

Abstract
Background
Genome-wide association studies (GWAS) have been extremely successful in the search for susceptibility risk factors for complex genetic autoimmune diseases. As more studies are published, evidence is emerging of considerable overlap of loci between these diseases. In juvenile idiopathic arthritis (JIA), another complex genetic autoimmune disease, the strategy of using information from autoimmune disease GWAS or candidate gene studies to help in the search for novel JIA susceptibility loci has been successful, with confirmed association with two genes, PTPN22 and IL2RA. Rheumatoid arthritis (RA) is an autoimmune disease that shares similar clinical and pathological features with JIA and, therefore, recently identified confirmed RA susceptibility loci are also excellent JIA candidate loci.
Objective
To determine the overlap of disease susceptibility loci for RA and JIA.
Methods
Fifteen single nucleotide polymorphisms (SNPs) at nine RA-associated loci were genotyped in Caucasian patients with JIA (n=1054) and controls (n=3531) and tested for association with JIA. Allele and genotype frequencies were compared between cases and controls using the genetic analysis software, PLINK.
Results
Two JIA susceptibility loci were identified, one of which was a novel JIA association (STAT4) and the second confirmed previously published associations of the TRAF1/C5 locus with JIA. Weak evidence of association of JIA with three additional loci (Chr6q23, KIF5A and PRKCQ) was also obtained, which warrants further investigation.
Conclusion
All these loci are good candidates in view of the known pathogenesis of JIA, as genes within these regions (TRAF1, STAT4, TNFAIP3, PRKCQ) are known to be involved in T-cell receptor signalling or activation pathways.
Introduction
Complex genetic diseases are caused by the effects of multiple genes in combination with lifestyle and environmental factors. In the past couple of years, with the emergence of genome-wide association studies (GWAS), there has been an explosion of genetic discoveries for many complex genetic autoimmune diseases such as Crohn's disease, type 1 diabetes, multiple sclerosis and rheumatoid arthritis (RA). Indeed, more genetic risk factors were identified for common diseases in 2007 than had been collectively reported before that date.1 It is also interesting to note that evaluating the results from the study of one disease in other complex diseases can disclose common risk factors.1 Thus there has been detectable overlap of loci for autoimmune diseases, with examples such as a region on chromosome 4q27 that encodes the interleukin 2 (IL2) and IL21 genes, which is associated with susceptibility to coeliac disease, multiple sclerosis, type 1 diabetes and RA,2 3 and the IL23R region on 1p31 which has been associated with Crohn's disease,4 ulcerative colitis,4 psoriasis5 and ankylosing spondylitis.6
Juvenile idiopathic arthritis (JIA), another complex genetic autoimmune disease, is the most common chronic inflammatory rheumatic disease in children and is an important cause of short- and long-term disability.7 It is defined as arthritis of unknown aetiology that starts before the age of 16 and persists for at least 6 weeks. JIA can be subdivided into seven clinically more homogeneous subtypes, using the International League of Associations for Rheumatology (ILAR) classification system.8 As in other autoimmune diseases there are well-established associations of the HLA region with JIA.9
In JIA, only a single, comparatively small 100k single nucleotide polymorphism (SNP) study has been published to date.10 This is largely owing to the low prevalence and clinically heterogeneous nature of JIA, meaning that collection of sufficiently large and thus powerful, sample sizes has been protracted compared with other complex autoimmune diseases. However, this does not mean that data from GWAS cannot be exploited to help elucidate the underlying genetic basis of this disease. Indeed, the strategy of using information from autoimmune disease GWAS or candidate studies to help identify JIA susceptibility genes has already been successful in identifying two JIA susceptibility genes, protein tyrosine phosphatase non-receptor 22 (PTPN22)11 and more recently, IL2RA,12 which have been validated in independent cohorts.
