

Abstract
The GnRH gene uses two well-defined regions to target expression to a small population of hypothalamic GnRH neurons: a 173-bp proximal promoter and a 300-bp enhancer localized at approximately −1800 to −1500 bp from the start site. Interaction of multiple factors with the GnRH enhancer and promoter is required to confer neuron-specific expression in vivo and in cells in culture. In addition, the expression of the GnRH gene is regulated by numerous neurotransmitters and hormones. Several of these effectors act through membrane receptors to trigger the protein kinase C pathway, and 12-O-tetradecanoyl phorbol-13-acetate (TPA), a modulator of this pathway, has been shown to suppress GnRH gene expression through the promoter. We find that TPA suppresses expression through the GnRH enhancer as well as the promoter. In the enhancer, an Oct-1 binding site, a Pbx/Prep binding site, Msx/Dlx binding sites, and a previously unidentified protein-binding element at −1793, all contribute to TPA suppression. TPA treatment leads to decreased binding of Oct-1 and Pbx1a/Prep to their sites. However, a complex formed by GT1–7 nuclear extracts on the −1793 site is not affected by TPA treatment. It is known that cooperative interaction among multiple factors is necessary for GnRH gene expression; thus, one mechanism by which TPA suppresses GnRH gene expression is to disengage some of these factors from their cis-regulatory elements.
GnRH, A DECAPEPTIDE that is key to reproductive function, is produced by several hundred neurons scattered throughout the rostral hypothalamus and forebrain. These cells originate near the olfactory placode and then migrate into the brain (1). During developmental migration, these cells increase their expression of GnRH, suggesting that these cells respond to environmental cues. Through studies using a cultured cell model of GnRH neurons, GT1–7 cells (2), a 300-bp neuron-specific enhancer from the rat GnRH regulatory region, has been identified and shown to confer cell specificity expression to GT1–7 cells, in conjunction with the 173-bp rat GnRH gene promoter (3, 4). This combination of small regulatory regions is also sufficient to target the GnRH neurons in vivo in transgenic mice (5). Multiple transcription factors including Oct-1 (6), GATA-4 (7, 8), Prep1/Pbx (9), Dlx2, Msx1 (10), Otx2 (11), and CCAAT/enhancer binding protein-β (12) have been shown to bind to the regulatory elements in the GnRH enhancer and promoter and contribute to GnRH expression in GT1–7 cells.
The level of GnRH mRNA is subjected to regulation during development (13), aging (14), the estrous cycle (15), and by various hormones (16) and neurotransmitters (17, 18). In rat hypothalamic tissues, the activation of cAMP and diacylglycerol-dependent pathways was shown to enhance GnRH mRNA steady-state level (19). The results from these studies are in contrast to those obtained with a mouse hypothalamic cell line, GT1–7 cells; where forskolin, an activator of the protein kinase A pathways, does not affect GnRH mRNA levels, whereas and 12-O-tetradecanoyl phorbol-13-acetate (TPA), an activator of protein kinase C (PKC) pathways, decreases GnRH mRNA levels (20, 21). This difference is most likely due to the effects of interneurons and glia in rat hypothalamic tissue. The down-regulation of GnRH mRNA occurs at the transcriptional level, and transcriptional activity from a transfected reporter driven by the rat GnRH regulatory region is also decreased by TPA treatment (22, 23). This down-regulation is likely due to the acute activation of PKC rather than the long-term degradation of PKC by TPA treatment (20, 21), and cotransfection of PKCα is sufficient to down-regulate a GnRH reporter gene (24). Attempts to map the TPA-responsive elements in the rat GnRH regulatory region have yielded some conflicting results. Eraly et al. (23) showed that a region defined by deoxyribonuclease I footprinting termed footprint 2 (−82 to −22 bp from the mRNA start site) in the proximal GnRH promoter was able to confer TPA suppression to a heterologous thymidine kinase promoter, although the exact transcription factor binding sites and the corresponding factors were not identified. Bruder et al. (22) mapped the TPA-responsive elements to a region that corresponds to footprint 5 through footprint 2 (−126 and −73) of the rat GnRH promoter, and found that there was a correlation between c-Fos expression and the suppression of the GnRH gene expression by TPA. However, direct binding of AP-1 proteins to this region was not detected. Therefore, although it is evident that GnRH gene expression is down-regulated by the PKC pathway, it was not clear which transcriptional factors are involved. Here we show that not only the promoter, but also the enhancer region participate in PKC repression. Furthermore, within the enhancer, Oct-1, Prep/Pbx, Dlx2/Msx1, and a previously unidentified protein participate in the regulation of GnRH transcription by the PKC pathway.
RESULTS
Suppression of the GnRH Enhancer through Activation of the PKC Pathway
Previous work suggested that footprint 2 (−82 to −22) of the rat GnRH promoter was the target of suppression by TPA in GT1–7 cells (23). Several transcriptional factors, including Oct-1 (25) and Prep/Pbx (9), have subsequently been shown to activate GnRH transcription through this promoter proximal region. These same transcription factors also act on the GnRH enhancer (Fig. 1A). Therefore, we examined whether TPA also suppresses the transcriptional activity of the GnRH enhancer on a heterologous promoter [Rous sarcoma virus long-terminal repeat promoter (RSVp)]. Treatment with 100 nM TPA for 16 h led to a significant suppression of luciferase activity, whereas a reporter driven by the RSV promoter alone was not suppressed (Fig. 1B). Therefore, the GnRH enhancer is able to confer suppression by TPA to a RSV promoter. Similar results were obtained with a reporter using a short GnRH promoter. This reporter lacks most of the cis elements in the GnRH promoter, including footprint 2 that is suppressed by TPA. In this case, the promoter activity of a DNA fragment starting from −28 to +112 of the GnRH promoter (−28p) was increased by TPA treatment, whereas addition of the GnRH enhancer produced a significant suppression (Fig. 1C), again suggesting that GnRH enhancer contains cis elements that can be suppressed by TPA. TPA caused a rapid and sustained suppression of GnRH enhancer, the decrease was observed as early as 2 h (the earliest sampling time point) and lasted up to 17 h (Fig. 1D).
Fig. 1.

The GnRH Enhancer Is Suppressed by TPA Treatment
A, An overview of cis elements on the GnRH enhancer and the corresponding transcription factors. B, The GnRH enhancer (GnRHe) confers TPA suppression to a RSV promoter (RSVp). C, The GnRH enhancer confers TPA suppression to a short GnRH promoter −28p (a DNA fragment spanning −28 to +112 of the GnRH promoter). D, The time course for TPA suppression of the GnRH enhancer. GT1–7 cells were transfected with luciferase reporters, along with a RSV β-galactosidase reporter as internal control, in the absence of serum for 24 h, then treated with 100 nM TPA for 16 h (A–C) or times indicated (D) or with 0.1% ethanol as vehicle. RLU indicates relative luciferase units normalized to the internal control β-galactosidase. Results were normalized to the luciferase activity of the rGnRHe/RSVp reporter that received vehicle treatment. Data are mean ± SE of three experiments conducted in triplicates or quadruplicates. Groups labeled with different letters are significantly different from each other.
