ. In vivo regulation of [3H]acetylcholine recognition sites in brain by nicotinic cholinergic drugs. J Neurochem. 1985;45:427–33. doi: 10.1111/j.1471-4159.1985.tb04005.x. [DOI] [PubMed] [Google Scholar]
- 55.Buisson B, Bertrand D. Chronic exposure to nicotine upregulates the human (alpha)4((beta)2 nicotinic acetylcholine receptor function. J Neurosci. 2001;21:1819–29. doi: 10.1523/JNEUROSCI.21-06-01819.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marks MJ, Stitzel JA, Romm E, Wehner JM, Collins AC. Nicotinic binding sites in rat and mouse brain: comparison of acetylcholine, nicotine, and alpha-bungarotoxin. Mol Pharmacol. 1986;30:427–36. [PubMed] [Google Scholar]
- 57.Nashmi R, Lester H. Cell autonomy, receptor autonomy, and thermodynamics in nicotine receptor up-regulation. Biochem Pharmacol. 2007;74:1145–54. doi: 10.1016/j.bcp.2007.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kishi M, Steinbach JH. Role of the agonist binding site in up-regulation of neuronal nicotinic alpha4beta2 receptors. Mol Pharmacol. 2006;70:2037–44. doi: 10.1124/mol.106.029298. [DOI] [PubMed] [Google Scholar]
- 59.Marks MJ, Grady SR, Collins AC. Downregulation of nicotinic receptor function after chronic nicotine infusion. J Pharmacol Exp Ther. 1993;266:1268–76. [PubMed] [Google Scholar]
- 60.Kuryatov A, Olale FA, Choi C, Lindstrom J. Acetylcholine receptor extracellular domain determines sensitivity to nicotine-induced inactivation. Eur J Pharmacol. 2000;393:11–21. doi: 10.1016/s0014-2999(00)00027-3. [DOI] [PubMed] [Google Scholar]
- 61.Nguyen HN, Rasmussen BA, Perry DC. Binding and functional activity of nicotinic cholinergic receptors in selected rat brain regions are increased following long-term but not short-term nicotine treatment. J Neurochem. 2004;90:40–9. doi: 10.1111/j.1471-4159.2004.02482.x. [DOI] [PubMed] [Google Scholar]
- 62.Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002;33:905–19. doi: 10.1016/s0896-6273(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 63.Mao D, McGehee DS. Nicotine and behavioral sensitization. J Mol Neurosci. 2010;40:154–63. doi: 10.1007/s12031-009-9230-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xiao C, Nashmi R, McKinney S, Cai H, McIntosh JM, Lester HA. Chronic nicotine selectively enhances alpha4beta2* nicotinic acetylcholine receptors in the nigrostriatal dopamine pathway. J Neurosci. 2009;29:12428–39. doi: 10.1523/JNEUROSCI.2939-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Picciotto MR, Addy NA, Mineur YS, Brunzell DH. It is not “either/or”: activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog Neurobiol. 2008;84:329–42. doi: 10.1016/j.pneurobio.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hulihan-Giblin BA, Lumpkin MD, Kellar KJ. Acute effects of nicotine on prolactin release in the rat: agonist and antagonist effects of a single injection of nicotine. J Pharmacol Exp Ther. 1990;252:15–20. [PubMed] [Google Scholar]
- 67.Sharp BM, Beyer HS. Rapid desensitization of the acute stimulatory effects of nicotine on rat plasma adrenocorticotropin and prolactin. J Pharmacol Exp Ther. 1986;238:486–91. [PubMed] [Google Scholar]