RA is an autoimmune disease that has similar clinical and pathological features to those of JIA. In the past year a number of novel RA susceptibility genes have been identified, which have been confirmed in independent datasets, including the STAT4 gene,13 14 the TRAF1 and C5 region on chromosome 9,14–16 a region on chromosome 6q23,17 18 the KIF5A gene,19 a region on chromosome 10p15 close to the PRKCQ gene,19 MMEL,19 20 CD40,20 CDK620 and a region on chromosome 9p13 close to the CCL21 gene.20 Thus, these loci are also excellent JIA candidate susceptibility loci and, therefore, the aim of this study was to determine whether these confirmed that RA susceptibility loci are also associated with susceptibility to JIA.
Subjects and methods
Subjects
DNA was available for 1054 UK Caucasian patients with JIA (332 male, 715 female) from three sources: the British Society for Paediatric and Adolescent Rheumatology (BSPAR) National Repository of JIA (n=654); a cohort of UK Caucasian patients with longstanding JIA (n=201), described previously21; and a third cohort collected as part of the Childhood Arthritis Prospective Study (CAPS), a prospective inception cohort study of JIA cases from five centres across the UK (n=199).22 JIA cases were classified according to ILAR criteria,23 which are a combined set of all ILAR subtypes (online supplementary table 1).
Table 1.
Power calculations for the JIA study
| SNP | Locus | Allele frequency | Effect size (OR) | Power of JIA study (%)* |
|---|---|---|---|---|
| rs10910099†,‡ | MMEL | 0.66 | 0.9 | 53 |
| rs11889341§ | STAT4 | 0.22 | 1.12 | 48 |
| rs7574865§ | STAT4 | 0.22 | 1.17 | 76 |
| rs8179673§ | STAT4 | 0.22 | 1.13 | 55 |
| rs10181656§ | STAT4 | 0.22 | 1.13 | 55 |
| rs13207033¶ | TNFAIP3 | 0.79 | 0.86 | 67 |
| rs6920220‡ | TNFAIP3 | 0.22 | 1.22 | 93 |
| rs42041** | CDK6 | 0.24 | 1.15 | 69 |
| rs3761847§ | TRAF1 | 0.43 | 1.11 | 55 |
| rs10818488§ | TRAF1 | 0.43 | 1.12 | 62 |
| rs2900180§ | TRAF1 | 0.35 | 1.17 | 86 |
| rs2812378** | CCL21 | 0.34 | 1.12 | 59 |
| rs4750316‡ | PRKCQ | 0.80 | 0.85 | 72 |
| rs1678542‡ | KIF5A | 0.63 | 0.88 | 69 |
| rs4810485** | CD40 | 0.75 | 0.84 | 84 |
Sample size, 1054 JIA cases and 3531 controls.
At α value 0.05, assuming a log-additive model;
SNP in r2=0.95 with the SNP associated with RA in the WTCCC study;
Barton et al (2008)19;
Barton et al (2008)14;
Raychaudhuri et al (2008)20;
Orozco et al (2009) (submitted).
JIA, juvenile idiopathic arthritis; OR, odds ratio; RA, rheumatoid arthritis; SNP, single nucleotide polymorphism.
Healthy Caucasian control DNA samples were available from five centres in the UK as described previously17: Manchester, 832 controls (including 228 1958 birth cohort controls); Sheffield, 929 controls; Leeds, 422 controls; Aberdeen, 825 controls; Oxford, 523 controls, total control sample size=3531). All individuals were recruited with ethical approval and provided informed consent (North-West Multi-Centre Research Ethics Committee (MREC 99/8/84) and the University of Manchester Committee on the Ethics of Research on Human Beings).
SNP selection
In total, 15 SNPs were selected for genotyping in nine independent genetic regions, all of which have robust confirmed evidence for association with RA. These comprised:
STAT4: Four SNPs (rs11889341, rs7574865, rs8179673 and rs10181656) located in the third intron of STAT4, which showed the strongest association with North American and Swedish RA cases13; Korean RA cases24 were also selected.
