

Abstract
Here, we show that phosphatidylinositol 3-kinase (PI3K) is a key player in the establishment of central sensitization, the spinal cord phenomenon associated with persistent afferent inputs and contributing to chronic pain states. We demonstrated electrophysiologically that PI3K is required for the full expression of spinal neuronal wind-up. In an inflammatory pain model, intrathecal administration of LY294002 [2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one], a potent PI3K inhibitor, dose-dependently inhibited pain-related behavior. This effect was correlated with a reduction of the phosphorylation of ERK (extracellular signal-regulated kinase) and CaMKII (calcium/calmodulin-dependent protein kinase II). In addition, we observed a significant decrease in the phosphorylation of the NMDA receptor subunit NR2B, decreased translocation to the plasma membrane of the GluR1 (glutamate receptor 1) AMPA receptor subunit in the spinal cord, and a reduction of evoked neuronal activity as measured using c-Fos immunohistochemistry. Our study suggests that PI3K is a major factor in the expression of central sensitization after noxious inflammatory stimuli.
Keywords: phosphorylation, ERK, GluR1, CaMKII, NMDA, formalin
Introduction
Central sensitization is a complex phenomenon of synaptic plasticity characterized by the rapid-onset, activity-dependent increase in the excitability of nociceptive spinal dorsal horn neurons (DHNs). Physiologically, it is associated with a reduction in the activation threshold of DHNs, enhancement and enlargement of their responsiveness and receptive fields, respectively. It has recently become clear that there are distinct forms of central sensitization. Several other types of synaptic plasticity arise in response to noxious stimuli that modify nociceptive transmission by altering synaptic efficacy. These include other immediate-onset, transcription-independent phenomena, such as wind-up and long-term potentiation (LTP). Indeed, evidence suggests that central sensitization in the spinal cord and synaptic plasticity in the hippocampus (i.e., LTP) may share common signaling pathways (Ji et al., 2003; Malcangio and Lessmann, 2003; Sandkuhler, 2007). However, the intracellular mechanisms underlying this particular form of LTP at the spinal level are not well understood.
Phosphatidylinositol-3-kinase (PI3K) is a lipid kinase that phosphorylates the inositol ring of phosphoinositides (PtdIns) (Whitman et al., 1988), generating three distinct membrane-associated second messengers (Toker and Newton, 2000). These second messengers are able to activate various effectors [e.g., Akt/PKB and extracellular signal-regulated kinase (ERK)]. Therefore, PI3K is involved in several signaling cascades and cellular processes (Cantley, 2002; Koyasu, 2003; Fresno Vara et al., 2004; Patel and Mohan, 2005; Patton et al., 2007; Parcellier et al., 2008). In particular, PI3K inhibitors suppress NMDA-receptor-mediated ERK activation in hippocampal neurons (Perkinton et al., 2002; Chandler and Sutton, 2005). Furthermore, several reports demonstrated interestingly a requirement of PI3K activity in different forms of synaptic plasticity including LTP (Man et al., 2003; van der Heide et al., 2005). An electrophysiological study in rat hippocampal slices showed that PI3K activity was induced during LTP, and, conversely, LTP could be reduced by two PI3K specific inhibitors, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one [LY294002 (LY)] and wortmannin (Sanna et al., 2002). However, after drug washout, LTP recovered, suggesting a predominant role of PI3K in the expression rather than maintenance of LTP (Sanna et al., 2002). Additional studies reinforced the role of PI3K in LTP through both ERK-dependent and ERK-independent mechanisms (Man et al., 2003; Opazo et al., 2003). In contrast to Sanna et al. (2002), Opazo et al. (2003) concluded that PI3K is only involved in LTP induction. Despite these discrepancies about the timing of activation of PI3K during LTP, it is clear that PI3K plays a crucial role. At spinal levels, more physiological stimuli than the high frequency stimuli used to induce LTP in the forebrain can induce central sensitization, and a number of correlates such as wind-up or temporal summation can be related to human psychophysical measures and pain complaints in patients (Koltzenburg et al., 1994; rendt-Nielsen et al., 1995). Therefore, we hypothesized that PI3K might be involved in early-onset phenomena of central sensitization occurring in the spinal cord. We investigated the effect of PI3K inhibition on spinal wind-up, the behavioral formalin test, and subsequently examined the intracellular mechanisms involved. Our results suggest that PI3K is a key player in the early events of central sensitization.
Materials and Methods
All experiments were undertaken with approval of the United Kingdom Home Office. The guidelines of the Committee for Research and Ethical Issues of the International Association for the Study of Pain were followed.
In vivo electrophysiology
Electrophysiological recordings were conducted in male Sprague Dawley rats (200–250 g; Central Biological Services, University College London, London, UK). Rats were anesthetized using halothane and a laminectomy was performed at L1–L3 vertebral level. Extracellular recordings of single deep wide dynamic range (WDR) dorsal horn neurons were made using parylene-coated tungsten electrodes (A-M Systems, Carlsborg, WA). A train of 16 transcutaneous electrical stimuli (2-ms-wide pulses; 0.5 Hz) was delivered to the receptive field at three times the threshold current for C-fiber via two stimulating needles inserted under the skin of the ipsilateral hindpaw. A poststimulus histogram was constructed and Aβ- (0–20 ms), Aδ- (20–90 ms), and C-fiber (90–300 ms) evoked responses were separated and quantified on a basis of latency. Responses occurring after the C-fiber latency band were taken to be the postdischarge of the cell (300–800 ms). “Input” was calculated as the number of action potentials (APs) after the first stimulus × 16. Wind-up was calculated as follows: (number of APs after 16 stimuli) − input. Data were captured and analyzed by a CED 1401 interface coupled to a Pentium computer with Spike 2 software (Cambridge Electronic Design, Cambridge, UK; poststimulus time histogram and rate functions).
Drug administration.
Stable control responses to electrical stimuli were established at 10 min intervals before drug administration. LY294002 (Tocris Biosciences, Bristol, UK), a potent inhibitor of PI3K, was applied directly to the exposed surface of the spinal cord of rats (n = 6), in a volume of 50 μl using a Hamilton syringe as follows: 50 μl of 50 μm (0.8 μg) LY294002 was applied and the effects followed for 1 h with tests performed at 10, 20, 30, 40, 50, and 60 min. The remaining solution was then removed from the surface of the cord and replaced by 50 μl of 100 μm (1.6 μg) LY294002, with subsequent testing as above. Drug vehicle was 10% dimethyl sulfoxide (DMSO) and control experiments with this vehicle were also conducted (n = 6) (data not shown).
Statistical analysis.
Data are presented as mean number of action potentials ± SEM, unless otherwise stated. The effects of LY294002 versus control predrug values and assessed by repeated-measures ANOVA (one-way ANOVA), followed by Dunnett's multiple-comparison test, using GraphPad (San Diego, CA) Prism, version 4. Levels of statistical significance were set at the following: *p < 0.05 and **p < 0.01.
Release of substance P from dorsal horn slices
Horizontal dorsal horn slices (400 μm thick) with dorsal roots attached were obtained from the lumbar spinal cord of adult male Wistar rats (Harlan, Bicester, Oxon, UK) as described previously (Malcangio et al., 2000; Clark et al., 2006). Briefly, lumbosacral spinal cord was excised and longitudinally hemisected producing a horizontal slice with L4 and L5 dorsal roots attached. One slice was obtained from each rat, mounted in the central compartment of a three-compartment chamber, and continuously superfused (1 ml/min) with oxygenated (95% O2 plus 5% CO2) Krebs' solution (in mol/L: 118 NaCl, 4 KCl, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 2.5 CaCl2, and 11 glucose) containing 0.1% bovine serum albumin (BSA), 20 μg/ml bacitracin, 100 μm captopril, 1 μm phosphoramidon, and 6 μm dithiothreitol (Sigma-Aldrich, St. Louis, MO). BSA and protease inhibitors were added to minimize loss of detectable substance P (SP)-like immunoreactivity (SP-LI) through surface adhesion and to prevent degradation. The dorsal roots were placed in the lateral compartments and immersed in mineral oil to avoid dehydration. Before, during, and after dorsal root stimulation, 8 ml fractions of superfusates were collected from the central compartment in glass tubes containing acetic acid (0.1N final concentration; VWR, West Chester, PA) to stabilize SP. Three 8 ml fractions were collected before stimulation to measure basal levels of SP. The dorsal roots were then electrically stimulated (20 V, 0.5 ms, 10 Hz for 8 min). Three additional fractions were collected after electrical stimulation to assess recovery to basal levels of SP (R1–R3). LY294002 hydrochloride (50 μm; Tocris Biosciences) was added to the superfusion solution one fraction before and during electrical stimulation. To quantify SP-LI, samples were partially purified and desalted using Sep-pak C18 reverse-phase silica gel cartridges (Waters Associates, Milford, MA). The cartridges were conditioned with acetonitrile (100%; HPLC grade; VWR) and trifluoroacetic acid (TFA) (0.1%; HPLC grade; VWR). Samples were then loaded into the columns, and the peptide was eluted using acetonitrile/TFA (80:20) solution. The eluates were dried by evaporation under nitrogen. Dried samples were reconstituted in 300 μl of phosphate buffer and 100 μl assayed by radioimmunoassay in duplicate using scintillation proximity assay (GE Healthcare, Little Chalfont, Buckinghamshire, UK).
