

Abstract
The long circulation persistence of human serum albumin (HSA) is enabled by its domain III (DIII) interaction with the neonatal Fc receptor (FcRn). A protein scaffold based on HSA DIII was designed. To modify the serum half life of the scaffold, residues H535, H510, and H464 were individually mutated to alanine. HSA DIII wild type (WT) and variants were fused to the anti-carcinoembryonic antigen (CEA) T84.66 diabody (Db), radiolabeled with 124I and injected into xenografted athymic mice for serial PET/CT imaging. All proteins targeted the CEA-positive tumor. The mean residence times (MRT) of the proteins, calculated by quantifying blood activity from the PET images, were: Db-DIII WT (56.7 h), H535A (25 h), H510A (20 h), H464A (17 h), compared with Db (2.9 h). Biodistribution confirmed the order of blood clearance from slow to fast: Db-DIII WT > H535A > H510A > H464A > Db with 4.0, 2.0, 1.8, 1.6 and 0.08 %ID/g of remaining blood activity at 51 h, respectively. This study demonstrates that attenuating the DIII–FcRn interaction provides a way of controlling the pharmacokinetics of the entire Db-DIII fusion protein without compromising tumor targeting. H464 appears to be most crucial for FcRn binding (greatest reduction in MRT), followed by H510 and H535. By mutating the DIII scaffold, we can dial serum kinetics for imaging or therapy applications.
Keywords: blood clearance, FcRn, HSA DIII, PET imaging, protein scaffold
Introduction
Within the past decade, combinatorial library technology has yielded a large number of molecules, including peptides, aptamers and small chemical molecules, selected to bind various targets or tissues with great specificity and affinity (Aina et al., 2007; Barbas and White, 2009; Bembenek et al., 2009). Yet, when administered in vivo, these molecules often exhibit suboptimal pharmacokinetics characterized by transient serum persistence and inability to accumulate at the target site to sufficient levels for either imaging or therapy applications. To address this problem, we proposed the following hypothesis. If low molecular weight tumor targeting molecules are attached, grafted or displayed onto a protein scaffold characterized by intrinsic serum stability, then improved pharmacokinetic profile and target uptake can be achieved. Maximizing tumor accumulation could potentially translate into a stronger signal in imaging applications or a sufficient drug payload delivery in therapy. In addition, the scaffold can provide residues for conjugation of a functional group (e.g. radionuclide, cytotoxic drug, toxin) and may also enhance the solubility of hydrophobic-targeting molecules. An ideal scaffold should be (i) non-immunogenic, (ii) capable of providing optimal serum persistence for different applications (tunable), (iii) low in molecular mass, facilitating extravasation, tumor penetration and renal clearance (<60 kDa), which is preferable for imaging application, (iv) a platform for increasing the functional affinity of targeting molecules by using the avidity effect (2–3 targeting molecules on the same scaffold) or introducing multiple specificities, and (v) soluble in serum, rendering molecules attached to its surface also soluble.
Human serum albumin (HSA; 67 kDa) is the most abundant protein in the human blood (30–50 g/l) and has already been incorporated into an approved pharmaceutical (i.e. Albuferon®, Novartis). In preclinical studies, HSA has successfully been utilized as a carrier molecule for drug delivery (Kratz et al., 2000; Burger et al., 2001; Wosikowski et al., 2003) and a vector for gene delivery (Aina et al., 2007). As a fusion protein, HSA has demonstrated its ability to improve the pharmacokinetics of molecules, such as interferon-α (Osborn et al., 2002), interleukin-2 (Melder et al., 2005), recombinant bispecific antibody molecule (Muller et al., 2007) or scFv antibody fragment (Yazaki et al., 2008). Similar to IgG, HSA interacts with the neonatal Fc receptor (FcRn), also known as Brambell receptor (Chaudhury et al., 2003). This interaction is responsible for the extended serum persistence of albumin. It is likely that albumin traffics in the same way as IgG, according to the following model. Albumin molecules in the circulation are taken by vascular endothelial cells through fluid phase endocytosis. Albumin binds the FcRn in the early endosome (∼pH 6.0). Most FcRn bound albumin leaves the early endosome in tubulovesicular recycling compartments. These compartments transcytose or move to the cell surface and exocytose, releasing albumin back in blood (pH 7.4). The late endosome either fuses completely or partially to a lysosome, delivering its content for degradation (Gan et al., 2009). Specifically, HSA domain III (DIII; 23 kDa) has been shown to be both necessary and sufficient for FcRn binding in a pH-dependent manner (Chaudhury et al., 2006). Owing to the similarity in IgG1 and albumin interaction with FcRn, it is likely that the histidine residues in HSA DIII alike the histidine residues in IgG1 Fc region mediate this interaction (Medesan et al., 1997; Kim et al., 1999; Shields et al., 2001). In acidic environments, the imidazole side chain (pKa = 6.0) of histidine residues is protonated; thus positively charged and capable of binding acidic residues in FcRn. The DIII has four histidine residues, of which three (H535, H510 and H464) are conserved in humans, mice, rats and bovines. Therefore, it is likely that these three residues play a role in the pH-depended mechanism of albumin–FcRn binding and release. Only H464 has been shown to undergo pH-dependent protonation between pH 5.0 and pH 7.0 during neutral-to-base transition of HSA (Bos et al., 1989; Chaudhury et al., 2006). The role that H535 and H510 play in the FcRn receptor interactions has not been elucidated and individual mutation of all three (H535, H510 and H464) residues is therefore necessary to reveal their relative importance for maintaining the serum half life of albumin. This may also provide the opportunity for fine tuning of serum persistence, as the requirements for circulation half life are different in imaging and therapy applications. Since HSA DIII alone is a small protein of human origin, it fits the criteria for a scaffold that may eventually find clinical application.
In modern medicine, the ability to dial in a desirable pharmacokinetics for diagnostics or therapeutics is highly valued. Thus, our goal is not only to create a molecule that can extend serum persistence (e.g. polyethylene glycol, dextran), but to also provide a spectrum of circulation half lives without changing molecular mass. As a proof of concept that HSA DIII can be such molecule, we generated fusion proteins consisting of a divalent antibody fragment targeting carcinoembryonic antigen (CEA) and either the HSA DIII wild type (WT, non-mutated) or one of three HSA DIII variants, each incorporating a mutation of H535, H510 or H464 to alanine residue. Xenografted athymic nude mice were injected with 124I-labeled proteins, and serial small animal PET/CT imaging studies were performed to evaluate the ability of the HSA DIII to modulate the serum persistence of the antibody fragment in vivo. In addition, we were able to draw conclusions about the relative importance of the H535, H510 and H464 residues for FcRn binding and circulation persistence of albumin.