TRAF1/C5: Three SNPs located in the TRAF1/C5 region were identified for investigation, one of which (rs10818488) showed the strongest evidence for association with Dutch RA cases.15 The other two SNPs, rs3761847 and rs2900180, showed the strongest evidence for association in North American and Swedish RA cases.16
6q23: rs6920220 and rs13207033, located at the chromosome 6q23 RA susceptibility locus, were also investigated. The SNPs lie in an intergenic region between OLIG3 and TNFAIP3. Variants in this region have been reported to be independently associated with RA risk.17 18 The minor allele of rs6920220 is associated with increased risk of RA (minor allele frequency (MAF) cases > controls), whereas the minor allele of rs10499194 is associated with protection (MAF controls > cases). Although the latter was not directly genotyped, a perfect proxy SNP (rs13207033; r2=1) was.
KIF5A: Association with rs1678542, which maps to intron 15 of the KIF5A gene on chromosome 12q13 was initially identified in the Wellcome Trust case–control consortium (WTCCC) genome-wide association screen of RA cases and controls25 and then validated in an independent cohort.19
PRKCQ: rs4750316 maps to an intergenic region on chromosome 10p15, the closest gene, PRKCQ, being 76 kb away. Association with this SNP has been validated in independent datasets.19 20
MMEL: Association with rs10910099, which maps to intron 11 of the MMEL (membrane metallo-endopeptidase-like 1) gene on chromosome 1p36 was initially identified in the WTCCC genome-wide association screen of RA cases and controls25 and then validated in an independent cohort.19
CD40: Association of rs4810485 on chromosome 20q13 with RA was reported in a meta-analysis of two published GWAS. The SNP lies in intron 2 of the CD40 gene.20
CCL21: rs2812378, which maps to chromosome 9p13, is situated ∼0.1 kb from the CCL21 gene. Association of this SNP with RA was also reported in the meta-analysis of two published GWAS.20
CDK6: rs42041, which maps to chromosome 7q21, lies within intron 7 of the cyclin-dependent kinase 6 gene. Association of this SNP with RA was also reported in the meta-analysis of two published GWAS.20
Genotyping
All SNPs were genotyped in UK JIA cases and controls. Genotyping was performed using the Sequenom iPLEX platform. A 90% sample quality control rate and 90% SNP genotyping success rate was imposed on the analysis. The control genotype data have been reported previously.14 17 19
Statistical analysis
Genotype and allele frequencies were compared between cases with JIA and controls using STATA version 9 SE (StataCorp, Texas, USA) and PLINK.26 Associations were expressed as two-sided p values, allelic odds ratios (ORs) and their 95% CIs. Power calculations were performed using Quanto based on previous RA studies for these SNPs.14 17–19 JIA is a phenotypically heterogeneous disease and can be classified into more clinically homogeneous diseases using the ILAR classification criteria.8 However, comparing each of the ILAR subtypes separately against controls would result in a large number of hypothesis tests. Therefore we first examined whether there was evidence of a difference in allele frequencies between the seven ILAR subtypes. Differences between subtypes were assessed using χ2 tests on the 7×2 tables. Only when a difference was found (p<0.05) were separate ORs and 95% CIs calculated for the subgroups.
Results
Fifteen SNPs across nine loci were genotyped in JIA cases (n=1054) and controls (n=3531) and analysed for association with JIA.
Table 1 shows the power of this study to detect the effect sizes identified in the RA studies for these SNPs, at a 5% significance level, assuming a log-additive model. As expected, despite being the largest collection of JIA cases and controls investigated to date, for many of the SNPs this study had modest power to detect an effect. A Bonferroni correction of 15 was applied to correct for the number of SNPs studied, resulting in a p value threshold of 0.003 for claims of significance.
