

Abstract
Carbon–fluorine bond formation by transition metal catalysis is difficult and only few methods for the synthesis of aryl fluorides have been developed. All reported transition metal-catalyzed fluorination reactions for the synthesis of functionalized arenes are based on palladium. Here we present silver catalysis for carbon–fluorine bond formation. Our report is the first example of the use of the transition metal silver to form carbon–heteroatom bonds by cross-coupling catalysis. The functional group tolerance and substrate scope presented here have not been demonstrated for any other fluorination reaction to date.
Introduction
Fluorinated aromatic compounds are used as pharmaceuticals, agrochemicals, materials, and tracers for positron emission tomography (PET).1 Fluorine incorporation often improves the properties of functional molecules; for example, fluorine substituents can increase the metabolic stability and the rate and extent of blood-brain barrier penetration of pharmaceuticals.2 Selective fluorination is important but challenging, especially when the desired fluorinated molecules are complex, and carbon–fluorine bond formation must occur at a late stage of their synthesis.3 In this manuscript we present a silver-catalyzed late-stage fluorination reaction of complex small molecules, including polypeptides, polyketides, and akaloids (eq 1). Our report is the first example of silver catalysis for carbon–heteroatom bond formation by cross-coupling chemistry. We propose that silver-catalyzed late-stage fluorination proceeds by a mechanism distinct from conventional cross-coupling chemistry.
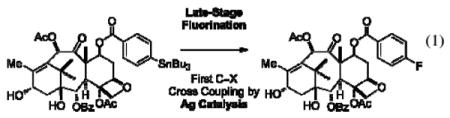 |
The synthesis of fluorinated arenes is challenging due to the properties of fluorine.4 Electrophilic fluorination is often unselective.5 Nucleophilic fluorination is complicated by strong hydrogen bonding and the high hydration energy of the fluoride anion, which results in low nucleophilicity of hydrated fluoride and high basicity of dry fluoride.6 Carbon–fluorine bond formation by transition metal catalysis is difficult,7 in part due to strong ionic metal–fluoride bonding, and only few examples have been reported.8 Reductive elimination, the product forming event in cross-coupling catalysis, becomes increasingly difficult and requires harsher reaction conditions in the bond formation series C–C, C–N, C–O, C–F.8d,9 Sanford8b and Yu8c have reported directed palladium-catalyzed electrophilic aromatic fluorination. The advantage of both transformations is the ability to convert C–H bonds into C–F bonds. Challenges include the high reaction temperature (120–150 °C), and the need for directing and blocking groups on the arene for regioselective fluorination. A breakthrough discovery in 2009 by Buchwald8d extended palladium-catalyzed cross-coupling chemistry to nucleophilic fluorination. The reaction temperature of 80–130 °C in combination with the basicity of fluoride, is a current challenge for the synthesis of molecules with protic functional groups and the synthesis of complex aryl fluorides by this method. In some cases, mixtures of regioisomers were observed. A general, functional group-tolerant late-stage fluorination of complex arenes has not yet been achieved by either nucleophilic or electrophilic fluorination reactions.
Silver is used in heterogeneous oxidative catalysis on industrial scale.10 In homogeneous catalysis, silver is typically used as non-redox active Lewis acid, and its redox catalysis is not well understood.11 Cross-coupling catalysis most commonly relies on mononuclear complexes derived from transition metals like palladium, with predictable two-electron redox pathways. Silver undergoes one-electron redox chemistry and previously has not been employed for carbon–heteroatom bond formation by cross-coupling catalysis.
Results and Discussions
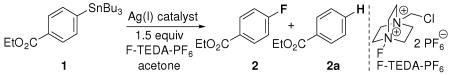
We have previously reported the electrophilic fluorination of arylsilver complexes, which required two to three equivalents of silver salt to obtain synthetically useful yields of aryl fluorides.12 Here, we address the conceptual disadvantage of our earlier results and present silver catalysis. The development of silver catalysis was complicated by protodestannylation as shown in Table 1. Addition of the electrophilic fluorinating reagent F-TEDA-PF6 to arylstannane 1 in the presence of 10 mol% silver catalyst produced 68% of the undesired protodestannylated product 2a and only 30% of the desired fluorinated product 2.
Table 1.
