

Abstract
Motivation: Single nucleotide polymorphisms are the most common form of variation in human DNA, and are involved in many research fields, from molecular biology to medical therapy. The technological opportunity to deal with long DNA sequences using shotgun sequencing has raised the problem of fragment recombination. In this regard, Single Individual Haplotyping (SIH) problem has received considerable attention over the past few years.
Results: In this article, we survey seven recent approaches to the SIH problem and evaluate them extensively using real human haplotype data from the HapMap project. We also implemented a data generator tailored to the current shotgun sequencing technology that uses haplotypes from the HapMap project.
Availability: The data we used to compare the algorithms are available on demand, since we think they represent an important benchmark that can be used to easily compare novel algorithmic ideas with the state of the art. Moreover, we had to re-implement six of the algorithms surveyed because the original code was not available to us. Five of these algorithms and the data generator used in this article endowed with a Web interface are available at http://bioalgo.iit.cnr.it/rehap
Contact: filippo.geraci@iit.cnr.it
1 INTRODUCTION
Recently, many researchers' focus has shifted from what individuals of a certain species have in common to their differences, and thus to DNA mutations. The single nucleotide polymorphism (SNP pronounced ‘snip’) is the most widespread form of variation in human DNA, and consists in the variation of the base present in a single fixed position of the DNA strand. The sequence of all SNPs in a given chromosome is called a haplotype. Humans are diploid organisms, this means that except for the sexual chromosomes of males, the chromosomes come in two copies: one inherited from the mother and one from the father. As a consequence, the haplotypes of a chromosome can be fully described by two sequences of SNPs: the mother's haplotype and the father's. Since haplotypes contain all the information about DNA variations, they play a crucial role in many studies about variations in gene expression and prediction of diseases. For this reason, several sequencing projects have been launched with the ultimate goal of building a complete map of the SNPs present in the human DNA (Frazer et al., 2007; Levy et al., 2007; Via et al., 2010). At the moment, the HapMap Consortium has produced the most complete map of SNPs in human DNA, consisting of over 3.1 million SNPs (Frazer et al., 2007) and has estimated that the overall number of SNPs in the human DNA is about 9–10 million. Single Individual Haplotype (SIH) reconstruction problem is one of the core problems in the reconstruction of whole genomes (Zhao et al., 2007). It consists in rebuilding the two haplotypes from a set of fragments obtained by the shotgun sequencing of the chromosomes. Current shotgun technology produces a very large set of fragments with lengths in the range of 200–900 bases with a certain degree of overlap between them (Morozova and Marra, 2008). This technology does not allow keeping track of the association of a fragment with its haplotype. An important characteristic of this problem is that, unlike the fragment assembly problem, the position and orientation of the fragments is known a priori; this means that fragments can be arranged in a matrix called SNP Matrix. In the absence of errors, it is easy to find a bipartition of the SNP matrix such that the fragments belonging to each partition do not conflict (two fragments are said to be conflicting if for a certain position not gap they have different values). In the real-world application, this is not the case and errors affect the SNP matrix. Errors can originate from various sources. Reading errors are typically due to chemical/optical errors in reading the SNPs and as a result lead to the insertion of a wrong base in a certain position. Ambiguous readings occur when the signal strength of a SNP in not enough to establish its correct value with a high degree of confidence. The effect of ambiguous readings is typically the insertion of a gap in the sequence.
The SIH problem has attracted considerable attention in the past few years and a large number of models and algorithms have been introduced in the literature. Many models have NP-hard solutions, thus heuristics are often used. However, a common framework to compare all those algorithms with each other is still lacking. We tested several algorithms for the SIH problem and from them, we selected the seven most effective heuristic algorithms. Moreover, we defined a common framework in which all the algorithms were evaluated. An exhaustive comparison of all the algorithms for the SIH problem is not feasible for many reasons. For example, many algorithms are designed for simplified models of the SNP matrix and are inadequate for the matrices generated according to the current shotgun sequencing technology. In this class of algorithms, there are those (like KMec; Xie et al., 2008) that do not allow the insertion of gaps inside the fragments or the insertion of mate pair sequences. Another class of algorithms (as in Wang et al., 2006) uses genotype information to solve the SIH problem. This information can improve the accuracy of the reconstructed haplotypes, but is not often available in practice.
We restrict our analysis to those algorithms designed for a model of the SNP matrix compatible with the current shotgun technology. For all the algorithms considered, we requested the software from the corresponding authors. In the cases in which the software was not provided, we checked whether the corresponding papers contain a description of the algorithms enough accurate, allowing us to carefully re-implement them. (We were unable to include in this review all the algorithms for which we had insufficient details). Re-implementing software raises the issue of validating its correctness and running time performance. To validate our implementations we tested each algorithm. For each test, we used exactly the same parameters used by the authors (i.e. the same error rate, coverage and haplotype length). Wherever possible we also used the same datasets. For example, this is the case of Wang et al. (2007) and Zhao et al. (2005) who generate their haplotypes from the freely available dataset described in Daly et al. (2001). Our tests reveal that all our implementations are comparable with the original algorithms both in accuracy and running time. We also excluded some algorithms for which we received the software due to their characteristics. For example, the Branch and Bound algorithm described in Wang et al. (2005) is much slower than its competitors, and its running time makes it unsuitable in practice even for small datasets. The same problem is present in HASH (Bansal et al., 2008). We also tested the PM-MFR algorithm described in Xie and Wang (2008) (in this case as well we received the software from the authors). The running time of this algorithm depends on many parameters. One of these parameters is the number of gaps in mate pair sequences. Our tests show that even for small values of this parameter, the running time of this algorithm becomes high.