Multiple cis-Regulatory Elements in the GnRH Enhancer Contribute to the Suppression of GnRH Transcription by TPA
Initial GnRH enhancer deletion experiments were carried out with reporter genes that had the GnRH enhancer and −178 to +112 GnRH promoter. However, because the −178 promoter itself was inhibited by TPA, most of the enhancer deletions did not change the suppression, although they did decrease the basal activity. Thus, to analyze the roles of enhancer elements in TPA suppression free of the influence of the −178 promoter, the GnRH enhancer fragments were cloned into the luciferase reporter plasmid driven by the RSV promoter. Truncation of the GnRH enhancer from −1876 to −1800 caused a decrease in basal expression but did not significantly affect the suppression by TPA (Fig. 2) indicating that even a dramatic decrease in basal activity of the enhancer is not sufficient to abrogate TPA suppression. However, truncation to −1780 dramatically relieves TPA suppression. No significant changes were observed upon further truncation. To determine whether other elements downstream of −1800 are also important for TPA suppression, truncations within this −1800 to −1576 fragment, from the 3′ end of the enhancer at −1576 were created (Fig. 3). Deletions from the −1576 end caused a gradual decrease in basal expression, as well as relief of TPA suppression, with changes upon removal of a region containing repeated CAATT sites that bind Dlx/Msx proteins (10), and a region that contains an Oct-1 and GATA-4 binding sites (6, 8). Further removal of a Prep/Pbx site also caused additional relief of suppression. Thus, multiple sites in this region are important for TPA suppression.
Fig. 2.

Truncations from the 5′-End of the Enhancer Indicate that the −1782 Oct-1 Site or an Upstream Site between −1800 and −1780 Is Important for Suppression of Transcriptional Activity by TPA
Reporter plasmids containing series of deletions were made using PCR approaches and transfected into GT1–7 cells. Reporter activities were assayed and analyzed as described in Fig. 1 and Materials and Methods. Data are mean ± SE of four experiments, with each conducted in triplicate or quadruplicate. Groups labeled with different letters are significantly different from each other.
Fig. 3.

Truncations from the 3′-End of the Enhancer Implicate Multiple cis Elements in Contributing to the Suppression of Transcriptional Activity by TPA
Reporter plasmids containing a series of deletions were created using PCR approaches and transfected into GT1–7 cells. Reporter activities were assayed and analyzed as described in Fig. 1 and Materials and Methods. Results were normalized to the luciferase activity of the −1800/−1657rGnRHe/RSVp reporter that received vehicle treatment. Data are mean ± SE of four experiments, with each conducted in triplicates. Groups labeled with different letters are significantly different from each other.
The Role of the −1782 Oct-1 Site in TPA Suppression
An AT-rich site beginning at −1782 was previously shown to bind Oct-1 and to play an important role for GnRH gene expression within the enhancer (6). Because deletion from −1800 to −1780, which partially destroys this site, completely relieves TPA suppression, we considered the possibility that this site and its binding partner, Oct-1, are regulated by TPA. A previously described mutation of this site that interrupts Oct-1 binding (6) was introduced into the luciferase reporter driven by the GnRH enhancer and RSV promoter. As demonstrated previously, introduction of this mutation decreased the basal expression of luciferase activity dramatically, although it remains significantly above background. This mutation almost completely eliminated the effect of TPA (Fig. 4A). This effect was also observed when the same mutation was introduced into the reporter driven by the GnRH enhancer and a short sequence of the GnRH promoter starting at −28 (data not shown). In contrast, mutation of this site did not affect the suppression by TPA of a reporter driven by the GnRH enhancer and −178-bp promoter (data not shown). In this case, we speculate that Oct-1 binding sites in the GnRH promoter may have compensated for the one mutated in the enhancer. Correlating with a role of the −1782 Oct-1 site for TPA suppression, there was a decreased binding of Oct-1 to a probe that contains this site using nuclear extracts from GT1–7 cells treated by TPA (Fig. 4B). The decrease was observed as early as 30 min and lasted for at least 1 h, after which it returned to the control level. The decrease in Oct-1 binding seemed to result from the decreased binding affinity of Oct-1, as TPA treatment did not change the level of Oct-1 in the nuclear extracts (Fig. 4C).
Fig. 4.
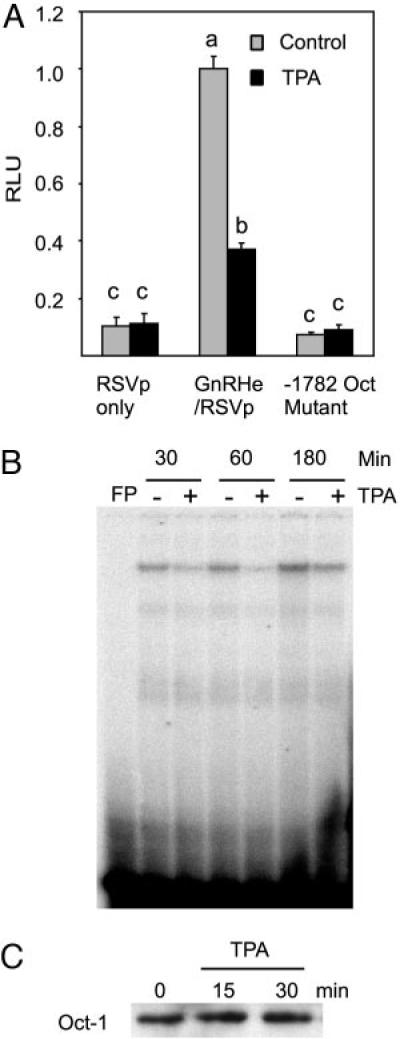
The −1782 Oct-1 Site Is Involved in TPA Suppression
A, Mutation of the Oct-1 site reverses the suppression of the enhancer activity by TPA. Data are mean ± SE of three experiments conducted in quadruplicates. Groups labeled with different letters are significantly different from each other. B, TPA decreases the binding of Oct-1 to the −1782 Oct-1 site. GT1–7 cells were starved for 24 h in serum-free DMEM, then treated with 100 nM TPA for the time indicated and harvested for nuclear extract preparation and subjected to EMSA as described in Materials and Methods. FP, Free probe without nuclear extracts. Representative results are shown. C, TPA does not change Oct-1 protein level in the nuclear extracts. A Western blot of nuclear extracts from TPA-treated and untreated GT1–7 cells is shown using an Oct-1 antibody.