Despite the modest power in this study, all of the four STAT4 SNPs were associated with JIA, with the strongest association for rs8179673 (allelic OR=1.25, 95% CI 1.11 to 1.4, trend p=0.0002) (table 2). These SNPs are in strong linkage disequilibrium with each other (r2>0.96). One of the three TRAF1/C5 SNPs was significantly associated with JIA, rs2900180 (allelic OR=1.21, 95% CI 1.09 to 1.35, trend p=0.0003). The associated SNP was the one for which the sample size tested had most power to detect statistical evidence for association.
Table 2.
Association analysis in JIA cases and controls of SNPs previously associated with RA
| Marker | Chromosome | Locus | HWE controls | MAF cases | MAF controls | Genotype frequency cases (%) | Genotype frequency controls (%) | Trend p value | Allelic OR (95% CI) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs10910099 | 1 | MMEL | 0.27 | 0.34 | 0.33 | 90 (10.3) | 412 (47.2) | 371 (42.5) | 401 (11.0) | 1572 (43.1) | 1672 (45.9) | 0.28 | 1.06 (0.95 to 1.19) |
| rs11889341 | 2 | STAT4 | 0.69 | 0.26 | 0.22 | 54 (5.7) | 376 (54.4) | 512 (54.4) | 162 (4.6) | 1200 (34.4) | 2125 (60.9) | 0.0004 | 1.24 (1.1 to 1.39) |
| rs7574865 | 2 | STAT4 | 0.62 | 0.26 | 0.22 | 52 (5.5) | 378 (40.1) | 513 (54.4) | 161 (4.6) | 1208 (34.3) | 2151 (61.1) | 0.0004 | 1.24 (1.1 to 1.39) |
| rs8179673 | 2 | STAT4 | 0.62 | 0.26 | 0.22 | 55 (5.9) | 378 (40.2) | 507 (53.9) | 162 (4.7) | 1198 (34.6) | 2103 (60.7) | 0.0002 | 1.25 (1.11 to 1.4) |
| rs10181656 | 2 | STAT4 | 0.52 | 0.26 | 0.22 | 54 (5.7) | 377 (40.1) | 509 (54.1) | 159 (4.6) | 1200 (34.5) | 2115 (60.9) | 0.0002 | 1.24 (1.11 to 1.4) |
| rs6920220 | 6 | 6q23/TNFAIP3 | 0.04 | 0.24 | 0.21 | 57 (6.5) | 297 (34.0) | 519 (59.5) | 140 (3.8) | 1251 (34.3) | 2253 (61.8) | 0.02 | 1.16 (1.02 to 1.31) |
| rs13207033 | 6 | 6q23/TNFAIP3 | 0.5 | 0.25 | 0.28 | 64 (7.1) | 327 (36.3) | 511 (56.7) | 229 (7.6) | 1237 (40.8) | 1563 (51.6) | 0.02 | 0.87 (0.77 to 0.98) |
| rs42041 | 7 | CDK6 | 0.66 | 0.26 | 0.25 | 65 (7.0) | 347 (37.5) | 514 (55.5) | 182 (6.1) | 1125 (38.0) | 1655 (55.9) | 0.59 | 1.03 (0.92 to 1.16) |
| rs3761847 | 9 | TRAF1/C5 | 0.39 | 0.44 | 0.43 | 199 (21.1) | 438 (46.5) | 305 (32.4) | 607 (18.8) | 1557 (48.2) | 1063 (33.0) | 0.27 | 1.06 (0.96 to 1.18) |
| rs10818488 | 9 | TRAF1/C5 | 0.81 | 0.45 | 0.43 | 201 (21.3) | 451 (47.8) | 291 (30.9) | 645 (18.5) | 1721 (49.3) | 1126 (32.2) | 0.1 | 1.09 (0.98 to 1.21) |
| rs2900180 | 9 | TRAF1/C5 | 0.97 | 0.39 | 0.35 | 149 (15.8) | 437 (46.4) | 356 (37.8) | 407 (11.9) | 1548 (45.3) | 1464 (42.8) | 0.0003 | 1.21 (1.09 to 1.35) |