Materials and methods
Generation of Db-DIII constructs
HSA DIII genes were amplified by polymerase chain reaction using commercial HSA cDNA (OriGene Technologies, Rockville, MD, USA) as a template and primers introducing 5′ SpeI and 3′ EcoRI restriction sites. The primer sequences were as follows:
Forward: SpeI-DIII: 5′-CCACTAGTGGCGAAGAGCCTCAGAATTTAATC-3′
Reverse: DIII-EcoRI: 5′-GAGAATTCTATTATAAGCCTAAGGCAGCTTGAC-3′
Mutations of histidine residues H535, H510 or H464 to alanine in the DIII were introduced by site-directed mutagenesis, using a Quick-Change mutagenesis kit (Stratagene, La Jolla, CA, USA) with the appropriate mutagenesis primers (only forward primers are listed):
H464A (exchanging histidine residue in position 464 with an alanine residue): 5′-CTGAACCAGTTATGTGTGTTGGCTGAGAAAACGCCAGTAAGTGAC-3′
H510A: 5′-GTTTAATGCTGAAACATTCACCTTCGCTGCAGATATATGCACAC-3′
H535A: 5′-CTGCACTTGTTGAGCTCGTGAAAGCCAAGCCCAAGGCAAC-3′
The complete DIII (WT, H535A, H510A and H464A) genes were cloned in pCR2.1-Topo vector (Invitrogen, Carlsbad, CA, USA) and then transferred into the pUC18 vector (New England Biolabs, Ipswich, MA, USA), already containing the anti-CEA T84.66 diabody (Db; Wu et al., 1999). The Db is a cross-paired dimer where the variable light (VL) region of one antibody single-chain Fv (scFv) monomer associates with the variable heavy (VH) region of the second scFv monomer, and the second VL pairs with the first VH. The parental T84.66 monoclonal antibody, whose VL and VH domains have been used to engineer the anti-CEA Db, has demonstrated high affinity (2.6 × 1010 M−1) and specificity for CEA (Neumaier et al., 1990). The complete Db-DIII genes were excised from the pUC18 vector and ligated into the pEE12 mammalian expression vector (Bebbington et al., 1992), using XbaI and EcoRI sites.
Expression, selection and purification
NS0 murine myeloma cells (Sigma-Aldrich, St Louis, MO, USA) were maintained in non-selective glutamine-free Dulbecco's modified Eagle's Medium (DME/High Modified; SAFC Biosciences, Lenexa, KS, USA), supplemented with 5% heat inactivated fetal bovine serum (FBS; Omega Scientific Inc., Tarzana, CA, USA), 1% v/v of 200 mM l-glutamine (Mediatech, Inc., Manassas, VA, USA) and 1% v/v of Penicillin–Streptomycin (10 000 IU/ml penicillin, 10 000 μg/ml streptomycin; Mediatech Inc.). A total of 1 × 107 NS0 cells in the logarithmic growth phase were transfected by electroporation with 10 μg of pEE12-Db-DIII DNA, linearized by digestion with SalI (New England Biolabs), as described previously (Kenanova et al., 2005).
Db-DIII production was assayed by ELISA and confirmed by Western blot. For ELISA, Protein A (Thermo Fisher Scientific, Rockford, IL, USA) was used to capture the Db-DIII proteins. Alkaline phosphatase (AP)-conjugated anti-mouse Fab-specific antibody (Sigma-Aldrich) served for detection in both ELISA and Western blot. Transfected NS0 cells were maintained in selective glutamine-free DME/High-Modified medium (SAFC Biosciences), supplemented with 5% heat inactivated, dialyzed FBS (Omega Scientific Inc.), 2% v/v of ×50 GS supplement (SAFC Biosciences) and 1% v/v Penicillin–Streptomycin (Mediatech Inc.). Selected clones, expressing high amounts of Db-DIII proteins, were gradually expanded into triple flasks (Nunclon, Rochester, NY, USA), containing 300 ml selective media, supplemented with 2% heat inactivated, dialyzed FBS and 1% v/v Penicillin–Streptomycin.
When cultures reached terminal state (∼3 weeks), harvested supernatants were centrifuged, filter sterilized and concentrated, using a Lab Scale tangential flow filtration system (Millipore, Billerica, MA, USA) with a 30 000 Da molecular weight cut-off (mwco) filter. Db-DIII proteins were purified on a Protein A column (Thermo Fisher Scientific, Waltham, MA, USA), using an AKTA Purifier (GE Healthcare, Piscataway, NJ, USA). The bound protein was eluted at 15% of 0.2 M Citrate buffer (pH 2.1) in 1× PBS, and pH was immediately neutralized by adding 80% v/v of 1 M Tris base (pH 8.2) directly to the eluted proteins. Fractions containing pure Db-DIII protein were pooled, dialyzed against 1× PBS and concentrated by Vivaspin 20 (mwco: 30 000; Sartorius Stedim Biotech Gmbh, Goettingen, Germany). The final concentration of purified Db-DIII proteins was determined by A280, using an extinction coefficient ε = 1.5, which was determined using a BCA Protein Assay Kit (Thermo Scientific, Rockford, IL, USA) according to the manufacturer's instructions.
Characterization of Db-DIII proteins
Purified Db-DIII proteins were analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) under non-reducing and reducing conditions, Western blot, mass spectrometry and size exclusion chromatography. To reduce the protein, 1 M dithiothreitol (DTT) was added to a final concentration of 0.2 M and boiled for 10 min. For the SDS–PAGE, 4–20% gradient Tris–HCl ready gels (Bio-Rad Laboratories, Hercules, CA, USA) were run and developed in Instant Blue Coomassie-based solution (Expedion Protein Solutions, Cambridge, UK). Detection of the Db-DIII proteins in Western blots was accomplished with (i) AP-conjugated goat anti-mouse Fab-specific Ab (Sigma-Aldrich) using nitro blue tetrazolium (NBT) and 5-bromo-4-chloro-3-indolyl-phosphate (BCIP) (Promega, Madison, WI, USA) AP substrates, or (ii) horseradish peroxidase (HRP)-conjugated Protein L (Sigma-Aldrich) developed with the 4-chloro-1-naphthol/3,3’-diaminobenzidine (CN/DAB) substrate kit (Thermo Scientific, Rockfort, IL, USA).
Mass spectrometry using an LTQ-FT Ultra Linear Ion Trap Fourier Transform Ion Cyclotron Resonance (FT-ICR) mass spectrometer (Thermo Fisher) was performed to confirm the identity of the purified proteins. Briefly, Db-DIII proteins were isolated following an in-gel trypsin digestion procedure, described in detail at http://massspec.wiki.zoho.com/In-Gel-Trypsin-Digest.html. Nano-liquid chromatography with tandem mass spectrometry and collisionally activated dissociation fragmentation was performed on an LTQ-FT (Thermo Fisher) integrated with an Eksigent nano-LC. Spectra were searched against the most up-to-date International Protein Index database (Version 3.54 with 39 925 entries) using the Mascot (Matrix, Boston, MA, USA) and Sequest (Thermo Fisher) programs. The results were filtered with a strict score filtering criterion and a 10 ppm mass resolution filter. Identified peptides were also matched to the Db-DIII sequence.
Determination of Db-DIII protein purity after purification, Db-DIII protein conformation under native, non-denaturing conditions (1× PBS, pH 7.4) and estimation of molecular size was accomplished through size exclusion chromatography using a Superdex 200 HR 10/30 column (GE Healthcare).