Surgery for intrathecal catheter and subsequent formalin test
One week before formalin experiments, adult male Wistar rats (Harlan) were deeply anesthetized by intraperitoneal injection of a mixture of medetomidine (0.25 mg/kg) and ketamine (60 mg/kg). A flexible SILASTIC catheter (external diameter, 1.14 mm; Merck, Essex, UK) was inserted subdurally to deliver LY294002 or vehicle at lumbar level. A small laminectomy was performed under sterile conditions at thoracic level and a thin catheter was inserted between the dura matter and the spinal cord. The tubing was secured to the atlanto-occipital bone with superglue and the external end guided under the skin and externalized over the head.
Formalin test experiments were performed 1 week after this surgery. Animals were placed in a Plexiglas box for 15 min to acclimatize. They then received intrathecal injections of 10, 50, or 100 μg of LY294002 in a volume of 10 μl or vehicle (10% DMSO) using the externalized catheter. Fifteen minutes later, rats received 50 μl of 5% formalin injected subcutaneously into the plantar surface of the right hindpaw. Lifting, shaking, biting, and licking of the injected paw were monitored by measuring the total duration of the response in seconds during 60 min after formalin administration. Data are presented in 5 min bins. At the end of the test, animals were terminally anesthetized using pentobarbitone and perfused intracardially with 100 ml of saline followed by 400 ml of 4% paraformaldehyde (PFA) with 15% of a saturated solution of picric acid for c-Fos immunohistochemistry.
For phospho-Akt, phospho-ERK, and phospho-calcium/calmodulin-dependent protein kinase II (CaMKII) immunohistochemistry, animals were first anesthetized with urethane (1 mg/kg), and then pretreated intrathecally with LY294002 (100 μg/rat) or vehicle. Fifteen minutes later, rats were injected subcutaneously with 50 μl of formalin into the plantar surface of the right hindpaw. Five minutes after formalin administration, rats were perfused transcardially with 100 ml of heparinized saline followed by 400 ml of 4% paraformaldehyde with 15% of a saturated solution of picric acid.
For assessment of phospho (p)-NR2A and p-NR2B NMDA receptor subunits and translocation of glutamate receptor 1 (GluR1) AMPA receptor subunit, rats underwent the same process as above but were killed by decapitation at 5, 10, 45, or 90 min after formalin administration. Fresh tissues were collected and immediately frozen in liquid nitrogen for additional extraction and Western blotting.
Immunohistochemistry for phospho-Akt, phospho-ERK, phospho-CaMKII, and c-Fos
After perfusion (see above), spinal cords (L3–L6) were postfixed overnight in the same fixative and cryoprotected in 30% sucrose solution overnight. Tissues were embedded in OCT and frozen using liquid nitrogen. Transverse spinal cord sections (30 μm thickness) were cut using a cryostat and every section was collected and placed in PBS solution (phosphate buffer, 0.9% NaCl) for free-floating immunohistochemistry. Sections were washed with PBS and then incubated with primary antibody, either rabbit anti-phospho-Akt 1 S473 [1:300 in PBST-azide (PBS with 0.3% Triton X-100–azide); New England Biolabs, Beverly, MA], rabbit anti-phospho-ERK (1:300 in PBST-azide; New England Biolabs), rabbit anti-phospho-CaMKII T286 (1:300 in PBST-azide, ref. 5683; Abcam, Cambridge, UK), or rabbit anti-c-Fos (1:5000 in PBST-azide; Calbiochem, La Jolla, CA) overnight at room temperature. Immunostaining against c-Fos and p-ERK were performed using direct fluorescence as follows: after several washes with PBS, sections were incubated for 2 h at room temperature with secondary antibody (goat anti-rabbit IgG-conjugated Alexa Fluor 488; 1:1000 in PBST-azide; Invitrogen, Carlsbad, CA). Immunostaining against phospho-Akt S473 and phospho-CaMKII T286 required tyramide amplification: sections were incubated with the secondary antibody, goat anti-rabbit biotin (1:300 in PBST; Vector Laboratories, Burlingame, CA) for 1 h at room temperature. After several washes, sections were incubated in avidin:biotinylated enzyme complex (1:500 in PBS; Vector Laboratories) for 1 h, followed by several washes in PBS. The sections were then incubated in biotinyl tyramide (1:75 in amplification buffer; PerkinElmer Life and Analytical Sciences, Waltham, MA) for 10 min. After several washes in PBS, sections were finally incubated in Extravidin FITC (1:500; diluted in PBS; Sigma-Aldrich). For all immunostaining, sections were mounted onto slides and coverslipped with Vectashield mounting medium (Vector Laboratories). Slides were visualized under a Carl Zeiss (Jena, Germany) Axioplan 2 fluorescent microscope and L3, L4, and L5 sections were identified for quantification (five sections per level per animal, randomly chosen). For p-ERK and c-Fos, labeled spinal cord neurons were counted in laminas I–II and V–VI, for both ipsilateral and contralateral sides by one blinded investigator. For assessment of p-Akt and p-CaMKII upregulation, images of dorsal horn (ipsilateral and contralateral at the level of lamina II, in which changes are mostly observed) from five sections per animal, chosen randomly, were taken at magnification of 20×, using the same setup of acquisition for all animals. Densitometric analysis of medial p-Akt and p-CaMKII staining (because staining was too abundant to count individual neurons) was performed using Scion (Frederick, MD) Image software. Results for p-Akt and p-CaMKII expression are given as a percentage of increase in the ipsilateral side versus the contralateral side. Statistical significance was tested by the Mann–Whitney rank sum test using SigmaStat software.
Western immunoblotting for phospho-NR2A and phospho-NR2B
Spinal cord samples (L4–L5 levels) of LY294002-treated (n = 4) and vehicle-treated (n = 4) animals were homogenized in RIPA (radioimmunoprecipitation assay) buffer (50 mm Tris, pH 8, 150 mm NaCl, 1% NP-40, 0.5% deoxyxcholate, 0.1% SDS, 1 mm sodium orthovanadate, and Complete protease inhibitor mixture). The protein concentrations of lysates were determined using a BCA Protein Assay kit (Pierce, Rockford, IL). Proteins (30 μg/sample) were separated using 8% SDS-PAGE, and transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were then incubated with primary antibody, either rabbit phospho-NR2A Y1387 (1:500 in 20 mm Tris, pH 7.5, 500 mm NaCl, 0.1% Tween 20 (TBST); ref. 16647; Abcam) or rabbit phospho-NR2B Y1472 (1:1000 in TBST; Imgenex, Servento Valley, CA), overnight at 4°C. After several TBST washes, membranes were incubated with donkey anti-rabbit HRP-linked secondary antibody (1:5000; GE Healthcare) for 1 h at room temperature, and revealed using ECL-plus reagent (5 min) for detection of phosphorylation by autoradiography. Gels were scanned and bands were quantified by densitometric analysis using Scion Image software. Results are expressed as percentage phosphorylation in the ipsilateral dorsal horn compared with the contralateral dorsal horn for each animal (both ipsilateral and contralateral samples were run on the same gel). Mann–Whitney rank sum tests were performed using SigmaStat software to test the statistical significance of the results. Reprobe of the membranes with Ponceau red showed a lack of significant differences between the samples among the same gel.