The Role of a Prep/Pbx Site in TPA Suppression
Truncation experiments indicated that multiple cis elements between −1576 and −1800 contribute to the effect of TPA on GnRH gene expression. Therefore, we examined the possible role of additional cis elements in this region. A previously described GATA-A site (7) has been shown to bind to Prep/Pbx (9); thus, a mutation that destroyed the binding of Prep/Pbx to this site was introduced into the rGnRHe/RSVp reporter. This mutation lowered basal transcriptional activity significantly, and partially relieved TPA suppression (Fig. 5A). In EMSA, this site formed two bands with GT1–7 nuclear extracts. The slower band is known to contain Prep/Pbx1a, whereas the faster band contains Prep/Pbx1b (9). TPA treatment decreased the slower band, whereas it did not affect the faster band. The decrease was seen as early as 15 min after treatment (the earliest time point examined, data not shown) and was most obvious at 1 h, with still some decrease at 2 h (Fig. 5B). Immunoblotting experiments indicated a rapid decrease of Pbx1a but not Pbx1b in the nuclear extracts upon TPA treatment (Fig. 5C), implying that trafficking of Pbx1a between nucleus and cytoplasm might be regulated by TPA treatment (26, 27). Hence, there is a correlation between Pbx1a DNA binding and the transcriptional activity of the GnRH enhancer.
Fig. 5.
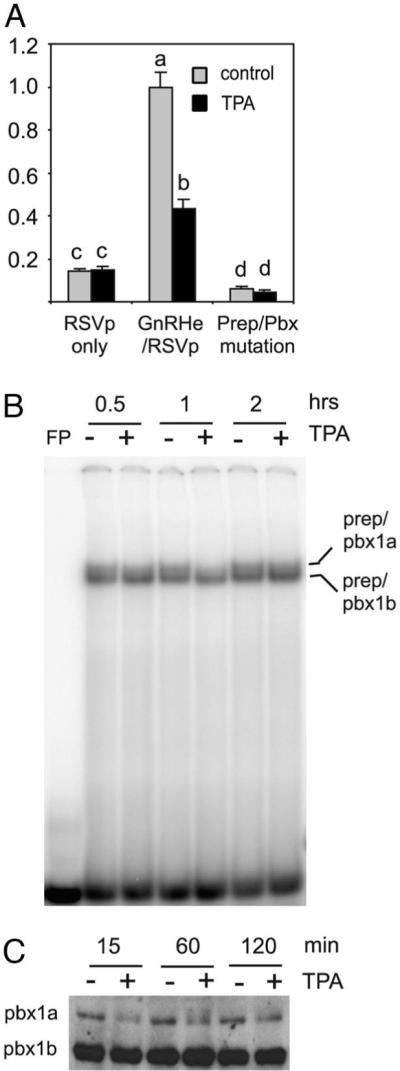
Prep/Pbx Is Involved in the TPA Suppression of the GnRH Enhancer Activity
A, Mutation of a Prep/Pbx site in the GnRH enhancer partially reverses the effect of TPA. RSVp, the reporter is driven by the RSV promoter only; GnRHe/RSVp, the reporter is driven by the GnRH enhancer and RSV promoter; Prep/Pbx mutation, includes a mutation from GATA to CTTA as described in Ref. 8 within the GnRHe/RSVp. Data are mean ± SE of three experiments, with each conducted in quadruplicate. Asterisks indicate P < 0.05 compared with GnRH enhancer. B, TPA treatment decreases the binding of Prep/Pbx1a but not that of Prep/Pbx1b to the −1758/−1735 probe in EMSA (top sequence: gttgttcacctatcattcaggaag). The identification of these transcription factors was previously described (9). C, TPA treatment decreases the level of Pbx1a, but not Pbx1b in the nuclear extracts. A Western blot of nuclear extracts from TPA-treated and untreated GT1–7 cells is shown using a Pbx1 antibody.
Role of CAATT Repeats in TPA Suppression
Truncation experiments in the −1576 to −1642 region caused a significant drop in suppression by TPA. CAATT repeats have been identified in this region and shown to be important for the transcriptional activity of the GnRH enhancer (28). These cis elements have recently been found to bind Dlx2 and Msx1 (10). A previously described reporter (28) that has three CAATT repeats deleted from the enhancer on the RSV promoter showed a lower basal expression as previously described but also showed a partial relief of TPA suppression comparing with wild-type reporter (Fig. 6A). TPA treatment produced a slight decrease in the binding of Dlx2 to a probe containing the CAATT site, whereas it also produced no change or a slight increase on the binding of Msx1 to the same probe (Fig. 6B). There appeared to be a correlation between the TPA suppression and a decrease in the binding of transcriptional activator Dlx2 to the CAATT sites.
Fig. 6.
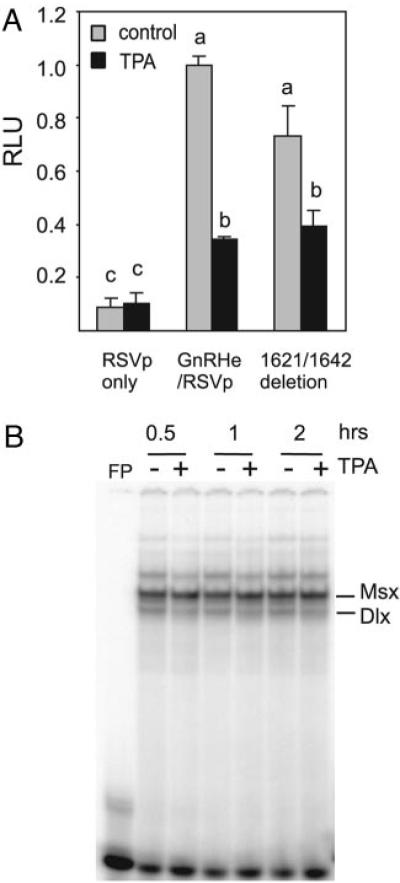
Contribution of Dlx/Msx Sites to the Suppression of the GnRH Enhancer by TPA
A, Deletion of three CAATT repeats that bind to Dlx/Msx proteins modestly relieves TPA suppression. Groups labeled with different letters are significantly different from each other. Results are presented as mean ± SE of three experiments with each conducted in quadruplicate. B, TPA treatment slightly decreases the binding of Dlx2 but not that of Msx to the −1642/−1623 probe (top sequence: ttgtgacaattataaagccc) as shown in EMSA.