| rs2812378 | 9 | CCL21 | 0.30 | 0.33 | 0.35 | 88 (9.5) | 433 (46.5) | 410 (44.0) | 377 (12.5) | 1339 (44.5) | 1294 (43.0) | 0.1 | 0.91 (0.82 to 1.02) |
| rs4750316 | 10 | PRKCQ | 0.91 | 0.18 | 0.2 | 38 (4.0) | 256 (27.2) | 649 (68.8) | 133 (3.8) | 1106 (31.6) | 2261 (64.6) | 0.05 | 0.88 (0.77 to 1.0) |
| rs1678542 | 12 | KIF5A | 0.39 | 0.35 | 0.37 | 109 (11.6) | 438 (46.5) | 394 (41.9) | 497 (14.1) | 1622 (45.9) | 1411 (40.0) | 0.08 | 0.91 (0.82 to 1.01) |
| rs4810485 | 20 | CD40 | 0.05 | 0.26 | 0.25 | 64 (6.9) | 351 (37.9) | 512 (55.2) | 173 (5.7) | 1189 (39.1) | 1681 (55.2) | 0.59 | 1.03 (0.92 to 1.16) |
SNPs with a trend p value <0.05 are highlighted in bold.
HWE, Hardy–Weinberg equilibrium; JIA, juvenile idiopathic arthritis; MAF, minor allele frequency; OR, odds ratio; SNP, single nucleotide polymorphism.
The chromosome 6q23 SNP, rs6920220, also showed weak association with JIA (allelic OR=1.16, 95% CI 1.02 to 1.31, trend p=0.02) (table 2). As with RA, the SNP, rs13207033, showed a weak protective association with JIA (allelic OR=0.87, 95% CI 0.77 to 0.98, trend p=0.02). Both the KIF5A SNP, rs1678542 and the SNP, rs4750316, close to PRKCQ, showed weak protective association with JIA (allelic OR=0.91, 95% CI 0.82 to 1.01, trend p=0.08 and allelic OR=0.88, 95% CI 0.77 to 1.0, trend p=0.05, respectively), although the evidence for association did not reach the corrected threshold for any of these loci.
The markers rs10910099 in the MMEL gene, rs42041 in the CDK6 gene, rs4810485 in the CD40 gene and rs2812378, close to the CCL21 gene, showed no association with JIA (table 2).
The associated SNPs were examined for differences in allele frequencies between the seven ILAR subtypes. In all cases this was not significant (p>0.05) (data not shown). Therefore further stratification by ILAR subtype was not performed.
Figure 1 shows a comparison between the association analysis results in RA and JIA. For all the JIA-associated SNPs the same allele is associated with JIA as was associated with RA and effect sizes are similar.
Figure 1.
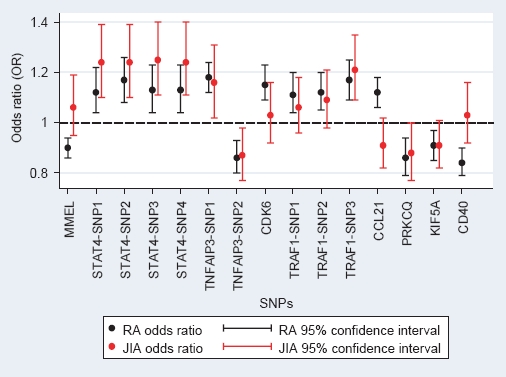
Plot of allelic odds ratios for minor allele for all previously associated rheumatoid arthritis (RA) single nucleotide polymorphisms (SNPs), comparison with juvenile idiopathic arthritis (JIA). Plots of allelic odds ratios and 95% CI for the association analysis of all SNPs, results in RA (black dots and lines) and in JIA (red dots and lines). STAT4-SNP1, rs11889341; STAT4-SNP2, rs7574865; STAT4-SNP3, rs8179673; STAT4-SNP4, rs10181656. TNFAIP3-SNP1, rs6920220; TNFAIP3-SNP2, rs13207033. TRAF1-SNP1, rs3761847; TRAF1-SNP2, rs10818488; TRAF1-SNP3, rs290018.