Translocation of AMPA-R subunit GluR1
As above (see Surgery for intrathecal catheter and subsequent formalin test), naive animals (n = 3 LY; n = 3 vehicle) and animals injected with formalin were killed by decapitation at 10 min (n = 3 LY; n = 3 vehicle), 45 min (n = 3 LY; n = 3 vehicle), and 90 min (n = 3 LY; n = 3 vehicle). Dorsal spinal cord, both ipsilateral and contralateral, at L4/L5 level, were dissected and frozen in liquid nitrogen for Western immunoblotting.
Subcellular fractioning (isolation of cytoplasm and plasma membranes) was performed as described by Galan et al. (2004). In brief, spinal cord samples were homogenized in CLB buffer (10 mm Tris, pH 7.5, 300 mm sucrose, 1 mm EDTA, 1 mm sodium orthovanadate, and protease inhibitor mixture), and then centrifuged at 7000 × g for 5 min. Supernatant containing cytoplasm (S1) was separated from the pellet containing nuclei and debris (P1), which was discarded. Supernatant (S1) was then centrifuged at 40,000 × g for 30 min to obtain pure cytoplasmic extract in the supernatant (S2) and crude membrane in the pellet (P2). This pellet (P2), which includes plasma and cellular organelles membranes, was resuspended in PBS with protease inhibitor mixture (Complete; Roche, Indianapolis, IN) and sodium orthovanadate. Protein titration was conducted using a BCA Protein Assay kit (Pierce) to determine protein concentrations in the cytoplasmic and membrane samples. Totals of 7.7 and 6.0 μg of proteins were loaded for each cytoplasmic (S2) and membrane (P2) samples, respectively. Proteins were separated using 7% SDS-PAGE, and transferred to PVDF membranes. Membranes were incubated with primary antibody, rabbit anti-GluR2 (1:1000 in TBST; ref. 1768; Millipore Bioscience Research Reagents, Temecula, CA) or rabbit anti-GluR1 (1:1000 in TBST; ref. 1504; Millipore Bioscience Research Reagents) overnight at 4°C. After several TBST washes, membranes were incubated with donkey anti-rabbit HRP-linked secondary antibody (1:5000; GE Healthcare) for 1 h at room temperature and revealed using ECL-plus reagent (5 min) for detection by autoradiography. Gels were scanned and bands were quantified by densitometric analysis using Scion Image software. Results are expressed as percentage of expression at each time point compared with naive rats (100%) in each group. Mann–Whitney rank sum tests were performed using SigmaStat software to test the statistical significance of the results.
Results
Spinal application of LY294002 reduces the “wind-up” of deep dorsal WDR neurons and inhibits electrically evoked responses
We first studied the potential involvement of PI3K on a form of short-term spinal plasticity, wind-up, which manifests as an increased response of spinal WDR neurons in response to repetitive stimulation of primary afferent fibers. Application of the PI3K inhibitor LY294002 directly onto the spinal cord produced a significant reduction of wind-up of deep dorsal horn WDR neurons with both 0.8 μg (55% reduction; p < 0.01) and 1.6 μg (50% reduction; p < 0.01) doses (Fig. 1A,B) compared with predrug control values. No change in wind-up was seen with vehicle (10% DMSO) alone (data not shown). Electrically evoked responses of deep dorsal horn WDR neurons were also markedly reduced by spinal application of LY294002 (Fig. 2). Statistically significant reductions were seen of A∂-fiber (1.6 μg, 24% reduction, p < 0.05), C-fiber evoked responses (0.8 μg, 23% reduction, p < 0.01; 1.6 μg, 27% reduction, p < 0.01), and also of postdischarge of deep dorsal horn neurons, a direct measure of increased excitability (0.8 μg, 39% reduction, p < 0.05; 1.6 μg, 51% reduction, p < 0.05) (Fig. 2) compared with predrug control values. In addition, Aβ-fiber evoked responses remained unaltered (Fig. 2). Importantly, neither LY294002, nor vehicle (10% DMSO), had any effect on input (number of action potentials after the first stimulus, which is an index of the effect of a drug on presynaptic inputs to the neuron), suggesting that effects of LY294002 were more likely attributable to a direct effect on dorsal horn neurons themselves rather than via presynaptic mechanisms. Administration of vehicle alone had no effect on any evoked response.
Figure 1.
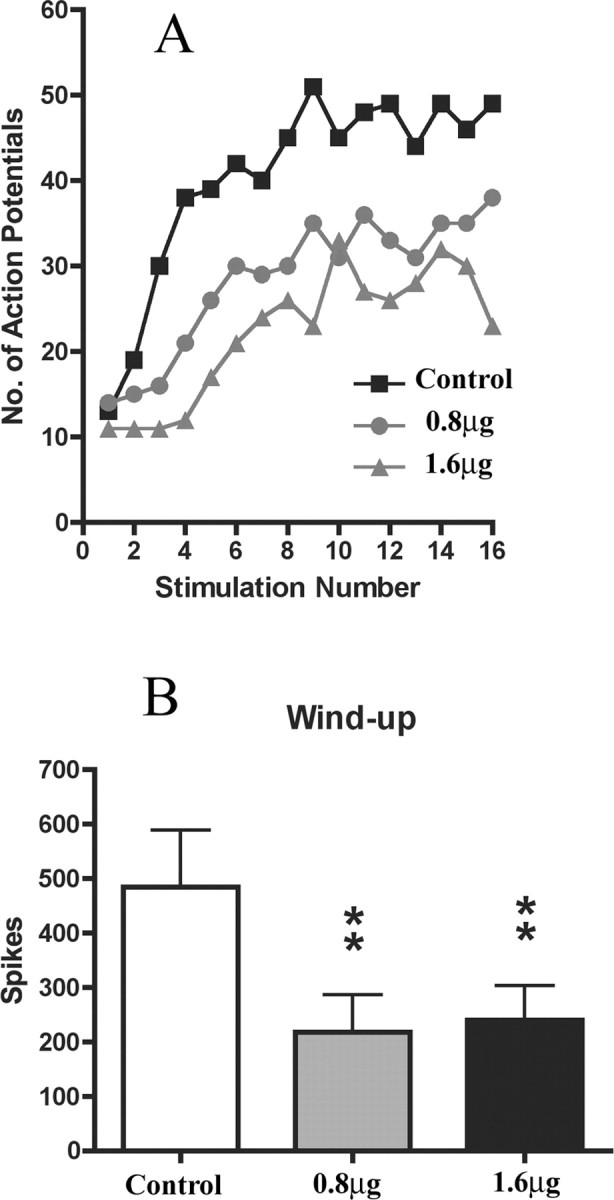
Spinal application of PI3K inhibitor (LY294002) inhibits wind-up of deep dorsal horn WDR neurons in naive rats. A, Example of the effect of spinal application of LY294002 (0.8 and 1.6 μg) on the wind-up of a single deep dorsal horn WDR neuron. Number of APs is plotted against the stimulus number. B, Both 0.8 and 1.6 μg doses of LY294002 significantly reduced wind-up of dorsal horn neurons compared with predrug control values. Data are presented as mean number of action potentials (spikes) ± SEM. **p < 0.01, one-way ANOVA followed by Dunnett's multiple-comparison test; n = 6 in each group.
Figure 2.

Spinal LY294002 does not modify the initial dorsal horn response or the release of substance P, but reduces the C-fiber- and Aδ-fiber-evoked responses of deep dorsal horn WDR neurons in naive rats. A–D, LY294002 significantly reduced Aδ- (B) and C- (C) fiber-mediated responses and postdischarge of dorsal horn neurons (D) compared with predrug control values. A, No effect was seen on Aβ-fiber-mediated responses. E, PI3K inhibition did not affect input. Data are presented as mean number of action potentials (spikes) ± SEM. *p < 0.05, **p < 0.01, one-way ANOVA followed by Dunnett's multiple-comparison test. F, LY294002 (100 μm) did not modify release of substance P induced by electrical stimulation of primary afferent fibers (20 V, 0.5 ms, 10 Hz for 8 min). *p < 0.05, **p < 0.01 compared with basal level, one-way ANOVA followed by Tukey's post hoc test; n = 5 in each group.