Computer models of DIII and Db-DIII molecules were generated using the PyMOL software (DeLano Scientific). Additionally, modeling of protein docking between FcRn and DIII was accomplished using the ZDOCK-FFT algorithm (Chen et al., 2003), available on a public server (http://zlab.bu.edu/~rong/dock).
Radioiodination of Db-DIII fusion proteins, xenograft imaging and biodistribution
Purified Db-DIII WT, H535A, H510A and H464A proteins were radioiodinated with the positron emitter 124I (sodium iodide in 0.02 M NaOH; IBA Molecular, Sterling, VA, USA) using the Iodogen method as described previously (Olafsen et al., 2006). Labeling reactions (0.114–0.130 ml) contained 0.1 mg of purified protein and 12.9–18.0 MBq Na124I. Labeling efficiency was measured by instant thin layer chromatography (ITLC) using the monoclonal antibody ITLC strips kit (Biodex Medical Systems, Shirly, NY, USA), as described previously (Olafsen et al., 2006). Immunoreactivity after labeling was determined by incubation of small amount of radiolabeled protein (∼50 000 cpm) with ∼1 × 107 LS174T cells. Percent immunoreactivity was calculated by the formula: [(activity associated only with the cells after washing)/(total activity of cells and washes)] × 100.
For in vivo studies, 7–8-week-old athymic nude mice (Charles River Laboratories, Wilmington, MA, USA) were injected subcutaneously in the left-shoulder region with 1 × 106–5 × 106 CEA-positive LS174T human colon carcinoma cells [American Type Culture Collection (ATCC), Manassas, VA, USA] and in the right-shoulder area with approximately the same number of CEA-negative C6 rat glioma cells (ATCC). Tumor masses were allowed to develop for an average of 10 days and reached a maximum of 200 mg weight. Four tumor bearing mice per construct were injected in the tail vein with 3.9–5.4 MBq 124I-labeled Db-DIII or Db in saline/1% HSA. At five different time points (4, 20, 28, 44 and 51 h), the injected mice were anesthetized using 2% isoflurane, placed on the bed and imaged for 10 min. A 10-min CT scan was completed following the final PET scan at 51 h. All imaging experiments utilized the Focus 220 small animal PET (Siemens Preclinical Solutions, Knoxville, TN, USA) and the small animal CAT II (Concorde Microsystems, Knoxville, TN, USA) scanners. Following the last scan (51 h), mice were euthanized. Blood, tumors (LS174T and C6), liver, spleen, kidneys, lungs and carcass were collected, weighed and counted in a Wallac WIZARD Automatic Gamma Counter (PerkinElmer Life and Analytical Sciences Inc., Wellesley, MA, USA). After decay correction, the percent injected dose per gram (%ID/g) for each tissue or organ was calculated, incorporating a correction for the labeling efficiency and immunoreactivity of each protein and a standard error (SE).
Image analysis and statistics
All images were reconstructed using a filtered back projection algorithm (Defrise et al., 1997) and displayed by the AMIDE software (Loening and Gambhir, 2003). The same color threshold was applied to all images. Regions of interest (ROIs; ellipsoid, 0.4 mm depth, n = 4) were drawn in the area of the CEA-positive tumor and in a low-activity, soft tissue region of the lower body (muscle). Tumor-to-soft tissue (T:ST) ratios were determined for individual mice and averaged for each time point and construct. ROIs (n = 4) were also drawn over the heart on each image, and %ID/g of blood was calculated by the AMIDE software after entering the injected dose (in MBq) and a cylinder factor (in MBq/cc/image units) as input functions. The ADAPTII software package was used for curve fitting and calculating the mean residence time (MRT) of each protein from its blood activity curve (D'Argenio and Schumitzky, 1979). Specifically, the blood time-activity curve of each protein was fitted with a new curve in order to calculate the area under the plasma concentration curve (AUC) from time 0 to infinity. AUC values for a specific agent can be used for determining pharmacokinetic parameters such as clearance and bioavailability. MRT, which is a measure of the agent average clearance time, was defined as the ratio of the area under the first moment curve (AUMC) to AUC (MRT = AUMC/AUC).
SE was calculated for all ratios and %ID/g values and expressed graphically (error bars). All T:ST ROI ratios and blood activity curves, respectively, were compared for a significant difference using an unpaired Student's t-test. A two-tailed P-value of ≤0.05 was considered statistically significant.
Results
Production and biochemical characterization of Db-DIII proteins
The assembled Db-DIII gene construct, shown in Fig. 1A, is ∼1.4kb pairs long and flanked by XbaI and EcoRI restriction sites. The engineered Db-DIII molecules were expressed at 10–16 µg/ml in terminal cultures of transfected NS0 cells, as determined by ELISA. T84.66 Db fusion proteins with Gaussia and Renilla luciferases were previously purified by Protein L affinity chromatography (Venisnik et al., 2006, 2007). However, we found that the Db-DIII proteins bound better to Protein A resin (Riechmann and Davies, 1995; Starovasnik et al., 1999). For this reason, Protein A affinity chromatography was selected for purification of all the Db-DIII proteins. Because the Db is a non-covalent dimer of two scFv molecules, each Db molecule has two DIII proteins attached to its C-termini, resulting in a fusion protein of ∼101 kDa calculated molecular mass (Fig. 1B).
Fig. 1.

(A) Gene assembly of the Db-DIII constructs. L, signal peptide leader for mammalian cell secretion; variable light (VL) and variable heavy (VH) antibody chains are joined through an 8 (glycine, serine rich) amino acid linker to form a single-chain fragment variable (scFv, 25 kDa). The scFv is connected to the HSA DIII gene by an 18 amino acid linker. DIII is flanked by SpeI and EcoRI restriction sites in a cassette to facilitate the exchange of one DIII with another (e.g. WT for H535A, etc.). (B) Cartoon representation of the Db-DIII protein, where two scFv-DIII molecules form a non-covalent dimer.
Purified Db-DIII WT and variants were analyzed by SDS–PAGE under non-reducing and reducing conditions (Fig. 2A). Under non-reducing conditions, the Db-DIII proteins produced a major band corresponding to a dimer [(scFv-DIII)2] of predicted molecular mass of ∼101 kDa (Fig. 2A, lanes 1–3). The presence of a dimer was unexpected as there are no covalent bonds between the two monomers. Several weaker bands of lower molecular mass that were probably degradation products were also noted, both on the SDS–PAGE Coomassie stained gel (Fig. 2A, lanes 1–3) and the Western blot, probed with an anti-mouse Fab-specific antibody (Fig. 2B, lane 2). Under reducing conditions, the (scFv-DIII)2 dimer divides into two bands corresponding to a scFv-DIII monomer (∼48 kDa) and a DIII molecule (∼23 kDa; Fig. 2A, lane 5). An attempt to detect the DIII portion of the fusion protein by Western blot with a polyclonal anti-HSA antibody was not successful. Therefore, HRP-conjugated Protein L was used instead for the detection of the Db component of the Db-DIII fusion protein in the Western blot (Fig. 2B, lane 3). After reduction, only (scFv-DIII)2 and scFv-DIII proteins were detected (Fig. 2B, lane 3, upper two bands). This suggests that the lower 23 kDa band on the reduced protein SDS–PAGE gel (Fig. 2A, lane 5) is the DIII alone. Detection of (scFv-DIII)2 after reduction in the Western blot implies that the reduction was not complete. Although less pronounced, higher molecular mass bands are also seen in the reduced Coomassie stained SDS–PAGE (Fig. 1A, lane 5). This difference in intensity can be explained by the inherently higher sensitivity of detection in Western blots. The observation that DIII was proteolytically cleaved from the scFv under reducing conditions, prompted the necessity of examining the non-reduced Db-DIII proteins by size exclusion chromatography, which would separate and differentially elute cleavage products such as the Db alone (∼55 kDa), and the DIII (∼23 kDa) protein. The size exclusion chromatogram shows that Db-DIII WT (101 kDa) elutes as a single peak with an elution time of 28.17 min (Fig. 2C). Under the same conditions, the Db-DIII H535A, H510A and H464A proteins were characterized by an average elution time of 28.2 min, with no extra peaks suggesting lack of degradation, aggregation or multimerization. Integration of the size exclusion chromatography peaks revealed ∼98% protein purity after a single step of Protein A affinity column purification. Apparently DIII cleavage only occurs when the Db-DIII protein is reduced, suggesting that this process may be mediated by the reducing agent and possibly high temperature forcing Db-DIII to break in the linker region as a result of stress.