Discussion
In this study we examined nine recently identified RA loci, which have been confirmed in independent datasets, for association with JIA. Using this approach we have identified two JIA susceptibility loci, of which one was a novel JIA association (STAT4); while the evidence for a true association for STAT4 is compelling, it will require validation in independent JIA case–control cohorts, to confirm it as a JIA susceptibility locus. The second association confirms previously published associations of the TRAF1/C5 locus with JIA susceptibility.27 28 However, previous studies of this locus have provided conflicting evidence for association. For example, a recent study of Dutch JIA cases and controls investigated the rs10818488 SNP in the TRAF1/C5 region,27 and found no evidence for association of this SNP in patients with JIA as a whole. Interestingly, in our UK study we also found no statistically significant evidence for association of this SNP in all JIA cases but there was a trend (allelic OR=1.09, 95% CI 0.98 to 1.21, trend p=0.1).
We performed a meta-analysis of the two studies which yielded significant evidence for association (OR=1.09, 95% CI 1.0 to 1.2, p=0.05) with no evidence for heterogeneity between the two cohorts (p=0.9). A second, small study of SNPs within TRAF1/C5 region in USA JIA cases also reported association with this region. The strongest effect in that study was with rs376187.28 This SNP was not associated in our current study, although there was association with another SNP (rs2900180). The lack of consistency between studies of TRAF1/C5 region and JIA susceptibility may be due to the lack of power of individual studies to detect the modest effect sizes reported at this locus. For the three SNPs in this study in the TRAF1/C5 region we had 86% power to detect an effect for the SNP rs2900180 and we found that this was the most strongly associated SNP with JIA for this region. For the other two SNPs we had ∼60% power to detect an association and therefore the lack of association may be a false-negative result due to small sample size. This is also compounded by the fact that, as yet, the causal SNP(s) in this region has not been determined.
Although not statistically significant, we found weak evidence of association with JIA of SNPs in the chromosome 6q23 region, within KIF5A and close to PRKCQ; in each case the effect sizes were similar to those seen in RA. Thus, given the relatively low power within our study, they warrant further investigation in independent datasets.
We found no significant association with the SNPs in the MMEL, CDK6, CCL21 or the CD40 genes, which may suggest that these genes are RA-specific loci. However, as the effect sizes for these genes in RA were modest, this study had limited power to detect an association with JIA and MMEL, CDK6 and CCL21 (53%, 69% and 59% respectively). For three of these SNPs, the direction of association was opposite in the JIA samples compared with the RA studies. Larger datasets and meta-analyses will be required to completely rule out association of these SNPs with JIA.
One limitation of the study may be the use of a common set of controls between this study and two of the previous RA validation studies.17 19 This is a conventional strategy, as used by the WTCCC GWAS,25 to minimise genotyping, but it may be a concern.29 All the control genotypes used in this study were compared with the control data from the WTCCC and there was no significant difference (p<0.05) in the frequencies, suggesting these controls accurately represent UK population frequencies.
It is interesting to note that for the JIA susceptibility loci identified, the associated allele is the same and effect size similar to that seen in studies of RA (figure 1).