Electrically evoked substance P release is not inhibited by LY294002
Using the isolated dorsal horn with dorsal roots attached preparation, we examined the contribution of PI3K to the release of primary afferent SP. We chose SP because it is a peptide contained in nociceptors and released by these fibers after increased activation of nociceptors in vitro (Malcangio and Bowery, 1994; Teoh et al., 1996) and painful stimulation in vivo (Lang and Hope, 1994; Yonehara et al., 1995). In dorsal horn superfusates, the levels of SP under basal conditions range from 15 to 23 pg/ml. After electrical stimulation of the dorsal roots (Elec, white bar), to recruit both A- and C-fibers, the levels of SP-LI in superfusates was significantly increased compared with basal levels (p < 0.001). This increase was still significant in the first recovery period (R1) after stimulation (p = 0.009). SP-LI levels returned to basal in the second and third recovery fractions (R2 and R3). After superfusion of LY294002 (100 μm) for a total of 16 min before and during electrical stimulation (Elec, black bar), SP-LI content in superfusates was also significantly enhanced compared with basal levels (p = 0.042). Furthermore, LY294002 did not inhibit electrically evoked SP-LI compared with control superfusates. SP-LI was elevated in the R1 fraction, although this increase failed to reach significance compared with basal levels, and returned to basal levels in the R2 and R3 fractions. These findings show that PI3K inhibition did not alter neurotransmitter release evoked by electrical stimulation of primary afferents, suggesting that LY294002 has postsynaptic effects on spinal neurons.
PI3K is involved in the development of central sensitization induced by intraplantar formalin
To investigate the role of PI3K in mechanisms of central sensitization in vivo, we investigated the ability of intrathecal administration of LY294002 to reduce pain-like behavior after intraplantar formalin injection. This test is typically characterized by two phases of pain behavior: a first phase mainly caused by direct peripheral activation (0–5 min after injection) and a second phase caused by spinal mechanisms of enhanced neuronal responsiveness, defined as central sensitization (15–40 min after injection).
Intrathecal vehicle treatment (10% DMSO) did not affect pain-like behavior at any time point after intraplantar formalin. In contrast, intrathecal administration of LY294002 15 min before formalin injection induced a significant dose-dependent decrease (10 μg, 6% decrease, p > 0.05; 50 μg, 23% decrease, p = 0.04; 100 μg, 31% decrease, p = 0.025) of the first phase of the formalin test (0–5 min) (Fig. 3A). More interestingly, the second phase (20–40 min) was also reduced in a dose-dependent manner (4% reduction with 10 μg, p > 0.05; 21% reduction with 50 μg, p > 0.05; 50% reduction with 100 μg, p = 0.017) (Fig. 3B,C), suggesting that PI3K is involved in the development of central sensitization induced by intraplantar formalin injection.
Figure 3.

Spinal blockade of PI3K activity reduced the development of central sensitization induced by peripheral inflammation. A, B, Intrathecally administered LY294002 statistically reduced nocifensive behavior (time spent licking and lifting of the injected paw), both during the first (0–5 min after injection) (A) and the second phase (20–40 min) (B) of the formalin test. C, Time course of nocifensive behavior induced by intraplantar formalin in animals receiving intrathecal vehicle (DMSO) or LY294002 (50 or 100 μg). The dose 10 μg had no effect on both phases. Data are presented as the mean time spent by the animal licking and/or lifting his injected paw. Error bars indicate ± SEM. *p < 0.05 compared with the vehicle group, Student's t test; n = 8 in each group.
Phosphorylation of Akt1 S473 in spinal neurons after intraplantar formalin
We used the phosphorylation of downstream kinase Akt1 (S473) as an indicator of the activation of PI3K pathway. We examined the effect of intrathecal administration of LY294002 (100 μg) on Akt1 S473 phosphorylation 5 min after formalin injection using immunohistochemistry. Formalin injection produced a large increase of p-Akt in the medial part of the superficial (I–II) layers of the ipsilateral dorsal horn (L4–L5 level) (Fig. 4B) (average increase of 82.2% ± 22.1) compared with the contralateral side (vehicle-treated) rats (Fig. 4A). Inhibition of PI3K by intrathecal injection of LY294002 caused a reduction in Akt phosphorylation in the ipsilateral dorsal horn (17.8% increase ± 21.5) compared with vehicle-treated rats (i.e., 78% reduction, p = 0.079, n = 5). However, this effect failed to reach statistical significance.
Figure 4.
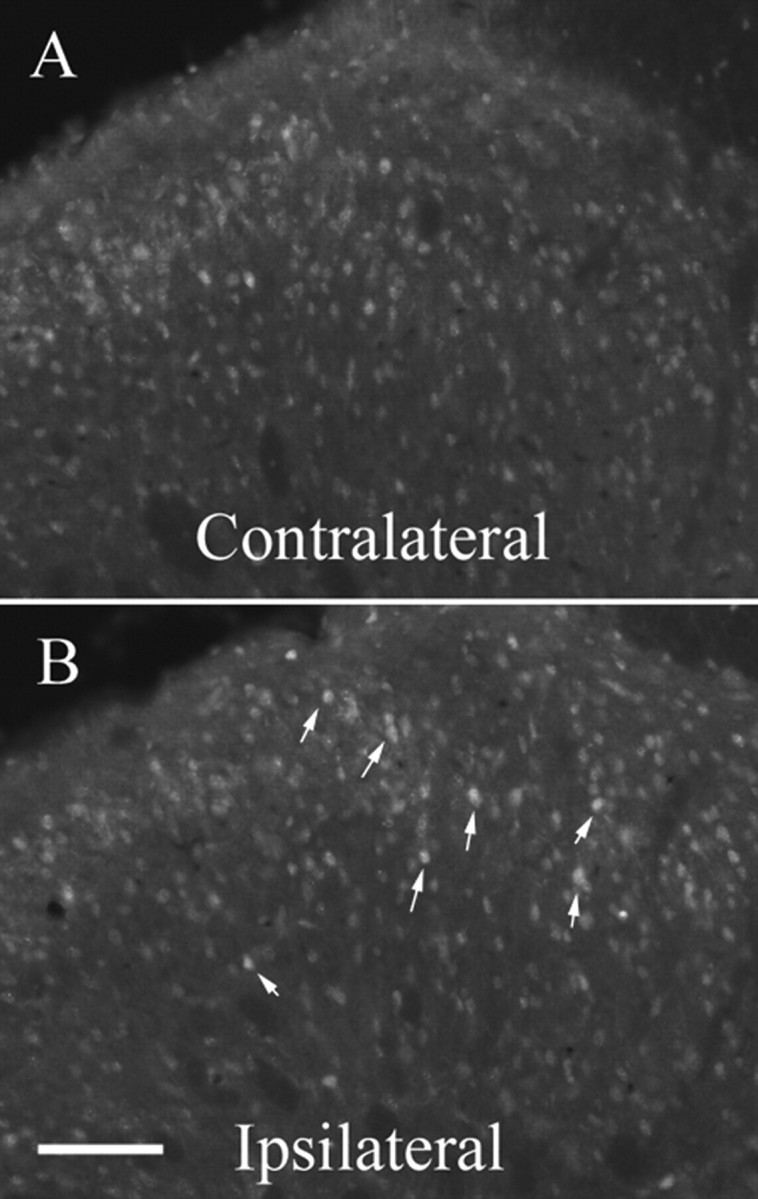
Effect of inhibition of PI3K on Akt phosphorylation induced by intraplantar formalin. A, B, Injection of formalin into the right hindpaw induced a significant increased density of p-Akt in the medial part of superficial laminas (I–II) in the ipsilateral spinal dorsal horn (B) compared with the controlateral side (A) 5 min after formalin injection in rats pretreated with vehicle (DMSO). Scale bar: (in B) A, B, 80 μm.