A Novel Site Spanning −1793 and −1785 Is Required for TPA Suppression
We also identified a cis element between −1793 and −1785 that is just upstream of the Oct-1 site. Nuclear extracts from GT1–7 cells formed a single complex with a probe spanning the region from −1802 to −1778 in the GnRH enhancer in EMSA. Competition experiments with scanning mutations (Fig. 7A) in this region indicated a 9-base sequence CTCCTGCTG is involved in the formation of this complex (Fig. 7B). A deletion of 4 bp (del in Fig. 7A) in this region destroyed the binding (Fig. 7B). When this deletion was introduced into a reporter driven by the GnRH enhancer and RSV promoter, it decreased basal transcriptional activity by more than 80%. In addition, the inhibitory effect of TPA on reporter gene transcription was abolished (data not shown). Under different binding conditions, this probe also binds to nuclear factor 1 (NF-1) (29). A two-base substitution mutation was created that does not affect NF-1 binding (29) but abolishes the formation of the novel complex. Importantly, this mutation (sequence is shown in Fig. 8A) did not affect the binding of Oct-1 to a probe that also contains the −1782 Oct-1 site (Fig. 8B). When this mutation was introduced into a reporter driven by the GnRH enhancer and RSV promoter, basal transcription activity decreased dramatically and the inhibitory effect of TPA was also reversed (Fig. 8C). Similar results were obtained when this mutation was introduced into the reporter driven by the GnRH enhancer and the GnRH promoter (data not shown). This complex also appeared to be unique to GT1–7 cells because only a weak band that migrated differently was formed between this probe and nuclear extracts from other types of cells such as αT3-1, LβT2, and NIH3T3 cells (data not shown). Consistent with its important role in the GnRH gene expression and suppression by TPA, the activity of GnRH enhancer and promoter in αT3-1 and LβT2 cells is low and was not suppressed by TPA. Even though mutation of this site alleviated suppression by TPA, protein binding in the GT1–7 nuclear extracts was not changed by TPA treatment (Fig. 8D)
Fig. 7.
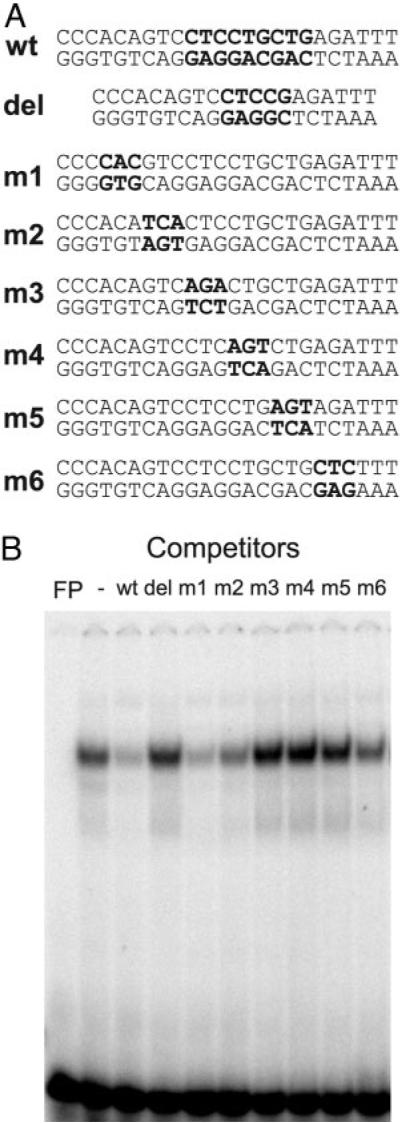
Characterization of a New Binding Site at −1793 in the GnRH Enhancer
A, Sequences for the region from −1802 to −1778 in the GnRH enhancer and mutations are shown. Bold letters in mutated oligonucleotides indicate mutated bases. B, EMSA competition experiments show that m3, m4, m5 as well as del (with deletion of four bases) do not compete with the wild-type sequence in binding to the GT1–7 nuclear extracts. Collectively, these experiments define a nine-base region CTCCTGCTG (bold in wild-type sequence) involved in binding to GT1–7 nuclear extracts.
Fig. 8.

The −1793 Site Is Necessary for TPA Suppression of the GnRH Enhancer
A, The sequences that were used for EMSA in B, mutated bases are bolded. B, A two-base mutation as depicted in A does not affect the binding to Oct-1, but causes the lost of the complex indicated by an asterisk. The wild-type probe forms two complexes with the GT1–7 nuclear extracts. One results from binding to Oct-1, another is the new complex identified in Fig. 7. The identities of these complexes were confirmed by EMSA competition experiments with oligos containing the −1782 Oct-1 site and the −1802/−1778 sequence respectively (data not shown). C, A two-base mutation as depicted in A abolishes the suppression of GnRH enhancer activity by TPA. Vectors were transfected into GT1–7 cells as described in Materials and Methods. Data are mean ± SE of three experiments conducted in triplicate. Groups labeled with different letters are significantly different from each other. D, TPA treatment does not affect the binding of GT1–7 nuclear extracts to the −1793 novel site (−1802/−1778 probe) in EMSA.
DISCUSSION
A minimal promoter and a 300-bp enhancer have been defined within the GnRH gene and have been shown to specifically target gene expression to the GT1 cells, as well as to the GnRH neurons in vivo (5). An array of transcription factors bind the promoter and/or enhancer, including Oct-1, Prep/Pbx, GATA-4, Dlx, Msx, Otx2, and CCAAT/enhancer binding protein-β. Previously, the footprint 2 region (−82 to −22) of the GnRH promoter was implicated in TPA-mediated suppression, although exactly which transcriptional factors were involved is not clear (23). Of note is that some of the factors we have studied in the enhancer, including Oct-1, Pbx/Prep, Dlx, and Msx, also each bind to two of their cognate binding elements within the −173-bp promoter (9, 10, 25, 29). In fact, the footprint 2 region alone has two sites for the Dlx/Msx family and one each for Oct-1 and Pbx/Prep. Therefore, it was not surprising that we have now found that the GnRH enhancer is also suppressed by TPA. Further analysis indicates that Oct-1, through binding to the −1782 Oct-1 site, is most important for the TPA suppression. Mutation of the Prep/Pbx site also eliminates the TPA suppression. In addition, mutation of another previously unidentified site at −1794 also totally eliminates suppression. We noticed that mutation of any one of them decreases transcriptional activity to 10 to 20% of basal levels; however, simple reduction in basal activity of the enhancer is not sufficient to abrogate TPA suppression as seen with the 5′ truncation from −1800 to −1571, which reduces enhancer activity to approximately 12% of wild-type but remains suppressed by TPA (Fig. 2). The involvement of multiple factors in TPA suppression might suggest cooperative interaction among these sites. Indeed, physical and functional interaction has been shown between Oct-1 and Prep/Pbx (9) and between Oct-1 and Msx1 as well (43). It is possible that the elimination of TPA suppression by mutation of these sites may reflect their importance in basal transcriptional activity of the GnRH enhancer rather than a specific role in TPA suppression. However, in the case of the Oct-1 and Prep/Pbx sites, TPA also decreased the binding of the corresponding factors to these sites in nuclear extracts from treated cells tested in vitro, suggesting a specific role for them in mediating TPA suppression. As some of these sites are also found in the GnRH promoter and the recently identified distal enhancer region (29), it is likely that TPA might also suppress GnRH expression through them. Consequently, elimination of one binding site for these factors in the GnRH enhancer in the context of intact GnRH promoter may have no apparent effect on the TPA suppression of the remained transcriptional activity. Indeed, whereas mutation of the −1782 Oct-1 site in a reporter that contains both GnRH enhancer and promoter almost eliminates enhancer activity, it does not affect TPA suppression of the remaining transcriptional activity. However, this does not mean that the Oct-1 site does not contribute to the TPA suppression in the context of intact GnRH promoter. For this reason, we focused on individual transcriptional factors, and how TPA might affect their interaction with their DNA binding sites and their activities in the GnRH enhancer, with the expectation that they might be regulated by TPA in a similar way on the GnRH promoter.