Our test data are available on request, which means that they can be used in the near future to compare new ideas with the actual state of the art.
Despite the high number of real human haplotypes freely available on the Web, there are no real SNP matrices. In this article, we made the effort to generate realistic data, writing a software program that gets in input a real haplotype and simulates the actual technologies of shotgun sequencing to produce realistic SNP matrices. The SNP matrices simulator is described in Section 4.1.
The article is organized as follows: in Section 2, we formally define the SIH reconstruction problem and its most common computational models. In Section 3, we describe all the compared algorithms. Section 4 reports quality and running time evaluation. We conclude in Section 5.
2 PROBLEM FORMULATION
Due to the diploid nature of humans cells, except for male sex chromosomes, each chromosome comes in two nearly identical copies, one inherited from the mother and one from the father. Current technology of shotgun sequencing is unable to keep track of the association between a fragment and its own chromosome. Thus, from the biological point of view, the Single Individual Haplotyping (SIH) problem consists in the reassignment of each fragment to the original haplotype.
From the computational point of view, the problem was first formalized in Lancia et al. (2001). A fragment is represented as a string of length m such that each character corresponds to a base in the alphabet Σ = {a, c, g, t} or a − in case of gap. The natural way to store all n fragments is a matrix M with n rows and m columns, such that, to each row corresponds a fragment fi = M[i]. The matrix M is called SNP matrix. The element M[i][j] stored in the j-th entry of the i-th row of M represents the j-th SNP of the haplotype for fragment fi. We will denote this element also as fi[j]. In case fi does not cover the j-th position of the haplotype, we have M[i][j] = −.
We say that a fragment f contains a gap (or is gapped) if for i, j, k ∈ [1, m] such that i < j < k we have f[i] ≠ −, f[j] = − and f[k] ≠ −. If no fragments in M are gapped, then the SNP matrix is said to be gapless otherwise it is gapped.
In absence of errors in the SNP matrix, each column of M can contain one or two distinct elements. The presence of only one element indicates that the SNP in the corresponding site is homozygous otherwise the SNP is heterozygous. We say that two fragments fi and fj are in conflict if the following condition is true: ∃k ∈ [1, n] such that fi[k] ≠ fj[k] ∧ fi[k] ≠ −∧fj[k] ≠ −. According to the definition of conflict between pairs of fragments, in Lancia et al. (2001) the conflict graph is defined. Let G = {V, E} be a graph such that each vertex corresponds to a fragment and there is an edge between two fragments if there is a conflict between them. If M does not contain errors, then G is bipartite (Fig. 1a). The bipartition is not necessarily unique, if the graph has several connected components. In practice, due to errors in the SNP matrix, the conflict graph is never bipartite (Fig. 1b). Thus, in this case, the SIH reconstruction problem can be formalized as the problem of removing a certain number of edges from G until the resulting graph becomes bipartite. The problem of reducing a graph to a bipartite graph is well studied in the literature where many models were proposed. Among them, in the context of the SIH problem the following formalizations are often used:
MEC (minimum error correction): determine a minimal set of entries of the matrix M whose correction to a different value induces a bipartite graph
MFR (minimum fragment removal): determine a minimal number of fragments whose removal from the input set induces a bipartite graph
MSR (minimum SNP removal): determine a minimal number of SNPs whose removal from the input set induces a bipartite graph
LHR (longest haplotype reconstruction): determine set of fragments whose removal from the input set induces a bipartite graph and the length of the induced haplotype is maximized.
Fig. 1.

(a) Two haplotype strings, six fragments (without errors) and the corresponding bipartite conflict graph. (b) The same two haplotypes, six fragments (with errors in gray) and the corresponding conflict graph (not bipartite).
In the presence of gaps, all the above problem formalizations are NP-hard. More details about the complexity of some of these problems can be found in Cilibrasi et al. (2007). Even if MEC is the most complex of the above models, it is the most commonly used in practice. Four of the algorithms described in this article approach the SIH problem using the MEC model (2d-mec, SHR, HapCUT) or a model derived from it (MLF). The other algorithms we considered do not follow the above problem formulations. Note that there is no proven relationship among the above problem formulations and the SIH problem (i.e. a more accurate solution of the above problems does not necessarily correspond to a better solution of the SIH problem). Since the goal of this review is to evaluate the accuracy of the algorithms with respect to the SIH problem, we derived the SNP matrices from pairs of real haplotypes from the HapMap project and used them as gold standard for evaluating the consensus haplotypes.