PI3K is implicated in the phosphorylation of ERK and CaMKII in spinal neurons after intraplantar formalin
To define the molecular mechanism by which PI3K is involved in the development of central sensitization, we studied the effect of PI3K inhibition on the activation of several intracellular signaling pathways previously involved in synaptic plasticity in the hippocampus and activated in the spinal cord after an inflammatory stimulus. Therefore, LY294002 (100 μg) was injected intrathecally 15 min before intraplantar injection of formalin and tissues were processed to study the activation of receptors (NMDA receptor subunits, GluR1) or ERK and CaMKII.
Using immunohistochemical methods, we observed an increase in the number of p-ERK-positive neurons in the superficial layers (I–II) of the L4–L5 ipsilateral dorsal horn in vehicle-treated rats (Fig. 5B) compared with the contralateral side, 5 min after formalin injection (Fig. 5, compare B, A). We also observed an increase in deeper laminas (V–VI), although to a lesser extent than in the superficial laminas (Fig. 5E). In contrast, intrathecal administration of LY294002 (100 μg) significantly attenuated this ERK phosphorylation in both superficial (31% reduction; p = 0.006) and deep laminas of the ipsilateral dorsal horn (63% reduction; p = 0.018) compared with vehicle-treated rats (Fig. 5D,E), 5 min after formalin injection.
Figure 5.

ERK phosphorylation induced by intraplantar formalin is reduced by inhibition of PI3K activity. A, B, Injection of formalin into the right hindpaw induced a significant increase in the number of p-ERK-positive neurons in the ipsilateral spinal dorsal horn 5 min after formalin injection (B) when compared with the contralateral side (A) in rats pretreated with vehicle (DMSO). C is a high-power magnification of B. This increase of ERK phosphorylation was most abundantly seen in laminas I–II but also in laminas V–VI (B, E). Pretreatment with the PI3K inhibitor LY294002 (100 μg) caused a significant reduction in the magnitude of ERK phosphorylation (D, E), both in superficial and deep laminas of the dorsal horn. Results are expressed as the mean number of p-ERK-positive neurons per section ± SEM. *p < 0.05 compared with the vehicle group, Student's t test; n = 5 in each group. Scale bar: (in C) A, B, 125 μm; C, D, 32 μm.
We also examined the effect of intrathecal administration of LY294002 on CaMKII phosphorylation 5 min after formalin injection using immunohistochemistry. Phosphorylation of CaMKII was quantified medially in the superficial (I–II) layers of the dorsal horn of L4–L5 region (Fig. 6B). Formalin injection produced a large increase in p-CaMKII in the ipsilateral dorsal horn of vehicle-treated rats (Fig. 6B,C) compared with the contralateral side (Fig. 6A) (12.85% increase ± 2.94). Inhibition of PI3K by intrathecal injection of LY294002 resulted in a significant reduction in CaMKII phosphorylation in the ipsilateral dorsal horn compared with vehicle-treated rats (3.87% ± 1.62) (i.e., 70% reduction, p = 0.013, n = 5) (Fig. 6D), suggesting that the phosphorylation of both ERK and CaMKII induced by peripheral inflammation are PI3K-dependent mechanisms.
Figure 6.

Increased CaMKII phosphorylation induced by intraplantar formalin is mediated by PI3K. A, B, Injection of formalin into the hindpaw induced an increase in the density of CaMKII phosphorylation in the medial part of laminas I–II in the ipsilateral spinal dorsal horn 5 min after formalin injection (B) compared with the contralateral side (A) in rats pretreated with vehicle (DMSO). C is a high-power magnification of B. Pretreatment with the PI3K inhibitor LY294002 (100 μg) caused a significant reduction in the density of p-CaMKII staining in lamina II (D). Scale bar: (in D) A, B, 130 μm; C, D, 32 μm.
PI3K is implicated in the phosphorylation of NR2B subunits of the NMDA receptor
We performed Western blot analysis to assess the role of PI3K in the phosphorylation of NMDA receptor subunits NR2A and NR2B in spinal dorsal horn of the L4–L5 spinal cord, 5 min after formalin injection. Vehicle-treated rats showed a marked increase in both NR2A and NR2B phosphorylation compared with the contralateral dorsal horn (100%) (Fig. 7A,B). Intrathecal administration of LY294002 significantly attenuated p-NR2B expression compared with vehicle-treated rats (Fig. 7A,B). Although a reduction in p-NR2A expression was also seen with LY294002, this effect did not reach statistical significance (Fig. 7A,B).
Figure 7.

Increased phosphorylation of NMDA receptor 2B and translocation of AMPA receptor GluR1 induced by inflammatory stimulation (intraplantar formalin) are PI3K-mediated mechanisms. A, B, Phosphorylation of NMDA receptor subunits 2A and 2B. Intraplantar injection of formalin in the ipsilateral hindpaw (A, C, Ipsi) induced an increased phosphorylation of NMDA receptors 2A and 2B compared with the contralateral side. Intrathecal injection of LY294002 (100 μg) significantly abolished the increased phosphorylation of NR2B (A, bottom panel; B) but not NR2A subunits (A, top panel; B). Results are expressed as the mean ratio of the densitometric analysis ipsilateral/contralateral ± SEM. The scattered line shows 100% (i.e., the lack of changes ipsilateral vs the contralateral side). White bars represent vehicle-treated group; black bars represent LY294002-treated group. C–F, Translocation of AMPA receptors GluR1 from the cytoplasm to the plasma membrane induced by peripheral inflammation is mediated by PI3K. Intraplantar injection of formalin in the right hindpaw induced a decrease in the relative amount of GluR1 in the cytoplasm 10 min after formalin injection (C, E) and an increase in the relative amount of GluR1 in the plasma membrane 45 and 90 min after formalin injection (not statistically significant) (D, F). Intrathecal injection of LY294002 (100 μg) abolished significantly both the decrease of GluR1 in the cytoplasm (C, E) and its increase in the membrane (D, F). Results are expressed as the mean ratio of the densitometric analysis ipsilateral/contralateral ± SEM. The scattered line shows 100% (i.e., the lack of changes ipsilateral vs the contralateral side). Mann–Whitney rank sum tests, n = 4 per group and time points for p-NR2A and p-NR2B, *p < 0.05 compared with the vehicle group; n = 3 for analysis of cytoplasmic GluR1, *p < 0.05 compared with naive animals.
PI3K is involved in the trafficking of AMPA-R GluR1 subunit after formalin injection
Using Western blot analysis, we tested whether inhibition of PI3K affects the trafficking of AMPA receptor GluR1 subunit from the cytoplasm to the plasma membrane in the dorsal horn of the spinal cord after intraplantar formalin injection. As previously demonstrated after visceral inflammatory stimulation (Galan et al., 2004), intraplantar injection of formalin (in vehicle-treated animals) induced an initial reduction of the relative amount of GluR1 receptor in the cytoplasm (10 min after formalin injection, 39% decrease; p = 0.029) (Fig. 7C,E), followed by an increase at 45 min and a significant increase at 90 min after injection in the amount of cytoplasmic GluR1 (42% increase; p = 0.039) (Fig. 7C,E). This was paralleled by increased GluR1 in the membrane fraction at 45 and 90 min after injection (Fig. 7D,F). PI3K inhibition by LY294002 (100 μg) prevented both the reduction of cytoplasmic GluR1 (10 min) (Fig. 7C,E) and the increase in both the cytoplasm (90 min) (Fig. 7C,E) and the membrane (45 and 90 min) (Fig. 7D,F). The level of GluR1 expression in the membrane after LY294002 was statistically not different from the control group (p > 0.05).
PI3K is involved in the increased synthesis of c-Fos after formalin injection
We examined the effect of intrathecal administration of LY294002 (100 μg) on the increased synthesis of c-Fos induced by formalin injection. As previously described (Presley et al., 1990), formalin injection induced an upregulation of c-Fos in superficial (laminas I–II) and deep laminas (V–VI) of the dorsal horn (Fig. 8A,B) in the lumbar cord L3 to L4. Inhibition of PI3K by intrathecal injection of LY294002 resulted in a significant reduction of c-Fos expression in the superficial (54% reduction in L3, p = 0.027; 61% reduction in L4, p = 0.049, compared with vehicle-treated group) (Fig. 8B–E) and to a smaller extent in deep laminas (in L3: 85% reduction in laminas V–VI, p = 0.15; 100% reduction in lamina X, p = 0.23; 100% reduction in lamina VII, p = 0.23, compared with vehicle-treated group) (Fig. 8B–E).