TPA appears to suppress the enhancer by decreasing the binding of transcription factors to the GnRH enhancer DNA. TPA treatment decreases Oct-1 binding to the −1782 site. The decreased binding of Oct-1 is also seen with other Oct sites in the GnRH gene regulatory regions (data not shown). It is possible that TPA might also suppress the GnRH promoter through the Oct-1 binding sites in the promoter and the recently identified distal enhancer (29). Because there is no decrease at the level of Oct-1 protein in the GT1–7 nuclear extracts after TPA treatment, the decreased binding to the Oct-1 binding site is possibly due to protein modification. It has been reported that Oct-1 binding can be modulated by phosphorylation. For example, Oct-1 is hyperphosphorylated upon mitosis with reduced binding to the octamer site and inhibition of transcription activity. The mitosis-specific phosphorylation is reversed as cells exit mitosis. Phosphorylation of a serine in the homeodomain of Oct-1 by protein kinase A mimics the mitosis-specific phosphorylation and is sufficient to inhibit its binding (30). In GT1–7 cells, Oct-1 binding to the −1680 AT-rich site in the GnRH enhancer is increased by the activation of cGMP pathway, apparently through phosphorylation of Oct-1 (12). Pit-1, a member of POU domain family, becomes phosphorylated at two distinct sites in the homeodomain in response to phorbol esters and cAMP. The effect of phosphorylation on its DNA binding activity is variable depending on the DNA sequence (31). We have not assessed the phosphorylation status of Oct-1 after TPA treatment, but it is possible that activation of PKC might lead to changes in phosphorylation of Oct-1 resulting in its decreased binding to the −1782 site. Finally, Subramaniam et al. (32) reported that glucocorticoids suppress GnRH gene expression by attenuation of Oct-1 binding to the GnRH promoter through glucocorticoid receptor. Therefore, the decreased binding of Oct-1 we observe in the EMSA could also be due to the actions of TPA on other transcription factors interacting with Oct-1.
A Prep/Pbx site also contributes to the suppression by TPA. Two complexes, Prep/Pbx1a and Prep/Pbx1b, bind to the Prep/Pbx site. Pbx1 belongs to the PBC subfamily of TALE (three-amino-acid-loop-extension) homeodomain proteins. PBC proteins form stable heterodimers with MEINOX proteins including Meis and Prep proteins (33). Pbx1a and Pbx1b are generated by differential splicing of the same gene so that Pbx1a has an additional 83 amino acids in the carboxyl terminal (34). The functional difference between the two isoforms has not been well characterized, although it was reported that Pbx1a, but not Pbx1b, is able to recruit the corepressors nuclear receptor corepressor (NCoR) and silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) (35). We find that TPA treatment decreases the binding of Prep/Pbx1a but not that of Prep/Pbx1b to its site. In addition, there is a decrease at the level of Pbx1a in GT1–7 nuclear extracts upon treatment by TPA as early as 15 min, potentially indicating that TPA could differentially affect trafficking of Pbx1a between nucleus and cytoplasm. The regulation of Pbx cellular distribution has been well documented, but how Pbx1a, vs. Pbx1b, is differentially regulated is not clear. Pbx1 contains sequences recognized by the nuclear importers as well as sequences by the nuclear exporters. Pbx contains two nuclear localization signals in its homeodomain that is thought to be masked by the Pbx N-terminal region (26). Pbx1 also contains two adjacent independent nuclear export signals of the leucine-rich type in the PBC-A domain (27). Thus binding of Pbx with MEINOX proteins in the PBC-A domain will mask the independent nuclear export signals in the PBA-A domain and expose nuclear localization signals in the homeodomain of Pbx, resulting in its nuclear location. Pbx1 is also subjected to phosphorylation. Phosphorylation of the PBC-B domain of Pbx1 by protein kinase A blocks the nuclear export, leading to its nuclear accumulation (27). Because these domains exist in both Pbx1a and 1b, this mechanism does not explain the differential regulation of Pbx isoforms by TPA. We noticed that there are some additional phosphorylation sites for PKC in the C-terminal of PBX1a and their role in export of Pbx1a from the nucleus needs further investigation.
The 3′ end of the GnRH enhancer contains several CAATT repeats binding homeodomain protein Dlx2 or Msx1. Dlx proteins are transcription activators, whereas Msx proteins are transcription repressors. In addition, dimerization between them blocks their DNA binding and results in reciprocal inhibition of their transcriptional activities (36). Our deletion and internal mutation experiments indicate that these CAATT repeats also may contribute somewhat to TPA suppression. In addition, TPA treatment appears to slightly decrease the binding of activator Dlx2 but not that of suppressor Msx1, a phenomenon consistent with TPA's moderate suppressive effect on transcription through these sites. Because the antibodies available do not work well for immunoblotting, we do not know whether this change in the binding pattern reflects the change at the protein level of Dlx2 in the nuclear extracts or change in Dlx2 DNA binding affinity. It has been reported that phosphorylation of Dlx3 in the N-terminal homeodomain by PKC decreases its binding to DNA (37). Because this region is highly conserved among Dlx proteins, it is possible that Dlx2 might also be phosphorylated by PKC upon TPA treatment, leading to reduced binding to its sites.