Silver catalysis for carbon–fluorine bond formation.
 | |||||
|---|---|---|---|---|---|
| Ag(I) catalyst |
Base (2.0 equiv) |
Additive | Temp time |
Yielda 2 |
Yielda 2a |
| 10 mol% AgOTf |
none | none | 65 °C 3 h |
30% | (68%) |
| 5 mol% Ag2O |
NaHCO3 | none | 65 °C 5 h |
87% | (9%) |
| 5 mol% Ag2O |
NaHCO3 | 1.0 equiv NaOTf | 65 °C 3 h |
90% | (5%) |
|
5 mol% Ag2O |
NaHCO3 |
1.0 equiv NaOTf 5.0 equiv MeOH |
65 °C 3 h |
92% | (2%) |
| 1 mol% Ag2O |
NaHCO3 | 1.0 equiv NaOTf | 90 °C 18 h |
92% | (2%) |
| 1 mol% Ag2O |
NaHCO3 | 1.0 equiv NaOTf 5.0 equiv MeOH |
90 °C 18 h |
75% | (20%) |
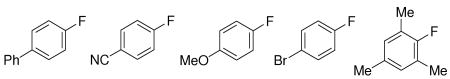 | |||||
| 3: 88%,b (4%) 4: 89%,b (2%) 5: 76%,b (3%) 6: 77%,b (5%) 7: 78%,b (5%) | |||||
Yields were determined by 19F NMR and 1H NMR with 1-fluoro-3-nitrobenzene as a standard. Yields of protodestannylated products are given in parentheses.
5.0 mol% Ag2O, 1.5 equiv F-TEDA-PF6, 2.0 equiv NaHCO3, 1.0 equiv NaOTf, 5.0 equiv MeOH, acetone, 65 °C, 4 h.
We hypothesized that tributyltin triflate, generated by transmetallation from arylstannanes to AgOTf, is hydrolyzed in situ to form triflic acid (TfOH),13 which is responsible for protodestannylation. Fluorination of purified arylsilver complexes, without tributyltin triflate contamination, did not afford protodestannylated byproduct. Because common drying reagents did not suppress protodestannylation, we evaluated bases to neutralize acids that may form during catalysis and identified sodium bicarbonate (NaHCO3), which, unlike many other bases (see Supporting Information), was compatible with F-TEDA-PF6 under the reaction conditions. The use of NaHCO3 had the additional, though unanticipated, benefit that most of the formed tributyltin bicarbonate precipitated and could be conveniently removed by filtration. Precipitation of the tin residue facilitated product purification, which is often cumbersome when organotin reagents are used.14
Several soluble Ag(I) salts catalyzed fluorination with equal efficiency but silver oxide (Ag2O) was selected as the catalyst of choice because it afforded the fluorinated products in the same range of yields as other silver salts and, in contrast to many soluble Ag(I) salts, is inexpensive ($1.0/g) and not hygroscopic. The addition of NaOTf to the Ag2O-catalyzed reaction increased the rate of fluorination, possibly due to the conversion of insoluble Ag2O into a soluble Ag(I) compound under the reaction conditions. While Ag2O is not soluble in acetone itself, we confirmed the presence of an active, soluble silver catalyst: Upon filtration of the reaction mixture after five hours, followed by addition of the same amount of all reagents except silver source, 80% of fluorinated product was observed, based on total added stannane. We determined gravimetrically by precipitation of AgCl that 92% of the silver remained in solution as Ag(I) after fluorination. In the absence of silver catalyst, less than 5% fluorination was observed at 90 °C. The catalyst loading could be reduced to 1 mol% Ag2O with no loss in yield; however, longer reaction time and higher reaction temperature were required.
Fluorination using 5 mol% of Ag2O afforded electron deficient (4), electron rich (5), halogenated (6), and ortho, ortho disubstituted arenes (7). The addition of five equivalents of methanol decreased the amount of protodestannylated product formed to 2–5% when 5 mol% of Ag2O was used; addition of MeOH reduced the yield of fluorinated product at 1 mol% catalyst loading. The role of MeOH is currently not understood. All fluorination reactions were performed under ambient atmosphere and in reagent grade acetone as solvent. The reaction temperature of 65 °C is slightly above the boiling temperature of acetone and therefore, reactions were performed in closed vessels. The reaction could also be performed at reflux with yields decreased by about 10% and reaction times three hours longer.
We selected a group of small molecules that contain several functional groups and found that the Ag2O-catalyzed fluorination reaction is general beyond simple arenes (Table 2). Biologically active molecules such as carbohydrates, peptides, polyketides, and alkaloids are compatible with fluorination, and molecules derived from complex natural products such as taxol and rifamycin were successfully fluorinated. It is worth mentioning that fluorination can proceed in the presence of several functional groups, including a vinyl ether, a dienone, alcohols, an allylic alcohol, ethers and esters, and an oxetane. To date, no other fluorination reaction has been shown to have a substrate scope as broad as shown in here. The practical reaction conditions allowed for the gram-scale synthesis of dipeptide 15 in 92% yield. For all fluorinated molecules shown in Table 2, carbon–fluorine bond formation was performed as the final synthetic step. The yield of protodestannylated byproducts was less than 10% in all cases, which simplified purification of the fluorinated molecules. Fluorination always occurred regiospecifically; no other regioisomers were detected. General regiospecificity of the silver-catalyzed fluorination is an additional advantage over the known palladium-catalyzed fluorination reactions.
Table 2.
Ag2O-catalyzed fluorination of complex small molecules.