We now introduce some definitions and notations that we will use later in the algorithms description. Let C be a set of rows of M and let xC[i] ∈ Σ be the character that appears most frequently at position i among the fragments in C (or xC[i] = − if all the fragments of C have a gap in position i), we define the haplotype consensus H(C) deduced by C as the string of m characters such that the character at position i is xC[i].
We define the generalized Hamming distance between two fragments fi and fj as:
| (1) |
where
If the generalized Hamming distance between two fragments is different from 0, we say that the two fragments are in conflict.
3 ALGORITHMS
3.1 2d-mec: a clustering algorithm for the MEC model
In Wang et al. (2007), a clustering algorithm is used to split the rows of M in two sets. The main contribution of the article consists in the combination of the two distance functions used by the clustering algorithm. As first distance the authors used the generalized Hamming distance Ham as defined in Equation (1). This distance takes into account only the number of mismatches between two fragments. The second distance is defined as follows: let fi and fj be two fragments
| (2) |
where
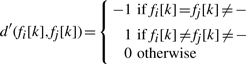 |
The definition of distance in Equation (2) also takes into account the number of matches between the two fragments. This means that given a certain fixed number of mismatches between two fragments, the more they overlap the closer they are according to D′. Note that D′ is not a distance in a strict sense: in fact it can be negative and has values in the range [−m, m]. Moreover, the triangular inequality does not hold.
Using the above distance functions, the authors proposed a simple iterative clustering procedure. To compare two fragments, the functions Ham and D′ are evaluated in cascade.
The algorithm proceeds as follows:
for each possible pair of fragments in the SNP matrix the generalized Hamming distance is computed. Let fi and fj be the two furthest fragments according to Ham, we initialize the two sets C1 = fi and C2 = fj.
Let H1 = H(C1) and H2 = H(C2) be the two consensus strings derived from C1 and C2: all the fragments are compared with H1 and H2 and assigned to the corresponding closer set. If a fragment is equidistant from the two consensus strings, the distance D′ is used to decide to which set assign the fragment.
Once all fragments are assigned, the consensus strings H1 and H2 are updated and the algorithm restarts from (2). The procedure loops until a stable haplotype pair is found (i.e. when the consensus haplotypes are the same before and after the update).
3.2 Clustering algorithm for the MLF problem
In Zhao et al. (2005), the authors raised a weighted variant of the MEC problem called Weighted Minimum Letter Flip (WMLF). Assuming we have a matrix W, such that each entry is a number in the range [0, 1] representing the degree of confidence of the SNP in the same position in the matrix M. We can define a weighted version of the generalized Hamming distance between two fragments fi and fj as:
where
The distance WHam is extended to deal with haplotypes, assuming that the weight associated with each character of the consensus string is 1.
The proposed algorithm is based on the well-known one-pass k-means clustering algorithm (McQueen, 1967). The procedure initialization consists in randomly partitioning the rows of M in two sets C1 and C2 deriving from them two consensus strings: H1 = H(C1) and H2 = H(C2). In the main procedure loop, at each iteration C1 and C2 are reinitialized and a new partition of the rows in M is done. For each fragment fi, we compare its distance from H1 and H2. If WHam(fi, H1) > WHam(fi, H2) then fi is assigned to C1, otherwise the fragment is assigned to C2. The procedure terminates when two stable haplotypes are detected. More details regarding the convergence of this method can be found in McQueen (1967). Due to the random initialization of sets C1 and C2, every run of this algorithm on a certain dataset can return a different haplotype consensus. To mitigate the effect of randomness, the authors run the algorithm 100 times and return as final result the consensus pair that minimizes the following target function:
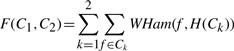 |
(3) |
The main drawback of this algorithm is that with actual shotgun sequencing technology, the information about the confidence level of each fragment is typically not available. In this case, the algorithm has to assume the same level of confidence for each fragment: thus WHam reduces to Ham and the target function to minimize in Equation (3) reduces to the target function of the MEC model. Even in Zhao et al. (2005), experiments are made assuming each entry of W as equal to 1.
3.3 Fast Hare
Fast Hare (Panconesi and Sozio, 2004) is one of the first heuristics for the SIH problem. Although it was designed to work in a gapless environment, our experiments and those reported in Genovese et al. (2008) confirm that this method still works nicely also in the more general case in which gaps are allowed. As a preliminary step, Fast Hare removes from the SNP matrix M all those columns where there is a character (not −) that is over-represented with respect to the others. These columns are considered as homozygous sites. We call this reduced matrix 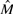 . More formally: let |xc| be the number of occurrences of the character x ∈ Σ in the column c of M. The probability to find x in c is
. More formally: let |xc| be the number of occurrences of the character x ∈ Σ in the column c of M. The probability to find x in c is
If a character x in c such that Pc(x) ≥ t exists, the column c is removed from the SNP matrix and the character x will be inserted in the final solution. In Panconesi and Sozio (2004) and in our experiments, the threshold t is set to 0.8. The intuition behind this filtering comes from the fact that when Pc(x) ≥ t column c represents a homozygous SNP (thus the column does not help in the reconstruction procedure) and the positions (not gaps) not containing x are errors. As the first step, Fast Hare sorts the fragments of 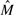 according to the following ordering criterion: let ki and kj be, respectively, the position of the first character not gap in fi and fj, thus
according to the following ordering criterion: let ki and kj be, respectively, the position of the first character not gap in fi and fj, thus 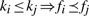 . After sorting the fragments of
. After sorting the fragments of  , Fast Hare initialize two sets C1 = C2 = ∅. At this point, according to their order, the fragments of
, Fast Hare initialize two sets C1 = C2 = ∅. At this point, according to their order, the fragments of 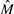 are scanned one at a time. The first fragment is assigned to C1. Considering the fragment fi, Fast Hare computes its similarity with the two partial consensus strings H(C1) and H(C2). As similarity score, Fast Hare uses −D′ where D′ is the distance we defined in Equation (2). The fragment is assigned to the set whose consensus shows higher similarity. Note that the values of −D stand in the range [−1, 1] and for all fragments holds −D′ (f, H(∅)) = 0.