Fig. 2.

(A) SDS–PAGE of four Db-DIII proteins: H535A, H510A, and H464A in lanes 1, 2, and 3, respectively, under non-reducing conditions, followed by an empty lane, protein standard in lane 4, and reduced Db-DIII WT protein in lane 5. (B) Western blot of Db-DIII WT under non-reducing conditions (lane 2; probed with AP-conjugated anti-mouse Fab Ab) and reducing conditions (lane 3; probed with HRP-Protein L). Lanes 1 and 4 are protein standards. (C) Size exclusion chromatography, using Superdex 200 column and 0.5 ml/min flow rate. The Db-DIII WT protein eluted at 28.17 min. Purity of ∼98% was estimated by integration of the peak.
A structural model of HSA DIII was generated based on the crystal structure of HSA (Sugio et al., 1999; Fig. 3A). DIII is comprised of 10 α-helices (six in DIIIa and four in DIIIb) connected to each other by loops. Residues H464 (in DIIIa), H535 and H510A (both in DIIIb) are depicted. A docking model of DIII and FcRn (Fig. 3B) was generated in order to examine the DIII–FcRn interface and to identify possible amino acid binding partners from both molecules that facilitate this interaction. The model was built using the crystal structures of HSA (Sugio et al., 1999) and FcRn (Martin et al., 2001). The ZDOCK algorithm was set to be biased toward interactions that included the FcRn residues H161 and H166 (Andersen et al., 2006), and the HSA DIII H535, H510 and H464. It produced 11 candidate structures. These structures were sorted and analyzed using PyMOL for potential strong pH-dependent binding occurring at pH 6.0 and release at pH 7.4. The majority of predicted structures suggested that the conserved aromatic residues surrounding FcRn amino acids H166 and H161 are likely to make contacts with two of the DIII histidines—H510 and H535. At the same time FcRn H166 and H161 could interact with glutamic acid residues on DIII, an interaction that would increase in affinity when the histidines are protonated in low pH environments. In addition, FcRn residues D102 and N101 paired with the H464 residue on DIII in many of the proposed structures. When protonated, H464 becomes positively charged and capable of forming an ionic bond with the negatively charged D102 acidic residue. N101 is likely to be part of the hydrophobic interaction between the two molecules, evidenced by the large positive change in entropy upon albumin–FcRn binding (Chaudhury et al., 2006). The tenth resultant structure provided by ZDOCK (Fig. 3B) fulfilled the requirement for strong interaction at pH 6.0 and no binding at pH 7.4 and therefore was deemed to be the most probable. This structure predicted potential interactions between DIII H535 and FcRn F157; DIII H510 and FcRn W51 and Y60; DIII H464 and either FcRn D101 and N102 or K123; FcRn H166 and DIII E505; and FcRn H161 and DIII E531. Finally, a space-filling model of the Db-DIII molecule was created using the crystal structure of the T84.66 Db (Carmichael et al., 2003; Fig. 3C). Two DIII molecules are attached to each dimeric Db through 18 amino acid linkers, which should produce a relatively flexible molecular structure.
Fig. 3.
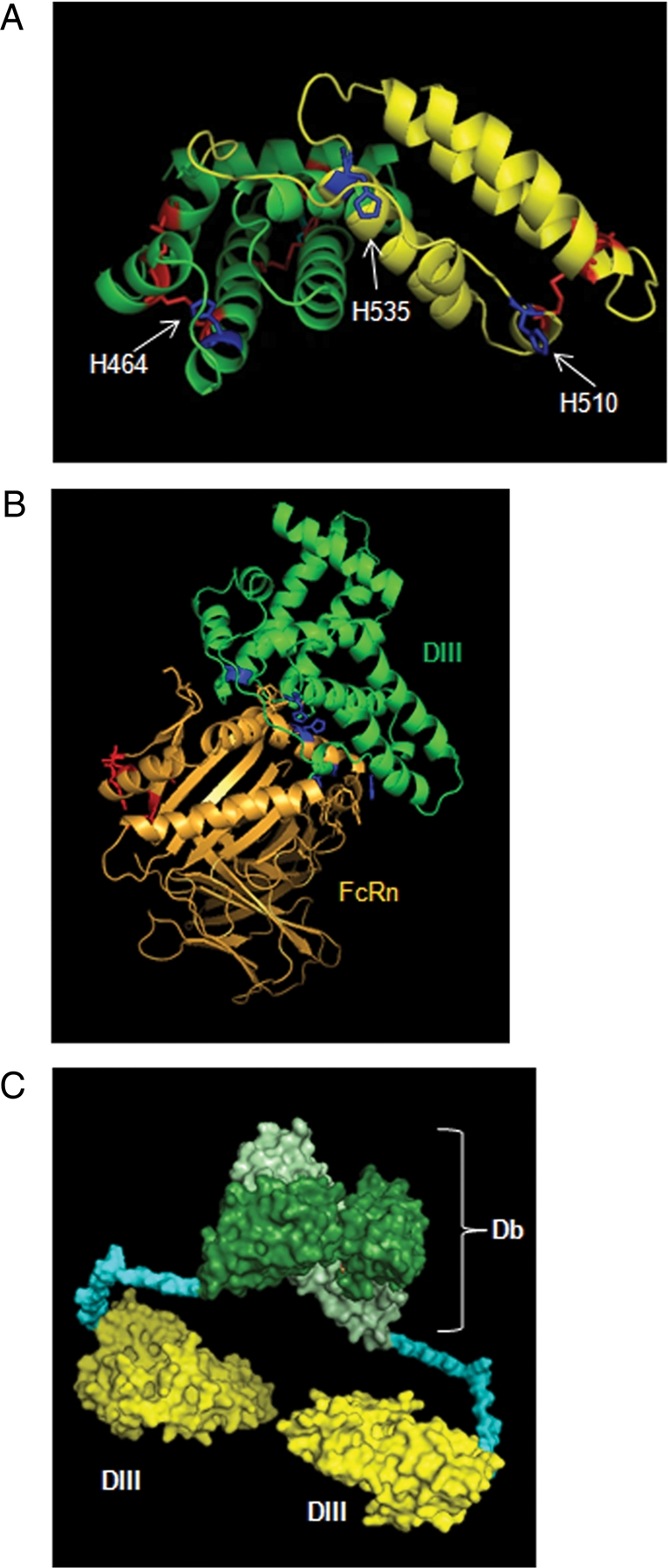
(A) The PyMOL model of HSA DIII composed of half domains DIIIa (green) and DIIIb (yellow). Six disulfide bridges are shown in red. The location of residues H535, H510, and H464 is pointed by the arrows. The H464 residue, located in DIIIa, was mutated to alanine to produce the DIII H464A variant. Amino acids H535 and H510, located in the DIIIb, were each exchanged with alanine to produce DIII H535A and DIII H510A variants. (B) The Docking model of the HSA DIII (green) and FcRn (gold) proteins. In red are the residues on FcRn that are involved in IgG binding. In blue are the constrained histidine residues in both FcRn and DIII. (C) The space-filling model of a divalent Db-DIII molecule. The scFv monomers are colored light and dark green, each attached to one DIII (yellow) via an 18 amino acid linker (blue).