In this study, we have also identified a novel site at − 1793 that is required for TPA suppression. This site binds to an as-yet-unidentified protein that is crucial for enhancer activity in GT1–7 cells. However, unlike the other transcription factors that are examined herein, the complex formed between this site and GT1–7 nuclear extracts is not affected by TPA treatment. In addition, the complex formed between this site and GT1–7 nuclear extracts is not seen in several other types of cells including LβT2 and NIH3T3 cells, indicating that this site may be important for cell-specific GnRH gene expression in GT1–7 cells. Consistent with the role of this unique complex in TPA suppression, the GnRH enhancer is not suppressed after transfection into LβT2 or αT3–1 cells (data not shown).
In conclusion, we have shown that activation of the PKC pathway requires multiple cis elements for down-regulation of GnRH enhancer activity and appears to act by decreasing the binding of Oct-1, Prep/Pbx1a, and Dlx2 to their sites. It is known that agents activating the PKC pathway stimulate GnRH secretion. By inhibiting GnRH transcription, the PKC pathway may provide a mechanism to prevent the GnRH neurons from excess secretion of GnRH. One of the physiological stimuli activating the PKC pathway in GT1–7 cells is GnRH itself (38), and GnRH has been shown to suppress its mRNA level in the GT1–1 cells (39), providing a intriguing possibility that the autocrine GnRH might prevent excess production of GnRH by suppressing its own expression. Melatonin, which activates several pathways in the GT1–7 cells including the PKC pathway, also inhibits GnRH transcription (40). Therefore, neurotransmitters or hormones could regulate GnRH gene expression through the PKC pathway. That this suppression is achieved by acting through multiple transcriptional factors underlies its importance.
Materials and Methods
Plasmid Cloning and Mutagenesis
The reporter plasmid rGnRHe/RSVp-luc contains the rat GnRH enhancer (positions −1571 to −1863 relative to the transcriptional start site) fused in reverse orientation to the RSV promoter (4). To generate deletion mutants of the enhancer from the −1863 end, desired regions were PCR-amplified with primer pairs additionally containing a Not1 site and a Xma1 site at each end. Amplicons were then cut with Not1 and Xma1 and placed in rGnRHe/RSVp-luc, between the Not1 and Xma1 sites, replacing the wild-type enhancer. Deletions from the −1571 end were similarly generated by amplifying the desired regions using primer pairs that in addition have Kpn1 site and SacI site at each end. The amplicons were then digested with Kpn1 and SacI and placed into rGnRHe/RSVp-luc between the Kpn1 and SacI sites. All amplicons were sequenced. The plasmid harboring the mutation of the GATA-A motif to CTTA-A in the GnRH enhancer was described (8) and the mutated GnRH enhancer was later moved to the RSVp-luc plasmid. Del 1642/1622 RSVp-luc was described in Ref. 28. Site-directed mutations were generated with QuikChange kit from Stratagene (La Jolla, CA) and confirmed by DNA sequencing.
Cell Culture and Transfections
GT1–7 cells were maintained in DMEM supplemented with 10% fetal bovine serum, 100 U/ml of penicillin and 0.1 mg/ml of streptomycin in an atmosphere with 5% CO2 at 37 C.
Transient transfections were carried out using FuGENE6 (Roche Biochemical, Indianapolis, IN) in 24-well plates. Briefly, 0.4 μg luciferase reporters and 0.1 μg RSV-β-gal plasmid were mixed with 2.5 μl FuGENE6 in 25 μl of DMEM, and added to each well containing 0.5 ml of DMEM. After 24 h, cells were treated with 100 nM TPA (diluted from 100 μM in ethanol) or 0.1% ethanol as vehicle control for 16 h. Cells were harvested and assayed for luciferase and β-galactosidase activities. Luciferase activity of each sample was normalized with β-galactosidase activity and normalized to the means of three to four replicates of the construct containing the wild-type GnRH enhancer treated with vehicle.
Statistical Analysis
All statistical analysis was performed using JMP version 5.1 software (SAS Institute, Cary, NC). Data are expressed as means ± SEM of at least three samples per group. Results were analyzed for significant differences using ANOVA. Post hoc group comparison was made using Tukey's honestly significant difference. Analysis was conducted using untransformed data or data optimally transformed by the method of Box and Cox as indicated (41). A P < 0.05 was the requirement for declaring significance.
EMSA
Nuclear extracts were prepared according to the method described by Schreiber et al. (42). One picomole of annealed oligonucleotides were phosphorylated with [γ32P] ATP (6000 Ci/mmol, DuPont NEN, Boston, MA) and polynucleotide kinase using standard procedures. Probes were passed over G-25 microcolumns (Amersham Biosciences, Piscataway, NJ), and radioactivity was counted with a scintillation counter. Binding reactions were carried out in conditions previously described (8). Each probe (1–5 fmol) was incubated with 3-μg crude nuclear extracts in 20-μl reactions. Reactions were incubated at room temperature for 20 min, and separated on 5% polyacrylamide gel in 0.5× TBE. Competitions were performed by preincubating the reactions with 10- to 200-fold excess of unlabeled oligonucleotides for 5 min before adding the probe. Supershift assays were performed by adding 1 μl of antibody or rabbit control IgG to the complete reaction and incubating as above.
Western Blotting
Ten micrograms of nuclear extracts were resolved on a 10% of sodium dodecyl sulfate-polyacrylamide gel. The proteins were transferred to a polyvinylidene difluoride membrane by electroblotting. The membranes were then blocked in 5% nonfat dry milk in TBST [10 mM Tris (pH 8.0), 150 mM NaCl, and 0.05% Tween 20] at room temperature for 1 h. Primary antibodies against Oct-1 or pbx (Santa Cruz Biotechnology, Santa Cruz, CA) were diluted at 1:1000 in 5% BSA in TBST. The membranes were incubated at room temperature for 2–3 h with antibodies and washed three times for 5 min each with TBST. The membranes were then incubated with horseradish peroxidase-conjugated secondary antibody diluted at 1:5000 in TBST at room temperature for 1 h and washed three times for 5 min each with TBST. Antigen-antibody complexes were detected with ECL Western blotting detection reagents (Amersham Biosciences).
Acknowledgments
We thank Naama Rave-Harel and Marjory Givens for helpful discussions and plasmids and Ravid Sasson, Nichol Miller, and Djurdjica Coss for careful reading of the manuscript.
This work was supported by National Institutes of Health (NIH) Grant R01 DK044838 (to P.L.M.). Q.T. was supported by NIH NRSA F32 DL62636.
Abbreviations
- NF-1
Nuclear factor 1
- PKC
protein kinase C
- RSV
Rous sarcoma virus
- TPA
12-O-tetradecanoyl phorbol-13-acetate
Footnotes
Molecular Endocrinology is published monthly by The Endocrine Society (http://www.endo-society.org), the foremost professional society serving the endocrine community.