are scanned one at a time. The first fragment is assigned to C1. Considering the fragment fi, Fast Hare computes its similarity with the two partial consensus strings H(C1) and H(C2). As similarity score, Fast Hare uses −D′ where D′ is the distance we defined in Equation (2). The fragment is assigned to the set whose consensus shows higher similarity. Note that the values of −D stand in the range [−1, 1] and for all fragments holds −D′ (f, H(∅)) = 0.
3.4 SpeedHap
SpeedHap (Genovese et al., 2007, 2008) approaches the haplotype assembly problem differently from previously described algorithms. Instead of considering each fragment as a whole, it attempts to solve n instances of the haplotyping problem on 1-base long fragments and combines results.
SpeedHap is a greedy heuristic. It builds its solution in a pre-processing phase and three main phases. The goal of each phase is to exploit the outcome solution of the preceding phase and improve it by relaxing some constraints.
Pre-processing: in this phase, SpeedHap performs a statistical analysis of the columns of the SNP matrix attempting to locate detectable errors and, if possible, correct them. In this phase, the heuristic also set up some data structures.
First phase: in this phase, the heuristic selects an initial set of columns such that they are likely to contain as few errors as possible. For each column, SpeedHap builds a set Gi (called profile) in which each element is the set of all the indices of the fragments containing the same character in position i. It is easy to observe that only profiles having two elements (i.e. columns of the SNP matrix containing exactly two distinct characters) are of interest since an empty profile corresponds to a hole in the haplotype, a profile with just one element corresponds to a homozygous site, a profile with more than two characters must contains errors. Let Pi = (Pi(1), Pi(2)) be the profile of column i such that it corresponds to a heterozygous site. Given two columns i and j, we can define the error matrix as
When the error matrix has positive values only in one diagonal and it is of full rank, there are no detectable conflicts between the two involved columns. Now, consider a graph such that it has a vertex for each column of M corresponding to a heterozygous site and there is an edge between two vertices if the corresponding error matrix does not reveal inconsistencies (i.e. the matrix is diagonal and of full rank). Using a DFS search, we can partition the graph in connected components. Each component corresponds to a set of columns not conflicting among themselves. The initial partitioning of the fragments of M is extracted from the largest set.
Second phase: in this phase, the algorithm works in a similar manner. The partitioning obtained from the previous step acts as a special profile (pivot). All the columns not involved in the previous phase are compared with the pivot and the error matrix is computed. If the error matrix does not show inconsistencies, the column is included in the solution. This procedure is repeated iteratively until it is no longer possible to add new columns to the pivot.
Third phase: in this phase, some constraints are relaxed: the insertion of the columns in the final solution no longer requires an error matrix of full rank.
Another important contribution of SpeedHap is the use of the context for resolving ambiguities in the final haplotype reconstruction. In Genovese et al. (2008), the authors parsed a large database of human haplotypes measuring the empirical entropy of order up to 2. As a result, they show that there seems to exist a statistical correlation between the base in a certain SNP site, its preceding SNP site and its succeeding one.
When the coverage (i.e. the number of fragments that cover a certain position) is low and the error rate is relatively high, it is not infrequent the case in which, building a haplotype, exactly half of the fragments in a certain position have a certain value and half of them have another value. In this case, the choice of which character should appear in the haplotype is arbitrary. To break ties, SpeedHap exploits the statistical correlation among contiguous sites. Consider the case in which the procedure has to choose for the site in position i whether select A or B. Let x be the character in position i − 1 and z the character in position i + 1. The procedure will decide for: A if the empirical entropy of the string xAz is lower than those of the string xBz, otherwise it will decide for B.
3.5 HapCUT
HapCUT (Bansal and Bafna, 2008) approaches the haplotype assembly as a MAX-CUT problem. The HapCUT algorithm considers the submatrix of the SNP matrix in which we remove all the columns corresponding to Homozygous SNPs and all the columns in which there are present more than two distinct bases (i.e. there must be at least a mistaken base). Let us call the resulting matrix X. Due to the fact that each column of X contains exactly two possible SNP values, it can be represented using the restricted alphabet 0, 1, −. The haplotype pair H associated with X is composed by a binary string h and its bit-wise complement ĥ.