This finding, combined with data from previous studies,30 31 suggests that for many complex diseases there will be some shared susceptibility loci while others will be disease specific. In JIA, given the clinical heterogeneity, the picture may be even more complicated. Current classification criteria divide JIA into seven subgroups and it would be interesting to determine whether the susceptibility loci identified are common to all subgroups or restricted to only some subgroups. However, stratified analysis leads to small sample sizes for many of the subgroups as well as introducing the problem of multiple testing and thus makes interpreting these results challenging. In our study, we tested whether there was an overall difference in allele frequency between the seven subgroups for each SNP; as no SNPs showed a significant difference between subgroups, further stratification analysis was not undertaken. This lack of difference between subgroups may reflect our hypothesis/strategy, in that we are looking for shared autoimmune or inflammatory arthritis susceptibility loci. Alternatively, as already stated, the study may have been simply underpowered to detect any differences between subgroups. As and when these findings are validated in other cohorts it will be important to combine data in order to improve power and accurately determine whether the associations seen are consistent between the different clinical subgroups of JIA.
In RA, there is some evidence to suggest that there are distinct genetic risk factors for anti-cyclic citrullinated peptide (anti-CCP)-positive and anti-CCP-negative disease. In the UK population the TRAF1/C5 association did appear stronger in the anti-CCP- positive subgroup than in the negative subgroup,14 however, the strength of association of the STAT4 locus was similar in both subgroups. There is little evidence for anti-CCP antibodies in JIA, apart from in the rheumatoid factor-positive polyarthritis subgroup, though antinuclear antibodies (ANA) are often observed. However, larger sample sizes will be required to investigate any specific associations with ANA-positive and ANA-negative subgroups.
It is important to note that for all these loci the causal variant, indeed in some cases the gene, has yet to be determined. Re-sequencing and fine-mapping studies are underway in order to further characterise the genetic effects before functional studies can be undertaken. However, all these loci represent good candidates for JIA as genes within these regions (TRAF1, STAT4, TNFAIP3, PRKCQ) known to be involved in T-cell receptor signalling or T-cell activation pathways.
In conclusion, by investigating the overlapping of RA and JIA susceptibility loci, we have identified a novel JIA susceptibility locus (STAT4) and confirmed association with a second, TRAF1/C5. It is clear that this strategy is useful in identifying autoimmune/arthritis-associated JIA susceptibility loci. However, it is likely that there are many more JIA-specific or JIA-subgroup-specific genes yet to be identified and well-powered GWAS will be required to identify these.
Acknowledgments
We thank David Strachan for facilitating access to the 1958 birth cohort. We acknowledge use of genotype data from the British 1958 Birth Cohort DNA collection, funded by the Medical Research Council grant G0000934 and the Wellcome Trust grant 068545/Z/02.
Footnotes
Competing interests: None.
Funding: This work was supported by the Arthritis Research Campaign: arc grant reference number 17552.
Ethics approval: This study was conducted with the approval of the North-West Multi-Centre Research Ethics Committee (MREC 99/8/84) and the University of Manchester Committee on the Ethics of Research on Human Beings.
Provenance and peer review: Not commissioned; externally peer reviewed.
Patient consent: Obtained.