REFERENCES
- 1.Schwanzel-Fukuda M, Pfaff DW. Origin of luteinizing hormone-releasing hormone neurons. Nature. 1989;338:161–164. doi: 10.1038/338161a0. [DOI] [PubMed] [Google Scholar]
- 2.Mellon PL, Windle JJ, Goldsmith P, Pedula C, Roberts J, Weiner RI. Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron. 1990;5:1–10. doi: 10.1016/0896-6273(90)90028-e. [DOI] [PubMed] [Google Scholar]
- 3.Whyte DB, Lawson MA, Belsham DD, Eraly SA, Bond CT, Adelman JP, Mellon PL. A neuron-specific enhancer targets expression of the gonadotropin-releasing hormone gene to hypothalamic neurosecretory neurons. Mol Endocrinol. 1995;9:467–477. doi: 10.1210/mend.9.4.7659090. [DOI] [PubMed] [Google Scholar]
- 4.Nelson SB, Lawson MA, Kelley CG, Mellon PL. Neuron-specific expression of the rat gonadotropin-releasing hormone gene is conferred by interactions of a defined promoter element with the enhancer in GT1–7 cells. Mol Endocrinol. 2000;14:1509–1522. doi: 10.1210/mend.14.9.0521. [DOI] [PubMed] [Google Scholar]
- 5.Lawson MA, MacConell LA, Kim J, Powl BT, Nelson SB, Mellon PL. Neuron-specific expression in vivo by defined transcription regulatory elements of the gonadotropin-releasing hormone gene. Endocrinology. 2002;143:1404–1412. doi: 10.1210/endo.143.4.8751. [DOI] [PubMed] [Google Scholar]
- 6.Clark ME, Mellon PL. The POU homeodomain transcription factor Oct-1 is essential for activity of the gonadotropin-releasing hormone neuron-specific enhancer. Mol Cell Biol. 1995;15:6169–6177. doi: 10.1128/mcb.15.11.6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lawson MA, Buhain AR, Jovenal JC, Mellon PL. Multiple factors interacting at the GATA sites of the gonadotropin-releasing hormone neuron-specific enhancer regulate gene expression. Mol Endocrinol. 1998;12:364–377. doi: 10.1210/mend.12.3.0082. [DOI] [PubMed] [Google Scholar]
- 8.Lawson MA, Whyte DB, Mellon PL. GATA factors are essential for activity of the neuron-specific enhancer of the gonadotropin-releasing hormone gene. Mol Cell Biol. 1996;16:3596–3605. doi: 10.1128/mcb.16.7.3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rave-Harel N, Givens ML, Nelson SB, Duong HA, Coss D, Clark ME, Hall SB, Kamps MP, Mellon PL. TALE homeodomain proteins regulate gonadotropin-releasing hormone gene expression independently and via interactions with Oct-1. J Biol Chem. 2004;279:30287–30297. doi: 10.1074/jbc.M402960200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Givens ML, Rave-Harel N, Goonewardena VD, Kurotani R, Berdy SE, Swan CH, Rubenstein JL, Robert B, Mellon PL. Developmental regulation of gonadotropin-releasing hormone gene expression by the MSX and DLX homeodomain protein families. J Biol Chem. 2005;280:19156–19165. doi: 10.1074/jbc.M502004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kelley CG, Lavorgna G, Clark ME, Boncinelli E, Mellon PL. The Otx2 homeoprotein regulates expression from the gonadotropin-releasing hormone proximal promoter. Mol Endocrinol. 2000;14:1246–1256. doi: 10.1210/mend.14.8.0509. [DOI] [PubMed] [Google Scholar]
- 12.Belsham DD, Mellon PL. Transcription factors Oct-1 and C/EBP β (CCAAT/enhancer binding protein-β) are involved in the glutamate/nitric oxide/cyclic guanosine 5′-monophosphate-mediated repression of gonadotropin-releasing hormone gene expression. Mol Endocrinol. 2000;14:212–228. doi: 10.1210/mend.14.2.0418. [DOI] [PubMed] [Google Scholar]
- 13.Gore AC, Roberts JL, Gibson MJ. Mechanisms for the regulation of gonadotropin-releasing hormone gene expression in the developing mouse. Endocrinology. 1999;140:2280–2287. doi: 10.1210/endo.140.5.6711. [DOI] [PubMed] [Google Scholar]
- 14.Gore AC, Oung T, Yung S, Flagg RA, Woller MJ. Neuroendocrine mechanisms for reproductive senescence in the female rat: gonadotropin-releasing hormone neurons. Endocrine. 2000;13:315–323. doi: 10.1385/ENDO:13:3:315. [DOI] [PubMed] [Google Scholar]
- 15.Zoeller RT, Young WS., 3rd Changes in cellular levels of messenger ribonucleic acid encoding gonadotropin-releasing hormone in the anterior hypothalamus of female rats during the estrous cycle. Endocrinology. 1988;123:1688–1689. doi: 10.1210/endo-123-3-1688. [DOI] [PubMed] [Google Scholar]
- 16.Selmanoff M, Shu C, Petersen SL, Barraclough CA, Zoeller RT. Single cell levels of hypothalamic messenger ribonucleic acid encoding luteinizing hormone-releasing hormone in intact, castrated, and hyperprolactinemic male rats. Endocrinology. 1991;128:459–466. doi: 10.1210/endo-128-1-459. [DOI] [PubMed] [Google Scholar]
- 17.He JR, Molnar J, Barraclough CA. Morphine amplifies norepinephrine (NE)-induced LH release but blocks NE-stimulated increases in LHRH mRNA levels: comparison of responses obtained in ovariectomized, estrogen-treated normal and androgen-sterilized rats. Brain Res Mol Brain Res. 1993;20:71–78. doi: 10.1016/0169-328x(93)90111-2. [DOI] [PubMed] [Google Scholar]
- 18.Liaw JJ, Barraclough CA. N-methyl-D,L-aspartic acid differentially affects LH release and LHRH mRNA levels in estrogen-treated ovariectomized control and androgen-sterilized rats. Brain Res Mol Brain Res. 1993;17:112–118. doi: 10.1016/0169-328x(93)90079-5. [DOI] [PubMed] [Google Scholar]
- 19.Lee BJ, Kim K, Cho WK. Activation of intracellular pathways with forskolin and phorbol ester increases LHRH mRNA level in the rat hypothalamus superfused in vitro. Brain Res Mol Brain Res. 1990;8:185-–191. doi: 10.1016/0169-328x(90)90015-6. [DOI] [PubMed] [Google Scholar]
- 20.Wetsel WC, Eraly SA, Whyte DB, Mellon PL. Regulation of gonadotropin-releasing hormone by protein kinases A and C in immortalized hypothalamic neurons. Endocrinology. 1993;132:2360–2370. doi: 10.1210/endo.132.6.8504741. [DOI] [PubMed] [Google Scholar]