Given a certain haplotype pair H, the authors defined a graph GX(H) such that there is a vertex for each column of the matrix X and there is an edge between two vertices of GX(H) if the corresponding columns in X are linked by at least one fragment. Consider the fragment Xi such that it covers both positions j and k. Let Xi[j, k] and H[j, k] represent the restriction of Xi and H to loci j and k. There are two cases: Xi[j, k] matches one of the two haplotype strings of H[j, k], or Xi[j, k] does not match any. The weight wH (j, k) associated with the edge between node j and k in the graph GX(H) is given by the number of fragments such that Xi[j, k] does not match any string in H[j, k] minus the number of fragments such that the match exists. The higher wH(j, k), the weaker is the correlation between the haplotype pair H and the SNP matrix restricted to columns j and k. Let (S, X − S) be a cut of G, the weight of the cut is defined as follows:
Consider the haplotype pair HS derived from H by flipping all the elements involved in S. The authors showed that if wH(S) is positive for the graph GX(H) then the following holds:
As a consequence of the above result, the problem of finding a haplotype pair minimizing the MEC score is reduced to the problem of finding a max-cut in GX(H). This problem is well known to be NP-complete (Karp, 1972), thus heuristic methods are often used.
The HapCUT procedure exploits the connection between the MEC optimization and the max-cut problem. Starting from a random haplotype pair, HapCUT iteratively attempts to refine the haplotype pair to reduce the MEC score. At each iteration, the algorithm proceeds as follows: (1) compute the graph GX(H), (2) compute a max-cut S using a greedy heuristic like that in Sahni and Gonzalez (1974), (3) if the MEC score of the pair HS is smaller than the score of H, keeps as new haplotype pair HS.
The procedure loops until it is no longer possible to reduce the MEC score.
3.6 DGS
In Levy et al. (2007), the authors described a large study on genome sequencing. In the paper they also described a good algorithm for the SIH problem. For lack of a better name, we call this algorithm DGS. As in HapCUT, this algorithm works with a submatrix of M in which we remove all the columns corresponding to homozygous sites and those with more than two distinct values. Let us again call this matrix X. Even in this case, X can be represented using the restricted alphabet 0, 1, −. The haplotype pair H associated with X is composed by a binary string h and its bit-wise complement ĥ. The DGS procedure works in two phases: an initialization in which we build a pair of initial haplotypes and a refinement step in which haplotypes are iteratively refined. The initialization works as follows:
the fragment with the minimal number of gaps is used to initialize a haplotype. The other haplotype is initialized with the complementary string;
until no more fragments share non-missing information with a haplotype, select the fragment such that the number of columns it has in common with one haplotype minus number of columns indicating the other haplotype is maximal and assign it to the corresponding haplotype. The other haplotype is updated with the complementary string.
The second phase iteratively refines the haplotype consensus strings and stops when, at the end of an iteration, the solution no longer changes. At each iteration, the haplotype consensus strings are determined by majority rule, then each fragment is associated with the closest haplotype.
3.7 SHR-three
In Chen et al. (2008), the authors proposed a probabilistic framework to approach the SIH problem. According to the proposed model, the authors designed a novel randomized algorithm [i.e. an algorithm that receives, in addition to its input data, a stream of random bits that it can use for the purpose of making random choices (Karp, 1991) ] and generalize it to handle reading errors and gaps. The most general variant of this algorithm is called SHR-three. The algorithm requires as input the SNP matrix and a parameter u that controls the number of iterations made by the main loop. As proposed by the authors in their experiments, we set u = 10.
The SHR-three main loop is as follows:
select at random two fragments f1 and f2 from M and assign to each of them an empty set (C1 to f1 and C2 to f2);
each fragment of M is compared through the generalized Hamming distance with f1 and f2 and inserted in the set related to the closest fragment;
for each of the two sets compute the MEC score (i.e. the sum of the distances among each fragment in Ci and fi for i = {1, 2}) and get as score the highest value; and
if the computed score is lower than the previous computed ones, than Ĉ1 = C1 and Ĉ = C2.
As consensus strings SHR-three returns H(Ĉ1) and H(Ĉ2).
4 RESULTS
4.1 Input data and fragment generation
The research project HapMap (HapMap, 2005) has produced a certain number of maps of the human haplotypes that are publicly available. In the first two phases of the project, the consortium used PHASE software for estimating haplotypes from population genotype data. Recently, in the third phase, the consortium has adopted sequencing for estimating haplotypes. For our experiments, we used the Phase I HapMap data1. A detailed description of the dataset characteristics can be found in HapMap (2005).
The Phase I HapMap dataset consists of all 22 chromosomes (for females, the haplotypes of the X chromosome are also available) of 269 different individuals coming from four different populations
CEU: Utah residents with ancestry from northern and western Europe (90 individuals)
YRI: Yoruba in Ibadan, Nigeria (90 individuals)
HCB: Han Chinese in Beijing, China (45 individuals)
JPT: Japanese in Tokyo, Japan (44 individuals).