Radiolabeling and murine xenograft imaging studies
The 124I labeling efficiency for the Db-DIII fusion proteins ranged from 63.9% to 81.5% and their immunoreactivity after radiolabeling was between 73.4% and 91.7%. The injected specific activities ranged from 13.0 to 18.0 GBq/μmol. By performing serial PET imaging, rather than a single time point image acquisition, we were able to follow the Db-DIII associated radioactivity decline as a function of time. This allowed us to directly compare the Db-DIII fusion proteins among each other and with the Db alone in vivo, in terms of tumor targeting and persistence in the circulation (Fig. 4). The images show that all five proteins target the LS174T (CEA-positive) tumor. The tumor anatomical location is clearly seen on the CT image. Targeting was noted as early as 4 h for all proteins; however, the background (circulation activity) in the abdominal and cardiac region dominated.
Fig. 4.

Small animal PET/CT imaging of athymic nude mice xenografted with CEA-positive LS174T (left) and CEA-negative C6 (right) tumors. Groups of four mice were injected with 124I-labeled Db-DIII proteins (WT, H535A, H510A or H464A) and the anti-CEA Db as a reference. Mice were imaged for 10 min at five different time points with coronal sections shown. Co-registered PET/CT images are included for the anatomical reference of the tumors and organs.
The signal in the CEA-positive tumor persisted throughout the entire study (51 h) for all proteins, whereas the background was variable. A statistical comparison of the T:ST ROI ratios of Db-DIII and Db proteins at different time points (Fig. 5A) showed that at 4 h all T:ST ratios were statistically the same (P> 0.05). At 20 and 28 h, the Db and H464A were not significantly different from each other but exhibited T:ST ratios that were significantly higher than the rest of the Db-DIII proteins (P-values ranging from 0.03 to 0.01). At 44 h, the Db T:ST ratio increased significantly compared with H464A (P= 0.01) and the rest of the Db-DIII proteins. H464A T:ST ratio was also significantly higher than the WT, H535A and H510A (P ranging from 0.01 to 0.04). At 51 h, all proteins' T:ST ratios were significantly different from each other (P ranging from 0.008 to 0.03), except H510A which was not significantly different from H464A (P= 0.07) and H535A (P= 0.1). However, H464A T:ST ratio was significantly higher than that of the H535A (P= 0.02). PET image quantification of the radioactivity in blood (%ID/g) for each time point allowed for the generation of blood activity curves (Fig. 5B) and calculation of the MRT for each protein in the blood (Table I). Db-DIII WT exhibited significantly slower blood clearance kinetics compared with all histidine mutants and the Db alone (P< 0.05). The Db-DIII H535A and H510A, as well as the H510A and H464A, blood activity curves were not significantly different from each other (P= 0.09 and P= 0.08, respectively), whereas H535A was characterized by significantly longer blood persistence, compared with the H464A variant (P= 0.01). Thus, the order from the longest to the shortest serum MRT was: Db-DIII WT > H535A ≥ H510A ≥ H464A > Db, where Db-DIII H535A had significantly longer circulation residence time compared with H464A. Biodistribution at 51 h confirmed the order of serum persistence (Table II). The measured activity in blood for the Db-DIII proteins ranged from 4.0 to 1.6 %ID/g, whereas the LS174T tumor uptake was between 2.5 and 1.3 %ID/g, compared with 0.5 %ID/g for the Db. Previous studies have shown that the radioiodinated T84.66 Db reaches maximum tumor uptake at 2 h after injection (13.68 ± 1.49 %ID/g), after which the activity in the tumor starts to decline (3.92 ± 1.68 %ID/g at 48 h; Wu et al., 1999). Tumor masses averaged 161 and 126 mg for LS174T and C6 tumors, respectively. It was noted that longer serum residence time was generally associated with higher LS174T tumor uptake. The CEA positive-to-negative tumor uptake ratios for the Db-DIII proteins ranged from 1.5 to 2.2, compared with 13 for the Db alone at 51 h. Tumor-to-blood ratios at 51 h were from 0.53 to 0.84, compared with 6.5 for the Db.
Fig. 5.

(A) Tumor-to-soft tissue ROI analysis of the PET images. Asterisks are indicative of significant difference. The T:ST bars under the same bracket are not significantly different from each other. Groups of T:ST bars under one bracket are significantly different from groups of T:ST bars under the other bracket. (B) Blood activity curves generated by quantitation of radioactivity (%ID/g) from the PET images at each time point.
Table I.
Estimated values of blood half-lives for the Db-DIII and Db
| Protein | Blood AUCa | First Mob (h) | MRTc (h) |
|---|---|---|---|
| Db-DIII WT | 1449 | 178 | 56.7 |
| Db-DIII H535A | 470 | 66 | 25 |
| Db-DIII H510A | 528 | 38 | 20 |
| Db-DIII H464A | 354 | 24 | 17 |
| Db | 104 | 26 | 2.9 |
aAUC is the area under the curve −∫u(t)dt from 0 to infinity.
bFirst Mo is the first moment: ∫t u(t)dt/∫u(t)dt, where u(t) is the measured (%ID/g) blood curve.
cMRT is the same integral format except du/dt replaces u(t).
Table II.