Figure 8.
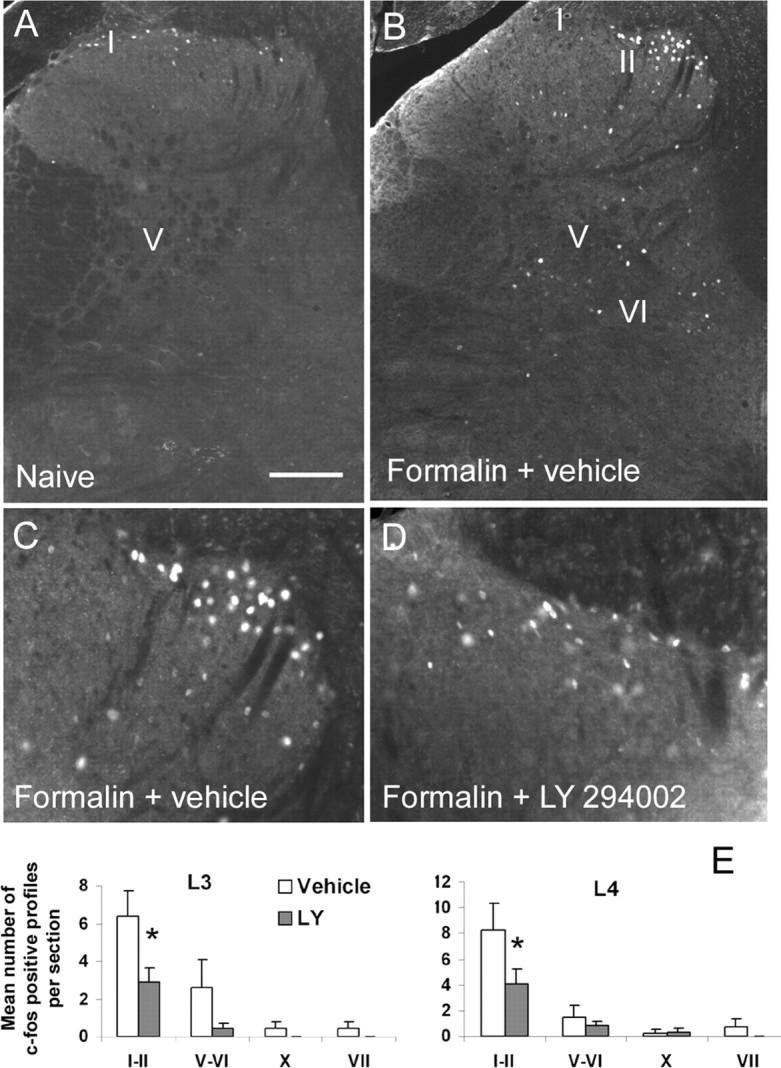
Long-term effects of PI3K inhibition after intraplantar formalin injection: reduction of c-Fos upregulation in spinal neurons. A, C, Intraplantar injection of formalin induced an upregulation of c-Fos in both superficial (I–II) and deep layers (V, VI, VII, X) of the spinal cord. B, D, This upregulation was significantly attenuated by pretreatment with the PI3K inhibitor LY294002 (100 μg). Results are expressed as mean number of c-Fos-positive profiles per section in the different groups of laminas ± SEM. *p < 0.05 compared with the vehicle group, Student's t test; n = 5 in each group. Scale bar: (in A) A, B, 155 μm; C, D, 78 μm.
Discussion
This study aimed at determining the potential involvement of PI3K in mechanisms of enhanced synaptic strength, such as wind-up and central sensitization, in the spinal dorsal horn, that contribute to pain hypersensitivity after peripheral inflammation. We showed that PI3K is involved in a transcription-independent and short-term form of spinal plasticity, termed “wind-up,” which may underlie central sensitization. We demonstrated a dose-dependent inhibitory effect of a specific PI3K inhibitor, LY294002, on Aδ- and, more potently, on C-fiber-mediated evoked responses. We also found an inhibitory dose-dependent effect of intrathecal LY294002 administration on the first phase and most interestingly on the second phase of the formalin test. This inhibitor also reduced the phosphorylation of NR2B subunits of NMDA receptor and long-term phenomenon such as c-Fos expression. This inhibitory effect of LY294002 was also correlated with a reduction of the phosphorylation of ERK and CaMKII as early as 5 min after formalin injection, as well as the abolishment of the translocation of GluR1 AMPA receptor subunit to the plasma membrane. This mechanism is an essential mediator of the establishment of LTP in hippocampus (Man et al., 2003) but has never been demonstrated before at the spinal cord level in a model of somatic inflammatory pain.
Development of wind-up is PI3K dependent
Wind-up in the spinal cord is a phenomenon whereby repetitive stimulation of deep dorsal horn WDR neurons induces an increase of their evoked responses and postdischarge with each stimulus (Dickenson and Sullivan, 1987). This form of spinal plasticity is NMDA-dependent, as is LTP, and seems to be the neuronal correlate of pain hypersensitivity. Despite being a fast-onset and short-lasting phenomenon, wind-up may contribute to long-term changes in the spinal cord leading to central sensitization as is produced by peripheral formalin injection (Haley et al., 1990). Our results indicate that PI3K is required for the full expression of wind-up, because the potent PI3K inhibitor, LY294002, was able to reliably reduce wind-up by 50%. In addition, we observed a more profound dose-dependent reduction of the Aδ- and C-fiber-mediated responses rather than Aβ-fiber-mediated responses, suggesting that PI3K blockade mainly interferes with nociceptive signaling. The ability to apply the drug locally to a restricted cord zone in this static system explains the low effective doses compared with the behavioral study in which CSF flow and dilution and the smaller volume make higher doses necessary. Nevertheless, the biochemical data establish the selectivity of even the highest dose of LY294002 for PI3K.
We found that the input to the dorsal horn was unaltered with both LY294002 and vehicle, indicating that the reductions observed in evoked neuronal response and wind-up were probably not attributable to an inhibition of primary afferent signaling, but rather a direct effect on the postsynaptic dorsal horn neurons themselves. This hypothesis was partially confirmed by the observation that the PI3K inhibitor did not alter SP-LI released from primary afferents after electrical stimulation of the dorsal roots compared with the control preparation. We cannot entirely exclude that LY294004 might affect glutamate release from primary afferent fibers. However, it seems more likely that PI3K is essentially involved at the postsynaptic level in the dorsal horn.
Spinal involvement of PI3K in an animal model of NMDA-dependent sustained neuronal activation
Despite differences between studies in the involvement of PI3K in the induction and/or expression of LTP, it is clear that PI3K has an important role to play in this phenomenon (Man et al., 2003; Opazo et al., 2003). Because of some similarities between the mechanisms of enhanced synaptic strength in the hippocampus and the spinal cord (Malcangio and Lessmann, 2003) and a study demonstrating an involvement of spinal PI3K on pain after peripheral capsaicin injection (Sun et al., 2006), we hypothesized that PI3K could be an important intracellular mediator for the development of central sensitization, and proceeded to reveal the mechanisms involved. We used an inflammatory pain model induced by intraplantar formalin injection to investigate intracellular mechanisms contributing to central sensitization. We found an activation of PI3K pathway after formalin administration reflected by increased phosphorylation of downstream p-Akt, like Sun et al. (2006) with the capsaicin model. Importantly, we observed a dose-dependent inhibitory effect of PI3K blockade on both the acute (first phase) and “tonic” pain (second phase) phases of the formalin test. The first phase is considered to be mainly A∂- and C-fiber mediated and activates second-order neurons in the superficial laminas of the dorsal horn. Inhibition of this acute phase could reflect the decreases in A∂-fiber- and C-fiber-mediated responses, as seen in our electrophysiological results. The second phase is known to be attributable to spinal mechanisms of enhanced synaptic strength, such as postdischarge and wind-up, reflecting central sensitization and requires spinal NMDA receptor activation (Haley et al., 1990). Because PI3K is a protein kinase and NMDA phosphorylation is associated with wind-up and central sensitization, we assessed the effect of LY294002 on NR2A and NR2B subunit phosphorylation after formalin administration. We found a significant inhibition of NR2B subunit phosphorylation in rats pretreated with LY294002. NMDA receptor subunit phosphorylation, especially of the NR2B subunit, has been correlated with pain behaviors in several inflammatory pain models and inhibition of phosphorylation of this particular subunit has produced a potent antinociceptive effect (Boyce et al., 1999; Malmberg et al., 2003; Tan et al., 2005). Finally, specific inhibition of phosphorylated NR2B subunit potently reversed electrophysiological wind-up activity (Kovacs et al., 2004). Therefore, it is reasonable to speculate that PI3K promotes the phosphorylation of NR2B subunits, directly or indirectly via ERK and/or CaMKII activation, and is involved in the establishment of central sensitization.