References
- 1.Xavier RJ, Rioux JD. Genome-wide association studies: a new window into immune-mediated diseases. Nat Rev Immunol 2008;8:631–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhernakova A, Alizadeh BZ, Bevova M, et al. Novel association in chromosome 4q27 region with rheumatoid arthritis and confirmation of type 1 diabetes point to a general risk locus for autoimmune diseases. Am J Hum Genet 2007;81:1284–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Heel DA, Franke L, Hunt KA, et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat Genet 2007;39:827–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duerr RH, Taylor KD, Brant SR, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 2006;314:1461–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cargill M, Schrodi SJ, Chang M, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet 2007;80:273–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burton PR, Clayton DG, Cardon LR, et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet 2007;39:1329–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet 2007;369:767–78 [DOI] [PubMed] [Google Scholar]
- 8.Petty RE, Southwood TR, Manners P, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol 2004;31:390–2 [PubMed] [Google Scholar]
- 9.Thomson W, Barrett JH, Donn R, et al. Juvenile idiopathic arthritis classified by the ILAR criteria: HLA associations in UK patients. Rheumatology (Oxford) 2002;41:1183–9 [DOI] [PubMed] [Google Scholar]
- 10.Hinks A, Barton A, Shephard N, et al. Identification of a novel susceptibility locus for juvenile idiopathic arthritis by genome-wide association analysis. Arthritis Rheum 2009;60:258–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hinks A, Worthington J, Thomson W. The association of PTPN22 with rheumatoid arthritis and juvenile idiopathic arthritis. Rheumatology (Oxford) 2006;45:365–8 [DOI] [PubMed] [Google Scholar]
- 12.Hinks A, Ke X, Barton A, et al. Association of the IL2RA/CD25 gene with juvenile idiopathic arthritis. Arthritis Rheum 2009;60:251–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Remmers EF, Plenge RM, Lee AT, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med 2007;357:977–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barton A, Thomson W, Ke X, et al. Re-evaluation of putative rheumatoid arthritis susceptibility genes in the post-genome wide association study era and hypothesis of a key pathway underlying susceptibility. Hum Mol Genet 2008;17:2274–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurreeman FA, Padyukov L, Marques RB, et al. A candidate gene approach identifies the TRAF1/C5 region as a risk factor for rheumatoid arthritis. PLoS Med 2007;4:e278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plenge RM, Seielstad M, Padyukov L, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis--a genomewide study. N Engl J Med 2007;357:1199–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomson W, Barton A, Ke X, et al. Rheumatoid arthritis association at 6q23. Nat Genet 2007;39:1431–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plenge RM, Cotsapas C, Davies L, et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet 2007;39:1477–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barton A, Thomson W, Ke X, et al. Rheumatoid arthritis susceptibility loci at chromosomes 10p15, 12q13 and 22q13. Nat Genet 2008;40:1156–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raychaudhuri S, Remmers EF, Lee AT, et al. Common variants at CD40 and other loci confer risk of rheumatoid arthritis. Nat Genet 2008;40:1216–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Packham JC, Hall MA. Long-term follow-up of 246 adults with juvenile idiopathic arthritis: functional outcome. Rheumatology (Oxford) 2002;41:1428–35 [DOI] [PubMed] [Google Scholar]
- 22.Adib N, Hyrich K, Thornton J, et al. Association between duration of symptoms and severity of disease at first presentation to paediatric rheumatology: results from the Childhood Arthritis Prospective Study. Rheumatology (Oxford) 2008;47:991–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petty RE, Southwood TR, Baum J, et al. Revision of the proposed classification criteria for juvenile idiopathic arthritis: Durban, 1997. J Rheumatol 1998;25:1991–4 [PubMed] [Google Scholar]
- 24.Lee HS, Remmers EF, Le JM, et al. Association of STAT4 with rheumatoid arthritis in the Korean population. Mol Med 2007;13:455–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.The Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007;447:661–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Albers HM, Kurreeman FA, Houwing-Duistermaat JJ, et al. The TRAF1/C5 region is a risk factor for polyarthritis in juvenile idiopathic arthritis. Ann Rheum Dis 2008;67:1578–80 [DOI] [PubMed] [Google Scholar]
- 28.Behrens EM, Finkel TH, Bradfield JP, et al. Association of the TRAF1-C5 locus on chromosome 9 with juvenile idiopathic arthritis. Arthritis Rheum 2008;58:2206–7 [DOI] [PubMed] [Google Scholar]
- 29.Seldin MF, Amos CI. Shared susceptibility variations in autoimmune diseases: a brief perspective on common issues. Genes Immun 2009;10:1–4 [DOI] [PubMed] [Google Scholar]
- 30.Smyth DJ, Plagnol V, Walker NM, et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med 2008;359:2767–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.IMSGC The expanding genetic overlap between multiple sclerosis and type I diabetes. Genes Immun 2009;10:11–14 [DOI] [PMC free article] [PubMed] [Google Scholar]