Individuals of the HCB and JPT populations are unrelated among each other. Individuals of CEU and YRI are parent-offspring trios. To prevent possible undesired effects due to the parental relationship, in our experiments, we consider only the haplotypes of the parents. This reduces the population to 209 unrelated individuals.
Thus, we were able to generate the fragments and the SNP matrices from real data instead of using synthetic haplotypes as input. Using real haplotypes, the Hamming distance between them is no longer a free parameter. We observed that haplotype pairs show a great variability in the Hamming distance. Typical values of Hamming distance are in the range [0.4m, m]. To evaluate whether Hamming distance produces effects in the outcome of the tested algorithms, we performed two sets of experiments: one in which we select haplotype pairs having Hamming distance < 0.7 m and one in which the Hamming distance between the haplotypes considered is > 0.7 m. Our tests show that Hamming distance does not affect the performance (both in terms of reconstruction rate and running time) of the algorithms considered.
Distilling realistic SNP matrices from the haplotypes is crucial for designing meaningful experiments. In this sense one should pay attention to three main aspects: the simulation of the shotgun sequencing process, the estimation of some technological parameters of sequencers and some intrinsic characteristics of human DNA. To simulate the shotgun sequencing process, we used the widely accepted algorithm described in Myers (1999). According to Li et al. (2003) and Metzker (2005), current shotgun sequencers are able to manage DNA fragments of hundreds of bases and the average distance in bp of two SNPs in human DNA is quantified in 300 bp on average. Thus, each DNA fragment covers roughly a number of SNPs in the range [3, 7]. The distribution of errors in the SNP matrices depends on the characteristics of sequencers. A hint about this distribution for different manufacturers can be found in Metzker (2005).
According to the above considerations, the generation of the SNP matrix is as follows: given a pair of haplotypes of length l, each haplotype is replicated c times (the parameter c is called coverage), then each copy is broken into non-overlapping fragments whose length is in the range [3, 7]. According to a certain probability some fragments are merged again in order to simulate matepair sequences (In our experiments, at the end of this phase, globally 50% of the fragments are 1-gapped). Once created, we arrange the fragments in a matrix and insert errors according to a uniform distribution. Note that the number of fragments is not determined a priori but depends on the length l, on the coverage c and on the distribution of the fragment lengths.
4.2 Quality evaluation
To evaluate the quality of the algorithms tested, we use a slightly modified version of the well-known error rate. Let H = (h1, h2) be the pair of correct haplotypes each of which has length m. Let Ĥ = (ĥ1, ĥ2) be the pair of consensus haplotypes returned by an algorithm. According to the standard definition, the reconstruction rate is
where D is the generalized Hamming distance and m the haplotype length. The main disadvantage of using the above formula is that the generalized Hamming distance assigns the same score to two matching characters and to characters matching with gaps. As a consequence of this, a pair of empty haplotypes (i.e. haplotypes in which each position contains a gap) receives the same positive evaluation of the correct solution. To remove the bias introduced dealing with gaps, we used in the computation of the error rate a variant of the Hamming distance in which gaps receive the same penalty of errors. We defined D in greater detail as follows:
where
For each parameter assignment, Table 1 reports the average reconstruction rate (over 100 runs on distinct instances of SNP matrices) of all the algorithms for the case in which the average Hamming distance between the input haplotypes is > 0.7 m. We do not report results for the case in which the Hamming distance is lower since the performances (both in terms of reconstruction rate and running time) of the algorithms are similar.
Table 1.
Each entry in the table represents the average, over 100 randomly selected HapMap strings, of the Reconstruction Rate when the Hamming distance is in the range [0.7m, m]
 |
The free parameters are: (i) the haplotype length l = 100, 350, 700; (ii) the coverage c = 3, 5, 8, 10; and (iii) the error rate e = 0%, 10%, 20%, 30%. In bold the algorithms with highest performance, in gray the algorithms with the second-best performance. We consider as equal to the performance of two algorithms when the difference between their Reconstruction Rate is in the range [0, 005].
To compare all the algorithms with the optimal haplotype reconstruction, Table 1 also reports the reconstruction rate for the naive baseline algorithm that can access the true fragment bipartition and simply reconstruct haplotypes by majority.
As shown in Table 1, when the error rate is low (up to 0.1) the DGS algorithm performs permanently better than the others. For higher error rate, there is no algorithm that works clearly better than the others. For small fragments (with l = 100) and coverage higher than three, MLF outperforms the others. For the other cases, there is no strong winner. If we consider the best and the second-best result (highlighted values in Table 1), we observe that Fast Hare should be considered reliable for an error rate up to 0.2. In the case in which the haplotype length is set to 100, MLF can be considered the most reliable algorithm. When the haplotype length is set to 350 bp, the most reliable algorithm is DGS followed by Fast Hare. It is possible to observe that for a low error rate DGS is always among the best algorithms. For low error rate SpeedHap performs quite well, while for high error rate MLF becomes reliable. The case in which the haplotype length is set to 700 is similar to the previous case. The DGS algorithm is reliable in all settings and outperforms the other algorithms for low error rate. Even in this case, Fast Hare is the second-best algorithm.