Intracellular mechanisms involved in PI3K-mediated central sensitization
Activation and phosphorylation of the NMDA receptor provokes increased calcium influx, which may induce sensitization-like mechanisms in the dorsal horn of the spinal cord. CaMKII, as well as having a crucial role in LTP, contributes to central sensitization in the spinal cord in several pain models (Fang et al., 2002; Choi et al., 2005, 2006). Influx of calcium via NMDA receptors leads to the phosphorylation and activation of CaMKII, and its inhibition prevents LTP as well as central sensitization using electrophysiological and behavioral methods (Malinow et al., 1989; Silva et al., 1992; Fang et al., 2002; Nagy et al., 2004). In our study, intrathecal administration of LY294002 produced a significant reduction in CaMKII phosphorylation in the dorsal horn induced by formalin injection. Our results suggest that the role of CaMKII in central sensitization and subsequently pain behavior might be dependent on upstream PI3K activity. Furthermore, the effect of CaMKII on pain transmission may be attributable to an alteration of the trafficking and/or activation of GluR1 subunit as previously observed after visceral inflammatory stimulation (Galan et al., 2004). Phosphorylation and trafficking of GluR1 by CaMKII is an essential component of the early phase of LTP (Derkach et al., 2007). We, indeed, observed changes in the trafficking of AMPA receptors (GluR1 but not GluR2/3) (data not shown) from the cytoplasm to the plasma membrane. Finally, CaMKII activation may also affect phosphorylation of NR2B subunit.
We also observed a reduced number of phospho-ERK-positive cells after intrathecal LY294002 injection. The phosphorylation of ERK has long been known to be involved in inflammatory pain (Silva et al., 1992; Obata and Noguchi, 2004). For example, ERK activation occurs rapidly in neurons of the dorsal horn after formalin injection and its inhibition blocks the resulting pain behavior (Ji et al., 1999, 2002). Interestingly, studies on LTP describe the role of PI3K through both ERK-dependent and ERK-independent mechanisms (Perkinton et al., 2002; Man et al., 2003; Opazo et al., 2003; Zhuang et al., 2004; Chen et al., 2005). In inflammatory pain conditions, Zhuang et al. (2004) demonstrated that PI3K and ERK signaling pathways were intricately associated at the periphery. ERK activation may participate in central sensitization through the phosphorylation of NR2B subunit and/or GluR1 trafficking. Because c-Fos upregulation is attributable to early increased activity of various intracellular pathways, the robust reductions of two important pathways, CaMKII and ERK, via PI3K inhibition, were also likely to contribute to the decreased incidence of c-Fos upregulation.
In conclusion, this study reinforces the idea of similarities between LTP in the hippocampus or amygdala and central sensitization in the spinal cord. However, although these two phenomena could be equivalent, they are not strictly similar and this may relate to the functional sequelae of prolonged painful inputs into spinal circuits as opposed to synaptic inputs that may induce memory in higher centers. For example, the effect of PI3K on CaMKII is intriguing because it has never been shown before in LTP in the hippocampus. Here, we show a direct and/or indirect relationship between these two pathways in spinal plasticity. The precise order, if any, by which PI3K activates different intracellular pathways that ultimately lead to central sensitization remains to be further clarified. Indeed, the effects on NR2B, CaMKII, and ERK after PI3K inhibition were observed as soon as 5 min after formalin injection. Thus, PI3K seems to sit at the junction of different pathways that interact and work in concert to induce the early phase of central sensitization (for diagram, see supplemental figure, available at www.jneurosci.org as supplemental material).
Footnotes
This work was supported by the Wellcome Trust London Pain Consortium (S.B.M., A.H.D., R.D'M., S.P.), the Biotechnology and Biological Sciences Research Council (A.K.C.), and the International Spinal Research Trust (F.M.).
References
- Arendt-Nielsen L, Petersen-Felix S, Fischer M, Bak P, Bjerring P, Zbinden AM. The effect of N-methyl-d-aspartate antagonist (ketamine) on single and repeated nociceptive stimuli: a placebo-controlled experimental human study. Anesth Analg. 1995;81:63–68. doi: 10.1097/00000539-199507000-00013. [DOI] [PubMed] [Google Scholar]
- Boyce S, Wyatt A, Webb JK, O'Donnell R, Mason G, Rigby M, Sirinathsinghji D, Hill RG, Rupniak NM. Selective NMDA NR2B antagonists induce antinociception without motor dysfunction: correlation with restricted localisation of NR2B subunit in dorsal horn. Neuropharmacology. 1999;38:611–623. doi: 10.1016/s0028-3908(98)00218-4. [DOI] [PubMed] [Google Scholar]
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Chandler LJ, Sutton G. Acute ethanol inhibits extracellular signal-regulated kinase, protein kinase B, and adenosine 3′:5′-cyclic monophosphate response element binding protein activity in an age- and brain region-specific manner. Alcohol Clin Exp Res. 2005;29:672–682. doi: 10.1097/01.alc.0000158935.53360.5f. [DOI] [PubMed] [Google Scholar]
- Chen X, Garelick MG, Wang H, Lil V, Athos J, Storm DR. PI3 kinase signaling is required for retrieval and extinction of contextual memory. Nat Neurosci. 2005;8:925–931. doi: 10.1038/nn1482. [DOI] [PubMed] [Google Scholar]
- Choi SS, Seo YJ, Kwon MS, Shim EJ, Lee JY, Ham YO, Lee HK, Suh HW. Increase of phosphorylation of calcium/calmodulin-dependent protein kinase-II in several brain regions by substance P administered intrathecally in mice. Brain Res Bull. 2005;65:375–381. doi: 10.1016/j.brainresbull.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Choi SS, Seo YJ, Shim EJ, Kwon MS, Lee JY, Ham YO, Suh HW. Involvement of phosphorylated Ca2+/calmodulin-dependent protein kinase II and phosphorylated extracellular signal-regulated protein in the mouse formalin pain model. Brain Res. 2006;1108:28–38. doi: 10.1016/j.brainres.2006.06.048. [DOI] [PubMed] [Google Scholar]
- Clark AK, D'Aquisto F, Gentry C, Marchand F, McMahon SB, Malcangio M. Rapid co-release of interleukin 1beta and caspase 1 in spinal cord inflammation. J Neurochem. 2006;99:868–880. doi: 10.1111/j.1471-4159.2006.04126.x. [DOI] [PubMed] [Google Scholar]
- Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci. 2007;8:101–113. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- Dickenson AH, Sullivan AF. Evidence for a role of the NMDA receptor in the frequency dependent potentiation of deep rat dorsal horn nociceptive neurones following C fibre stimulation. Neuropharmacology. 1987;26:1235–1238. doi: 10.1016/0028-3908(87)90275-9. [DOI] [PubMed] [Google Scholar]
- Fang L, Wu J, Lin Q, Willis WD. Calcium-calmodulin-dependent protein kinase II contributes to spinal cord central sensitization. J Neurosci. 2002;22:4196–4204. doi: 10.1523/JNEUROSCI.22-10-04196.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresno Vara JA, Casado E, de CJ, Cejas P, Belda-Iniesta C, Gonzalez-Baron M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004;30:193–204. doi: 10.1016/j.ctrv.2003.07.007. [DOI] [PubMed] [Google Scholar]