- 21.Bruder JM, Drebs WD, Nett TM, Wierman ME. Phorbol ester activation of the protein kinase C pathway inhibits gonadotropin-releasing hormone gene expression. Endocrinology. 1992;131:2552–2558. doi: 10.1210/endo.131.6.1446598. [DOI] [PubMed] [Google Scholar]
- 22.Bruder JM, Wierman ME. Evidence for transcriptional inhibition of GnRH gene expression by phorbol ester at a proximal promoter region. Mol Cell Endocrinol. 1994;99:177–182. doi: 10.1016/0303-7207(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 23.Eraly SA, Mellon PL. Regulation of GnRH transcription by protein kinase C is mediated by evolutionarily conserved, promoter-proximal elements. Mol Endocrinol. 1995;9:848–859. doi: 10.1210/mend.9.7.7476968. [DOI] [PubMed] [Google Scholar]
- 24.Sun W, Choe YS, Lee YJ, Kim K. Suppression of GnRH gene expression in GT1–1 hypothalamic neuronal cells: action of protein kinase C. Neuroreport. 1997;8:3541–3546. doi: 10.1097/00001756-199711100-00025. [DOI] [PubMed] [Google Scholar]
- 25.Eraly SA, Nelson SB, Huang KM, Mellon PL. Oct-1 binds promoter elements required for transcription of the gonadotropin-releasing hormone gene. Mol Endocrinol. 1998;12:469–481. doi: 10.1210/mend.12.4.0092. [DOI] [PubMed] [Google Scholar]
- 26.Berthelsen J, Kilstrup-Nielsen C, Blasi F, Mavilio F, Zappavigna V. The subcellular localization of PBX1 and EXD proteins depends on nuclear import and export signals and is modulated by association with PREP1 and HTH. Genes Dev. 1999;13:946–953. doi: 10.1101/gad.13.8.946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kilstrup-Nielsen C, Alessio M, Zappavigna V. PBX1 nuclear export is regulated independently of PBX-MEINOX interaction by PKA phosphorylation of the PBC-B domain. EMBO J. 2003;22:89–99. doi: 10.1093/emboj/cdg010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kelley CG, Givens ML, Rave-Harel N, Nelson SB, Anderson S, Mellon PL. Neuron-restricted expression of the rat gonadotropin-releasing hormone gene is conferred by a cell-specific protein complex that binds repeated CAATT elements. Mol Endocrinol. 2002;16:2413–2425. doi: 10.1210/me.2002-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Givens ML, Kurotani R, N R-H, Miller NLG, Mellon PL. Phylogenetic footprinting reveals functional upstream regions of the gonadotropin-releasing hormone gene that enhance cell-specific expression. Mol Endocrinol. 2004;18:2950–2966. doi: 10.1210/me.2003-0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Segil N, Roberts SB, Heintz N. Mitotic phosphorylation of the Oct-1 homeodomain and regulation of Oct-1 DNA binding activity. Science. 1991;254:1814–1816. doi: 10.1126/science.1684878. [DOI] [PubMed] [Google Scholar]
- 31.Kapiloff MS, Farkash Y, Wegner M, Rosenfeld MG. Variable effects of phosphorylation of Pit-1 dictated by the DNA response elements. Science. 1991;253:786–789. doi: 10.1126/science.1652153. [DOI] [PubMed] [Google Scholar]
- 32.Subramaniam N, Cairns W, Okret S. Glucocorticoids repress transcription from a negative glucocorticoid response element recognized by two homeodomain-containing proteins, Pbx and Oct-1. J Biol Chem. 1998;273:23567–23574. doi: 10.1074/jbc.273.36.23567. [DOI] [PubMed] [Google Scholar]
- 33.Bürglin TR. The PBC domain contains a MEINOX domain: coevolution of Hox and TALE homeobox genes? Dev Genes Evol. 1998;208:113–116. doi: 10.1007/s004270050161. [DOI] [PubMed] [Google Scholar]
- 34.Kamps MP, Look AT, Baltimore D. The human t(1; 19) translocation in pre-B ALL produces multiple nuclear E2A-Pbx1 fusion proteins with differing transforming potentials. Genes Dev. 1991;5:358–368. doi: 10.1101/gad.5.3.358. [DOI] [PubMed] [Google Scholar]
- 35.Asahara H, Dutta S, Kao HY, Evans RM, Montminy M. Pbx-Hox heterodimers recruit coactivator-core-pressor complexes in an isoform-specific manner. Mol Cell Biol. 1999;19:8219–8225. doi: 10.1128/mcb.19.12.8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang H, Hu G, Wang H, Sciavolino P, Iler N, Shen MM, Abate-Shen C. Heterodimerization of Msx and Dlx homeoproteins results in functional antagonism. Mol Cell Biol. 1997;17:2920–2932. doi: 10.1128/mcb.17.5.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park GT, Denning MF, Morasso MI. Phosphorylation of murine homeodomain protein Dlx3 by protein kinase C. FEBS Lett. 2001;496:60–65. doi: 10.1016/s0014-5793(01)02398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shah BH, Soh JW, Catt KJ. Dependence of gonadotropin-releasing hormone-induced neuronal MAPK signaling on epidermal growth factor receptor transactivation. J Biol Chem. 2003;278:2866–2875. doi: 10.1074/jbc.M208783200. [DOI] [PubMed] [Google Scholar]
- 39.Wu TJ, Mani SK, Glucksman MJ, Roberts JL. Stimulation of luteinizing hormone-releasing hormone (LHRH) gene expression in GT1–7 cells by its metabolite, LHRH-(1–5) Endocrinology. 2005;146:280–286. doi: 10.1210/en.2004-0560. [DOI] [PubMed] [Google Scholar]
- 40.Roy D, Belsham DD. Melatonin receptor activation regulates GnRH gene expression and secretion in GT1–7 GnRH neurons. Signal transduction mechanisms. J Biol Chem. 2002;277:251–258. doi: 10.1074/jbc.M108890200. [DOI] [PubMed] [Google Scholar]
- 41.Box GEP, Cox DR. An analysis of transformations. J Royal Stat Soc B. 1964;26:211–252. [Google Scholar]
- 42.Schreiber E, Matthias P, Müller M, Schaffner W. Rapid detection of octamer binding proteins with mini-extracts prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rave-Harel N, Miller NLG, Givens ML, Mellon PL. The Groucho-related gene family regulates the gonadotropin-releasing hormone gene through interaction with the homeodomain proteins MSX1 and OCT1. J Biol Chem. 2005;280:30975–30983. doi: 10.1074/jbc.M502315200. [DOI] [PMC free article] [PubMed] [Google Scholar]