The MLF performances highlight that the higher the error rate, the better the strategy of computing many independent haplotype pairs (and return the solution that minimizes the MEC score) works.
It is surprising that even in the cases in which the error rate is set to 0, the 2d-mec, MLF and SHR-three algorithms can introduce errors in their final solution. This can be explained by the fact that their initializations involve random choices that can heavily affect the final result. The other algorithms are almost always able to rebuild the haplotypes entirely without errors (sometimes introducing some gaps).
It should be observed that the SHR-three algorithm consistently has a reconstruction rate lower than that of the other algorithms. This can be imputed to the initialization step of the algorithm in which the two sets C1 and C2 are initialized with two random fragments that have high probability of being mates in the correct bipartition.
Another important observation is that when the error rate is 0.3 and the coverage is 3, the SpeedHap algorithm performs much worse than in the other cases. This is due to the fact that the high error rate and low coverage makes the first phase of SpeedHap to be unable to select the set of columns that are likely to contain few errors. The effect of this phenomenon is that SpeedHap is unable to assign most of the rows, hence a large part of the haplotypes is filled with gaps.
Looking at the algorithms that compute a certain number of solutions and return the one that explicitly minimize the MEC score (MLF and SHR-three), we can observe that their performances are not among the best ones. According to this observation, it seems that the target function of minimizing the MEC score performs worse than other approaches. We do not want to claim that this observation is necessarily true, in fact (even if not explicitly) DGS minimize the MEC score and attains good results.
4.3 Running time evaluation
In this section, we show the running time of the algorithms compared. Except for HapCUT whose implementation is freely provided by the authors, we reimplemented all the other algorithms using Python v2.6. For each parameter assignment, we run the algorithms over 100 different instances and collect the average running time. For our tests, we used a Pentium D 3.2 GHz endowed with 3 Gb of RAM.
Figure 2 shows a comparison of the haplotype reconstruction times. The slowest algorithm is HapCUT, while the two fastest algorithms are Fast Hare and SHR-three. Except for HapCUT, it is possible to observe that all the algorithms are able to solve all the instances of the reconstruction problem for each parameter assignment in <5 s in the worst case, making all of them suitable for real applications. Instead, HapCUT running time does not scale and requires tens of seconds for large instances of the reconstruction problem.
Fig. 2.

Average running time expressed in seconds over 100 instances for different settings of the haplotype length (a) and coverage (b). The error rate is set to 0.2.
Figure 2 also shows that both haplotype length and coverage affect the final running time of all the algorithms in different ways. Coverage involves a linear increase of the running time for all algorithms, while haplotype length involves a quadratic increase. Due to lack of space, in this article we do not report results in the case in which we vary the error rate or the Hamming distance because we observed that they have no effect on the running time of all the algorithms.
5 CONCLUSIONS
The SIH problem is one of the core problems in the whole genome sequencing. Due to the presence of various types of errors and missing data in the fragments, the problem is very hard to solve. In recent years, many algorithms and heuristics were proposed in the literature, but a systematic comparison between them is still missing. In this article, we survey seven algorithms that are among the most commonly used for the SIH problem. We also developed a common framework to compare them. Our framework simulates the actual technology for shotgun sequencing to generate realistic SNP matrices from real human haplotypes collected from the HapMap project. The web-based interface of the framework allows to control all the generation parameters. The Hamming distance and fragment size can be specified only as a range. Due to the fact that we use real haplotypes, it is possible (for too-small ranges) that the selected chromosome does not contain a portion of haplotype with the desired Hamming distance. In this case an error is raised. The choice of the generation parameters is heavily influenced by the sequencing hardware, and it can have considerable impact on the final results. To facilitate the choice of parameters, our framework suggests default settings compatible with actual standards. Our experiments show that the DGS algorithm can be considered the best choice, especially in those experiments in which some parameters cannot be estimated in advance. The data we used for the comparison are available upon request, and thus can be used in the near future to compare novel algorithmic ideas with the actual state of the art.
ACKNOWLEDGEMENTS
We would like to thank Rui-Sheng Wang, Jianxin Wang, Minzhu Xie, Zhixiang Chen and Zhiyu Zhao who provided us with their software and allowed us to test them.
Funding: Italian Registry of.it ccTLD (partially); the EU funded 7FP Virtual Physiological Human Network of Excellence (VPH NoE) (contract number 223920) (partially).
Conflict of Interest: none declared.