Biodistribution, showing the 124I-labeleled Db-DIII proteins in descending order (top to bottom), in terms of their persistence in the circulation (blood %ID/g)
| Protein | Organ/tissue (%ID/g) at 51 h |
|||||||
|---|---|---|---|---|---|---|---|---|
| Blood | Liver | Spleen | Kidneys | Lungs | LS174T (+) | C6 (−) | Carcass | |
| Db-DIII WT | 4.00 (0.22) | 0.79 (0.03) | 0.89 (0.11) | 1.01 (0.11) | 2.05 (0.20) | 2.46 (0.19) | 1.67 (0.30) | 0.83 (0.12) |
| Db-DIII H535A | 2.04 (0.08) | 0.67 (0.05) | 0.63 (0.05) | 0.75 (0.06) | 0.91 (0.08) | 1.08 (0.11) | 0.69 (0.12) | 0.33 (0.03) |
| Db-DIII H510A | 1.77 (0.14) | 0.54 (0.06) | 0.58 (0.03) | 0.53 (0.08) | 0.98 (0.07) | 1.10 (0.07) | 0.72 (0.14) | 0.40 (0.02) |
| Db-DIII H464A | 1.56 (0.15) | 0.44 (0.06) | 0.50 (0.07) | 0.58 (0.08) | 0.85 (0.12) | 1.31 (0.15) | 0.59 (0.06) | 0.28 (0.03) |
| Db | 0.08 (0.01) | 0.19 (0.01) | 0.10 (0.01) | 0.33 (0.02) | 0.10 (0.02) | 0.52 (0.06) | 0.04 (0.001) | 0.03 (0.001) |
Groups of four mice per protein were analyzed. Organ uptake is expressed as %ID/g [mean (SE)].
Discussion
Currently, the most successful targeting agents used for cancer therapy in the clinic are intact antibodies (e.g. Trastuzumab, Rituximab, Bevacizumab). The advantage of using antibodies is that in addition to their superb target affinity and specificity, and good safety profile, they also possess the necessary pharmacokinetics to achieve therapeutic effect. The disadvantages include certain limitations with target accessibility, but predominantly the lengthy, highly laborious process of production, which also increases antibody drug cost. Other targeting moieties, including peptides and aptamers can also be selected to exhibit nanomolar affinity and high specificity for various targets, are much faster and cheaper to make. However, a major drawback is that these low molecular weight targeting agents clear very rapidly from the circulation, with typical serum half-lives in the order of minutes. This leads to low target uptake and limits their potential for clinical use.
In order to take advantage of what small targeting molecules can offer, we designed a potentially non-immunogenic HSA DIII protein scaffold. This scaffold can provide tailored phamacokinetics, opportunity for multivalence and/or multiple specificities through grafting or conjugation of more than one target binding sequence onto the scaffold, and residues for attachment of functional groups. The closest counterpart of the HSA DIII in terms of the ability to control its pharmacokinetics is the Fc region (∼50 kDa) of human IgG (Kenanova et al., 2005). In addition to being twice the size of HSA DIII, Fc domains of antibodies interact with various endogenous Fc receptors which may lead to unwanted side effects in clinical applications. Before expressing the HSA DIII WT and variants alone, two practical questions needed to be answered. (i) When attached to antigen binding moiety(s), would DIII interfere with the ability to bind targets in vivo? (ii) In the context of a fusion, would DIII maintain its ability to control the circulation persistence of the entire molecule? In addition, determining subtle differences in circulation persistence among the DIII variants could be difficult because of the small size of DIII and its first pass renal clearance. Therefore, as a first step and a proof of principle, we designed an antibody–HSA DIII fusion protein of molecular mass above the renal elimination threshold. The antibody moiety used was the anti-CEA T84.66 Db, which is a small divalent antibody fragment that has been extensively studied in vivo. The anti-CEA Db exhibits terminal β half life ranging from 2.89 h (123I) to 3.04 h (111In) in LS174T (CEA-positive) tumor bearing mice (Yazaki et al., 2001). This Db has already been successfully fused to other proteins (i.e. Renilla or Gaussia luciferases), while retaining its in vivo targeting capacity (Venisnik et al., 2006, 2007). Therefore, the T84.66 Db is a good targeting molecule for a proof of concept study. The second component of the fusion protein is the one with unknown characteristics, namely the HSA DIII WT or one of its variants with mutated H535, H510 or H464 residue.
The Db-DIII fusion proteins were expressed in mammalian cells to ensure proper folding. Expression levels were reasonable and affinity purification yielded proteins of molecular mass consistent with the calculated 101 kDa (Fig. 2A). The Db is a non-covalent dimer of two scFv molecules, which separate from each other under SDS–PAGE conditions and migrate around 25 kDa (Wu et al., 1999). We expected that the Db-DIII molecules would migrate as scFv-DIII (∼48 kDa) species, as all cysteine residues, both in the DIII and scFv, are paired (Dugaiczyk et al., 1982; Curry et al., 1998; Wu et al., 1999). Interestingly, under non-reducing conditions, the bulk of the protein remained in its dimeric form [i.e. (scFv-DIII)2 of 101 kDa], exhibiting increased dimeric structural stability under SDS conditions. After closer examination of the Db-DIII computer model (Fig. 3C), the linker length between the Db and DIII was reduced from 18 to 5 amino acids (data not shown). However, this alteration did not affect the migration pattern of the (scFv-DIII)2 protein (Fig. 2A). Because of this unexpected behavior, the Db-DIII protein bands from the SDS–PAGE gel (Fig. 2A) were excised and the extracted protein was analyzed by mass spectrometry (data not shown). The results confirmed that the protein of ∼101 kDa was indeed the Db-DIII. One explanation for the enhanced dimer stability may be attributed to a network of polar, ionic interactions between the two scFv-DIII molecules, mediated by the presence of DIII. Such interactions and similar behavior was reported for β-glycosidase (Gentile et al., 2002). The molecular size of Db-DIII proteins was confirmed by size exclusion chromatography under non-reducing and non-denaturing conditions. The elution time of Db-DIII was close to another protein of similar molecular mass (scFv-Fc, 105 kDa), which elutes at ∼27.3 min, under the same conditions (Kenanova et al., 2005). Being smaller, the Db-DIII eluted at around 28.2 min, whereas the Db alone elutes at 38.2 min (Kenanova et al., 2005). In addition to high purity, the single peak on the chromatogram (Fig. 2C) revealed the integrity of the Db-DIII protein and that it exists as single species. In addition, the fact that the Db-DIII proteins remained immunoreactive after radiolabeling suggests that the protein is stable and most likely does not undergo rapid proteolytic degradation.
Analysis of the DIII–FcRn docking model (Fig. 3B) was very useful in defining the possible FcRn interaction partners of H535, H510 and H464 residues in DIII, as well as proposing other DIII amino acids that may play a role (e.g. E505 and E531). However, it did not predict the ranking of DIII residues in importance for FcRn binding. This is because the DIII and FcRn molecules were not docked in an unconstrained manner. Instead, a bias was introduced toward FcRn residues H161 and H166, and DIII residues H535, H510 and H464 as the critical amino acids, in order to narrow down the number of possible docking configurations and to select the interaction that is most likely to occur. The ranking of DIII residues H535, H510 and H464 was determined experimentally by in vivo molecular imaging.