- Galan A, Laird JM, Cervero F. In vivo recruitment by painful stimuli of AMPA receptor subunits to the plasma membrane of spinal cord neurons. Pain. 2004;112:315–323. doi: 10.1016/j.pain.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Haley JE, Sullivan AF, Dickenson AH. Evidence for spinal N-methyl-d-aspartate receptor involvement in prolonged chemical nociception in the rat. Brain Res. 1990;518:218–226. doi: 10.1016/0006-8993(90)90975-h. [DOI] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci. 2002;22:478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Koltzenburg M, Torebjork HE, Wahren LK. Nociceptor modulated central sensitization causes mechanical hyperalgesia in acute chemogenic and chronic neuropathic pain. Brain. 1994;117:579–591. doi: 10.1093/brain/117.3.579. [DOI] [PubMed] [Google Scholar]
- Kovacs G, Kocsis P, Tarnawa I, Horvath C, Szombathelyi Z, Farkas S. NR2B containing NMDA receptor dependent windup of single spinal neurons. Neuropharmacology. 2004;46:23–30. doi: 10.1016/s0028-3908(03)00339-3. [DOI] [PubMed] [Google Scholar]
- Koyasu S. The role of PI3K in immune cells. Nat Immunol. 2003;4:313–319. doi: 10.1038/ni0403-313. [DOI] [PubMed] [Google Scholar]
- Lang CW, Hope PJ. Evidence for localized release of substance P within rat spinal cord evoked by physiological and electrical stimuli. Neuropeptides. 1994;26:413–419. doi: 10.1016/0143-4179(94)90027-2. [DOI] [PubMed] [Google Scholar]
- Malcangio M, Bowery NG. Spinal cord SP release and hyperalgesia in monoarthritic rats: involvement of the GABAB receptor system. Br J Pharmacol. 1994;113:1561–1566. doi: 10.1111/j.1476-5381.1994.tb17174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcangio M, Lessmann V. A common thread for pain and memory synapses? Brain-derived neurotrophic factor and trkB receptors. Trends Pharmacol Sci. 2003;24:116–121. doi: 10.1016/S0165-6147(03)00025-7. [DOI] [PubMed] [Google Scholar]
- Malcangio M, Ramer MS, Jones MG, McMahon SB. Abnormal substance P release from the spinal cord following injury to primary sensory neurons. Eur J Neurosci. 2000;12:397–399. doi: 10.1046/j.1460-9568.2000.00946.x. [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- Malmberg AB, Gilbert H, McCabe RT, Basbaum AI. Powerful antinociceptive effects of the cone snail venom-derived subtype-selective NMDA receptor antagonists conantokins G and T. Pain. 2003;101:109–116. doi: 10.1016/s0304-3959(02)00303-2. [DOI] [PubMed] [Google Scholar]
- Man HY, Wang Q, Lu WY, Ju W, Ahmadian G, Liu L, D'Souza S, Wong TP, Taghibiglou C, Lu J, Becker LE, Pei L, Liu F, Wymann MP, MacDonald JF, Wang YT. Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron. 2003;38:611–624. doi: 10.1016/s0896-6273(03)00228-9. [DOI] [PubMed] [Google Scholar]
- Nagy GG, Al-Ayyan M, Andrew D, Fukaya M, Watanabe M, Todd AJ. Widespread expression of the AMPA receptor GluR2 subunit at glutamatergic synapses in the rat spinal cord and phosphorylation of GluR1 in response to noxious stimulation revealed with an antigen-unmasking method. J Neurosci. 2004;24:5766–5777. doi: 10.1523/JNEUROSCI.1237-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Noguchi K. MAPK activation in nociceptive neurons and pain hypersensitivity. Life Sci. 2004;74:2643–2653. doi: 10.1016/j.lfs.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Opazo P, Watabe AM, Grant SG, O'Dell TJ. Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms. J Neurosci. 2003;23:3679–3688. doi: 10.1523/JNEUROSCI.23-09-03679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parcellier A, Tintignac LA, Zhuravleva E, Hemmings BA. PKB and the mitochondria: AKTing on apoptosis. Cell Signal. 2008;20:21–30. doi: 10.1016/j.cellsig.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Patel RK, Mohan C. PI3K/AKT signaling and systemic autoimmunity. Immunol Res. 2005;31:47–55. doi: 10.1385/IR:31:1:47. [DOI] [PubMed] [Google Scholar]
- Patton DT, Garcon F, Okkenhaug K. The PI3K p110delta controls T-cell development, differentiation and regulation. Biochem Soc Trans. 2007;35:167–171. doi: 10.1042/BST0350167. [DOI] [PubMed] [Google Scholar]
- Perkinton MS, Ip JK, Wood GL, Crossthwaite AJ, Williams RJ. Phosphatidylinositol 3-kinase is a central mediator of NMDA receptor signalling to MAP kinase (Erk1/2), Akt/PKB and CREB in striatal neurones. J Neurochem. 2002;80:239–254. doi: 10.1046/j.0022-3042.2001.00699.x. [DOI] [PubMed] [Google Scholar]
- Presley RW, Menetrey D, Levine JD, Basbaum AI. Systemic morphine suppresses noxious stimulus-evoked Fos protein-like immunoreactivity in the rat spinal cord. J Neurosci. 1990;10:323–335. doi: 10.1523/JNEUROSCI.10-01-00323.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandkuhler J. Understanding LTP in pain pathways. Mol Pain. 2007;3:9. doi: 10.1186/1744-8069-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna PP, Cammalleri M, Berton F, Simpson C, Lutjens R, Bloom FE, Francesconi W. Phosphatidylinositol 3-kinase is required for the expression but not for the induction or the maintenance of long-term potentiation in the hippocampal CA1 region. J Neurosci. 2002;22:3359–3365. doi: 10.1523/JNEUROSCI.22-09-03359.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Paylor R, Wehner JM, Tonegawa S. Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257:206–211. doi: 10.1126/science.1321493. [DOI] [PubMed] [Google Scholar]
- Sun RQ, Tu YJ, Yan JY, Willis WD. Activation of protein kinase B/Akt signaling pathway contributes to mechanical hypersensitivity induced by capsaicin. Pain. 2006;120:86–96. doi: 10.1016/j.pain.2005.10.017. [DOI] [PubMed] [Google Scholar]
- Tan PH, Yang LC, Shih HC, Lan KC, Cheng JT. Gene knockdown with intrathecal siRNA of NMDA receptor NR2B subunit reduces formalin-induced nociception in the rat. Gene Ther. 2005;12:59–66. doi: 10.1038/sj.gt.3302376. [DOI] [PubMed] [Google Scholar]
- Teoh H, Malcangio M, Fowler LJ, Bowery NG. Evidence for release of glutamic acid, aspartic acid and substance P but not gamma-aminobutyric acid from primary afferent fibres in rat spinal cord. Eur J Pharmacol. 1996;302:27–36. doi: 10.1016/0014-2999(96)00052-0. [DOI] [PubMed] [Google Scholar]
- Toker A, Newton AC. Cellular signaling: pivoting around PDK-1. Cell. 2000;103:185–188. doi: 10.1016/s0092-8674(00)00110-0. [DOI] [PubMed] [Google Scholar]
- van der Heide LP, Kamal A, Artola A, Gispen WH, Ramakers GM. Insulin modulates hippocampal activity-dependent synaptic plasticity in a N-methyl-d-aspartate receptor and phosphatidyl-inositol-3-kinase-dependent manner. J Neurochem. 2005;94:1158–1166. doi: 10.1111/j.1471-4159.2005.03269.x. [DOI] [PubMed] [Google Scholar]
- Whitman M, Downes CP, Keeler M, Keller T, Cantley L. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature. 1988;332:644–646. doi: 10.1038/332644a0. [DOI] [PubMed] [Google Scholar]
- Yonehara N, Saito K, Oh-ishi S, Katori M, Inoki R. Contribution of bradykinin to heat-induced substance P release in the hind instep of rats. Life Sci. 1995;56:1679–1688. doi: 10.1016/0024-3205(95)98574-y. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Xu H, Clapham DE, Ji RR. Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J Neurosci. 2004;24:8300–8309. doi: 10.1523/JNEUROSCI.2893-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]