Footnotes
1Original HapMap haplotypes can be downloaded from: http://hapmap.ncbi.nlm.nih.gov/downloads/phasing/2005-03_phaseI/full/
REFERENCES
- Bansal V, Bafna V. European Conference on Computational Biology. Cagliari, Italy: 2008. HapCUT: an efficient and accurate algorithm for the haplotype assembly problem; pp. 153–159. [DOI] [PubMed] [Google Scholar]
- Bansal V, et al. An MCMC algorithm for haplotype assembly from whole-genome sequence data. Genome Res. 2008;18:1336–1346. doi: 10.1101/gr.077065.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, et al. Linear time probabilistic algorithms for the singular haplotype reconstruction problem from SNP fragments. J. Comput. Biol. 2008;15:535–546. doi: 10.1089/cmb.2008.0003. [DOI] [PubMed] [Google Scholar]
- Cilibrasi R, et al. On the complexity of the single individual SNP haplotyping problem. Algorithmica. 2007;49:13–36. [Google Scholar]
- Daly MJ, et al. High-resolution haplotype structure in the human genome. Nat. Genet. 2001;29:229–232. doi: 10.1038/ng1001-229. [DOI] [PubMed] [Google Scholar]
- Frazer K, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese LM, et al. Workshop on Algorithms in Bioinformatics, Philadelphia, PA, Lecture Notes in Computer Science. Berlin/Heidelberg: Springer; 2007. A fast and accurate heuristic for the single individual SNP haplotyping problem with many gaps, high reading error rate and low coverage; pp. 49–60. [Google Scholar]
- Genovese LM, et al. SpeedHap: an accurate heuristic for the single individual SNP haplotyping problem with many gaps, high reading error rate and low coverage. EEE/ACM Trans. Comput. Biol. Bioinform. 2008;5:492–502. doi: 10.1109/TCBB.2008.67. [DOI] [PubMed] [Google Scholar]
- HapMap. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp RM. Reducibility among combinatorial problems. Complex. Comput. Comput. 1972:85–103. [Google Scholar]
- Karp RM. An introduction to randomized algorithms. Discrete Appl. Math. 1991;34:165–201. [Google Scholar]
- Lancia G, et al. Proceedings of the Ninth European Symposium on Algorithms, Aarhus, Denmark, Lecture Notes in Computer Science. Berlin/Heidelberg: Springer; 2001. SNPs problems, complexity, and algorithms; pp. 182–193. [Google Scholar]
- Levy S, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:2113–2144. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, et al. Proceedings of the Seventh International Conference on Computational Molecular Biology, Lisbon, Portugal, Lecture Notes in Computer Science. Berlin/Heidelberg: Springer; 2003. Haplotype reconstruction from SNP alignment; pp. 207–216. [Google Scholar]
- McQueen J. Fifth Berkeley Symposium on Mathematics, Statistics, and Probability, Statistical Laboratory of the University of California, Berkeley. Berkeley, CA: University of California Press; 1967. Some methods for classification and analysis of multivariate observations; pp. 281–298. [Google Scholar]
- Metzker ML. Emerging technologies in DNA sequencing. Genome Res. 2005;15:1767–1776. doi: 10.1101/gr.3770505. [DOI] [PubMed] [Google Scholar]
- Morozova O, Marra MA. Applications of next-generation sequencing technologies in functional genomics. J. Genomics. 2008;5:255–264. doi: 10.1016/j.ygeno.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Myers G. Proceedings of the Seventh International Conference on Intelligent Systems for Molecular Biology. Heidelberg, Germany: AAAI; 1999. A dataset generator for whole genome shotgun sequencing; pp. 202–210. [PubMed] [Google Scholar]
- Panconesi A, Sozio M. Workshop on Algorithms in Bioinformatics, Bergen, Norway, Lecture Notes in Computer Science. Berlin/Heidelberg: Springer; 2004. Fast hare: a fast heuristic for single individual SNP haplotype reconstruction; pp. 266–277. [Google Scholar]
- Sahni S, Gonzalez T. P-complete problems and approximate solutions. Annu. Symp. Switching Automata Theory. 1974;23:28–32. [Google Scholar]
- Via M, et al. The 1000 genomes project: new opportunities for research and social challenges. Genome Med. 2010;2:3. doi: 10.1186/gm124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, et al. Haplotype reconstruction from SNP fragments by minimum error correction. Bioinformatics. 2005;21:2456–2462. doi: 10.1093/bioinformatics/bti352. [DOI] [PubMed] [Google Scholar]
- Wang R, et al. A markov chain model for haplotype assembly from SNP fragments. Genome Inform. 2006;17:162–171. [PubMed] [Google Scholar]
- Wang Y, et al. A clustering algorithm based on two distance functions for MEC model. J. Comput. Biol. Chem. 2007;31:148–150. doi: 10.1016/j.compbiolchem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Xie M, Wang J. An improved (and practical) parameterized algorithm for the individual haplotyping problem MFR with mate-pairs. Algorithmica. 2008;52:250–266. [Google Scholar]
- Xie M, et al. BMEI : Proceedings of the 2008 International Conference on BioMedical Engineering and Informatics. Sanya, Hainan, China: IEEE; 2008. A practical exact algorithm for the individual haplotyping problem MEC; pp. 72–76. [Google Scholar]
- Zhao Y, et al. Haplotype assembly from aligned weighted SNP fragments. J. Comput. Biol. Chem. 2005;29:281–287. doi: 10.1016/j.compbiolchem.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Zhao Y, et al. An overview of the haplotype problems and algorithms. Front. Comput. Sci. China. 2007;1:272–282. [Google Scholar]