The strength of molecular imaging, specifically PET, is that the same individual can be imaged tomographically multiple times after injection of the tracer to extract quantitative information about pharmacokinetics, tumor targeting and accumulation, as well as cross-reactivity. In this study, mice bearing CEA-positive and negative xenografts were injected with 124I-labeled Db-DIII or Db proteins and imaged at five different time points. This allowed for head-to-head comparison of the Db-DIII proteins with each other, as well as with the Db alone in terms of their persistence in the circulation and tumor targeting. Since the Db was the constant component, differences in pharmacokinetics among Db-DIII proteins were attributed to the function of the DIII. Thus, PET imaging enabled us to make conclusions about the behavior of the DIII protein in vivo. The Db alone achieved higher T:ST ratios at earlier time points due to its rapid blood clearance which is also the reason why the Db images demonstrate high contrast and little non-specific background at 20 h (Fig. 4). On the other hand, as circulation residence time increases, Db-DIII associated activity in the lungs, liver, spleen, kidneys and muscle is also enhanced due to the blood component in these organs and tissues. This ultimately translates into lower T:ST ratios. Therefore, based on the T:ST ROI ratios at 51 h (Fig. 5A), we were able to deduce the order of blood clearance from fastest (highest T:ST ratio) to slowest (lowest T:ST ratio) as: Db >> Db-DIII H464A > H510A > H535A > WT. The statistical analysis showed that Db-DIII H510A was not significantly faster clearing than the H535A variant. Both H535 and H510 residues are located in subdomain DIIIb. This finding could suggest that within albumin the H510 and H535 residues may be redundant, playing a backup role for each other in case one is non-functional or not available for binding to FcRn. This hypothesis remains to be elucidated. Simultaneous mutation of both residues can provide more insight. The same order of circulation clearance was also confirmed by the blood activity curves generated from quantifying the radioactivity in the mouse heart at each time point for every protein (Fig. 5B). Statistical comparison of the blood activity curves led to the same conclusion as above—Db-DIII H535A clears significantly slower than H464A, but not compared with H510A. Owing to the lack of more time points within the first 12h post-tracer injection, calculation of MRT, rather than α and β half lives was more feasible. The MRT ranged from ∼2.4 days for the Db-DIII WT to 17 h for the Db-DIII H464A, compared with 2.9 h for the Db alone. The overall size of the Db-DIII fusion proteins (101 kDa) is above the threshold for renal clearance (∼60 kDa). Therefore, Db-DIII proteins are eliminated through the hepatobiliary route, whereas the Db (55 kDa) is cleared through the kidneys. Thus, the difference in molecular mass between Db and Db-DIII proteins is largely responsible for the difference in MRT. However, the fact that the Db-DIII pharmacokinetics in vivo can be modulated through single amino acid mutation (same molecular mass) suggests that there is an additional molecular mechanism that governs serum pharmacokinetics in vivo apart from increase in molecular size (e.g. FcRn interaction). Furthermore, the mutations (H535A, H510A or H464A) allow for finer tuning of the overall protein serum residence time. One can choose from a spectrum of circulation half lives ranging from days to hours. This is advantageous when selecting for diagnostic or therapeutic agents with specific serum pharmacokinetics requirements.
The imaging studies clearly demonstrate the ability of the HSA DIII to increase the circulation persistence of the Db, while retaining tumor targeting. All Db-DIII proteins remained in blood significantly longer than the Db alone (P < 0.05). Direct count of the radioactivity (%ID/g) remaining in blood for the mice injected with the Db-DIII fusion proteins was from 50-fold (WT) to 20-fold (H464A) higher than that for the Db injected mice at 51 h. Collectively, our findings suggest that the HSA DIII WT and mutants alone should be capable of tailoring the serum residence time of the moiety which they are attached to. Comparison of the blood activity curves of our T84.66 Db-DIII WT (Fig. 5B) to the monovalent T84.66 scFv fragment fused to full-length HSA (Yazaki et al., 2008) reveals that the Db-DIII WT persists in the circulation longer than the scFv–HSA fusion protein. Specifically, 48 h post-injection of 125I-labeled anti-CEA scFv–HSA fusion protein (∼90 kDa) in LS174T xenografted athymic nude mice, the remaining activity in blood was 2.79 %ID/g, compared with 4.00 %ID/g for the 124I-labeled Db-DIII WT at 51 h (Table II). This difference can be explained by the larger molecular mass of Db-DIII (∼101 kDa). Nevertheless, it confirms the finding that DIII is both necessary and sufficient for maintaining the serum half life of the entire HSA molecule. H464 (located in DIIIa) appears to have the biggest effect on FcRn binding and attenuation of circulation persistence. Additionally, since H535A and H510A mutations produce significantly faster blood clearances compared with the WT, we can conclude that both subdomains DIIIa and DIIIb participate in maintaining serum persistence.
The purpose of engineering the Db-DIII fusion proteins was to elucidate the potential of the HSA DIII for use as a single domain scaffold with controlled pharmacokinetics. Expression of the DIII WT and variants without a targeting moiety, and evaluation of their pharmacokinetics in vivo, is the next step toward the selection of DIII scaffolds, exhibiting properties optimal for imaging or therapy applications. The DIII scaffolds described in this work may be used for grafting or chemically conjugating tumor targeting molecules (peptides, aptamers, small chemical molecules) or for directed evolution in display libraries. DIII contains both α-helices and loops suitable for diversification. The targeted scaffolds with appropriate pharmacokinetics for imaging may be used for diagnostic purposes. Alternatively, cytotoxic drugs could be conjugated to the targeted scaffolds with optimal characteristics for cancer treatment.
Funding
This work is supported by the 2008 SNM Postdoctoral Molecular Imaging Award (0708pmi – award) and an ICMIC grant (NIH CA086306). A.M.W. is a member of the UCLA Jonsson Comprehensive Cancer Center (CA16042). This work was supported by the National Institute of Health (PO1 CA43904 and CA33572).
Acknowledgments
We thank the staff of the Crump Institute for Molecular Imaging small animal PET imaging facility (National Institute of Health grant R24 CA92865) and the UCLA Pasarow Mass Spectrometry Laboratory (National Institute of Health grant S10 RR16793). Special acknowledgment is also given to Juni Samos who assisted with the cloning and mutagenesis of DIII.
Footnotes
Edited by Valerie Daggett
References
- Aina O.H., Liu R., Sutcliffe J.L., Marik J., Pan C.X., Lam K.S. Mol. Pharm. 2007;4:631–651. doi: 10.1021/mp700073y. [DOI] [PubMed] [Google Scholar]
- Andersen J.T., Dee Qian J., Sandlie I. Eur. J. Immunol. 2006;36:3044–3051. doi: 10.1002/eji.200636556. [DOI] [PubMed] [Google Scholar]
- Barbas A.S., White R.R. Curr. Opin. Investig. Drugs. 2009;10:572–578. [PubMed] [Google Scholar]
- Bebbington C.R., Renner G., Thomson S., King D., Abrams D., Yarranton G.T. Biotechnology (NY) 1992;10:169–175. doi: 10.1038/nbt0292-169. [DOI] [PubMed] [Google Scholar]
- Bembenek S.D., Tounge B.A., Reynolds C.H. Drug Discov. Today. 2009;14:278–283. doi: 10.1016/j.drudis.2008.11.007. [DOI] [PubMed] [Google Scholar]
- Bos O.J., Labro J.F., Fischer M.J., Wilting J., Janssen L.H. J. Biol. Chem. 1989;264:953–959. [PubMed] [Google Scholar]
- Burger A.M., Hartung G., Stehle G., Sinn H., Fiebig H.H. Int. J. Cancer. 2001;92:718–724. doi: 10.1002/1097-0215(20010601)92:5<718::aid-ijc1257>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Carmichael J.A., Power B.E., Garrett T.P., Yazaki P.J., Shively J.E., Raubischek A.A., Wu A.M., Hudson P.J. J. Mol. Biol. 2003;326:341–351. doi: 10.1016/s0022-2836(02)01428-6. [DOI] [PubMed] [Google Scholar]
- Chaudhury C., Brooks C.L., Carter D.C., Robinson J.M., Anderson C.L. Biochemistry. 2006;45:4983–4990. doi: 10.1021/bi052628y. [DOI] [PubMed] [Google Scholar]
- Chaudhury C., Mehnaz S., Robinson J.M., Hayton W.L., Pearl D.K., Roopenian D.C., Anderson C.L. J. Exp. Med. 2003;197:315–322. doi: 10.1084/jem.20021829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R., Li L., Weng Z. Proteins. 2003;52:80–87. doi: 10.1002/prot.10389. [DOI] [PubMed] [Google Scholar]
- Curry S., Mandelkow H., Brick P., Franks N. Nat. Struct. Biol. 1998;5:827–835. doi: 10.1038/1869. [DOI] [PubMed] [Google Scholar]
- D'Argenio D.Z., Schumitzky A. Comput. Programs Biomed. 1979;9:115–134. doi: 10.1016/0010-468x(79)90025-4. [DOI] [PubMed] [Google Scholar]
- Defrise M., Kinahan P.E., Townsend D.W., Michel C., Sibomana M., Newport D.F. IEEE Trans. Med. Imaging. 1997;16:145–158. doi: 10.1109/42.563660. [DOI] [PubMed] [Google Scholar]
- Dugaiczyk A., Law S.W., Dennison O.E. Proc. Natl Acad. Sci. USA. 1982;79:71–75. doi: 10.1073/pnas.79.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Z., Ram S., Vaccaro C., Ober R.J., Ward E.S. Traffic. 2009;10:600–614. doi: 10.1111/j.1600-0854.2009.00887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile F., Amodeo P., Febbraio F., Picaro F., Motta A., Formisano S., Nucci R. J. Biol. Chem. 2002;277:44050–44060. doi: 10.1074/jbc.M206761200. [DOI] [PubMed] [Google Scholar]
- Kenanova V., Olafsen T., Crow D.M., et al. Cancer Res. 2005;65:622–631. [PMC free article] [PubMed] [Google Scholar]
- Kim J.K., Firan M., Radu C.G., Kim C.H., Ghetie V., Ward E.S. Eur. J. Immunol. 1999;29:2819–2825. doi: 10.1002/(SICI)1521-4141(199909)29:09<2819::AID-IMMU2819>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Kratz F., Muller-Driver R., Hofmann I., Drevs J., Unger C. J. Med. Chem. 2000;43:1253–1256. doi: 10.1021/jm9905864. [DOI] [PubMed] [Google Scholar]
- Loening A.M., Gambhir S.S. Mol. Imaging. 2003;2:131–137. doi: 10.1162/15353500200303133. [DOI] [PubMed] [Google Scholar]
- Martin W.L., West A.P., Jr, Gan L., Bjorkman P.J. Mol. Cell. 2001;7:867–877. doi: 10.1016/s1097-2765(01)00230-1. [DOI] [PubMed] [Google Scholar]
- Medesan C., Matesoi D., Radu C., Ghetie V., Ward E.S. J. Immunol. 1997;158:2211–2217. [PubMed] [Google Scholar]
- Melder R.J., Osbom B.L., Riccobene T., et al. Cancer Immunol. Immunother. 2005;54:535–547. doi: 10.1007/s00262-004-0624-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller D., Karle A., Meissburger B., Hofig I., Stork R., Kontermann R.E. J. Biol. Chem. 2007;282:12650–12660. doi: 10.1074/jbc.M700820200. [DOI] [PubMed] [Google Scholar]
- Neumaier M., Shively L., Chen F.S., Gaida F.J., Ilgen C., Paxton R.J., Shively J.E., Riggs A.D. Cancer Res. 1990;50:2128–2134. [PubMed] [Google Scholar]
- Olafsen T., Kenanova V.E., Wu A.M. Nat. Protoc. 2006;1:2048–2060. doi: 10.1038/nprot.2006.322. [DOI] [PubMed] [Google Scholar]
- Osborn B.L., Olsen H.S., Nardelli B., Murray J.H., Zhou J.X., Garcia A., Moody G., Zaritskaya L.S., Sung C. J. Pharmacol. Exp. Ther. 2002;303:540–548. doi: 10.1124/jpet.102.037002. [DOI] [PubMed] [Google Scholar]
- Riechmann L., Davies J. J. Biomol. NMR. 1995;6:141–152. doi: 10.1007/BF00211778. [DOI] [PubMed] [Google Scholar]
- Shields R.L., Namenuk A.K., Hong K., et al. J. Biol. Chem. 2001;276:6591–6604. doi: 10.1074/jbc.M009483200. [DOI] [PubMed] [Google Scholar]
- Starovasnik M.A., O'Connell M.P., Fairbrother W.J., Kelley R.F. Protein Sci. 1999;8:1423–1431. doi: 10.1110/ps.8.7.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugio S., Kashima A., Mochizuki S., Noda M., Kobayashi K. Protein Eng. 1999;12:439–446. doi: 10.1093/protein/12.6.439. [DOI] [PubMed] [Google Scholar]
- Venisnik K.M., Olafsen T., Loening A.M., Iyer M., Gambhir S.S., Wu A.M. Protein Eng. Des. Sel. 2006;19:453–460. doi: 10.1093/protein/gzl030. [DOI] [PubMed] [Google Scholar]
- Venisnik K.M., Olafsen T., Gambhir S.S., Wu A.M. Mol. Imaging Biol. 2007;9:267–277. doi: 10.1007/s11307-007-0101-8. [DOI] [PubMed] [Google Scholar]
- Wosikowski K., Biedermann E., Rattel B., Breiter N., Jank P., Loser R., Jansen G., Peters G.J. Clin. Cancer Res. 2003;9:1917–1926. [PubMed] [Google Scholar]
- Wu A.M., Williams L.E., Zieran L., Padma A., Sherman M., Bebb G.G., Odom-Maryon T., Wong J.Y.C., Shively J.E., Raubitschek A.A. Tumor Target. 1999;4:47–58. [Google Scholar]
- Yazaki P.J., Wu A.M., Tsai S.W., Williams L.E., Ikler D.N., Wong J.Y., Shively J.E., Raubitschek A.A. Bioconjug. Chem. 2001;12:220–228. doi: 10.1021/bc000092h. [DOI] [PubMed] [Google Scholar]
- Yazaki P.J., Kassa T., Cheung C.W., Crow D.M., Sherman M.A., Bading J.R., Anderson A.L., Colcher D., Raubitschek A. Nucl. Med. Biol. 2008;35:151–158. doi: 10.1016/j.nucmedbio.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]