 R′ = Me, Bu |
 |
 |
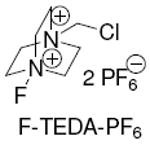 F-TEDA-PF6 |
|
|---|---|---|---|---|
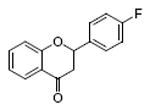 flavanone 90%, 8 |
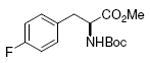 Boc-Tyr-OMe 85%, 9 |
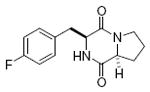 maculosin 78%, 10 |
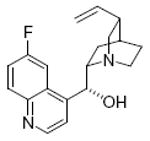 quinine 75%, 11a |
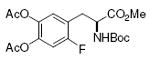 DOPA 78%, 12 |
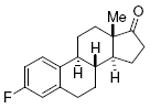 estrone 81%, 13 |
Leu-enkephalin 83%, 14 |
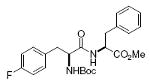 Boc-Tyr-Phe-OMe 92%, 15 |
||
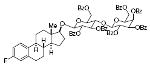 estrol-17-β-D-lactosde heptabenzoate 80%, 16 |
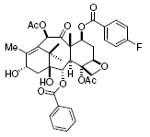 taxol 72%, 17 |
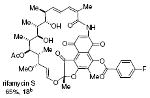 rifamycin S 65%, 18b |
||
20 mol% of AgOTf, 2 equiv of NaOTf and 2 equiv of F-TEDA-PF6 were used at 90 °C.
20 mol% of AgOTf, 2 equiv of NaOTf, and 5 equiv of MeOH was used. The names under the molecules in Table 2 refer to the parent molecules and not to fluorinated analogs shown.
Limitations of the Ag-catalyzed fluorination reaction are shown in Chart 1. We identified that basic functional groups that react with the electrophilic fluorinating reagent unproductively are often not tolerated. Certain amines (19) may react with F-TEDA-PF6 to form N-fluoro compounds that subsequently eliminate HF. Amines that lack β-hydrogen atoms for E2 elimination of HF participate successfully in fluorination: For example, both the quinoline and the bridgehead tertiary amine of quinine (11) did not impede aryl fluorination. In contrast to sulfides, which likely react as nucleophiles with F-TEDA-PF6, sulfones are compatible with the reaction conditions (20, 21). Fluorination of substrates with protic functional groups that can be deprotonated by NaHCO3 proved problematic (22). Carboxylate anions generated by the deprotonation of carboxylic acids may react with AgOTf to form silver carboxylates, which are less effective silver salts for silver-mediated fluorination of arylstannanes. Alcohols, on the other hand, did not affect successful fluorination (23).
Chart 1.
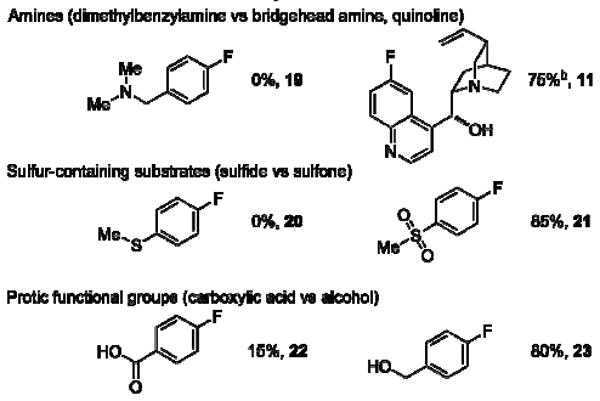
Limitations of the presented fluorination reaction.a a) The fluorination was attempted from the corresponding arylstannane: 5.0 mol% Ag2O, 1.5 equiv F-TEDA-PF6, 2.0 equiv NaHCO3, 1.0 equiv NaOTf, 5.0 equiv MeOH, acetone, 65 °C, 4 h. b) 20 mol% of AgOTf, 2 equiv of NaOTf and 2 equiv of F-TEDA-PF6 were used at 90 °C.
Late-stage fluorination is valuable for complex target molecules that are otherwise more challenging to obtain. We selected the pharmaceutical ezetimibe and the natural product strychnine to illustrate the advantage of late-stage fluorination compared to conventional synthesis to access complex fluorinated molecules. Fluoro-deoxy-ezetimibe (26) was prepared in a three-step sequence from available ezetimibe as shown in Scheme 1. De novo synthesis of the fluorinated molecule 26 would require several more synthetic steps.15 The structure of strychnine (27) is a challenge for the Ag-catalyzed fluorination reaction because it does not possess a phenol functionality, which is a convenient functional group for late-stage fluorination as shown for ezetimibe (24). In addition, strychnine is an alkaloid, which contains a tertiary amine that cannot be readily protected as an amide or carbamate. Indeed, fluorination of strychnine-derived arylstannane 29, obtained via known iodination of strychnine16 followed by stannylation, did not afford any fluoro-strychnine (30), likely due to competing N-fluorination. However, in situ N-benzylation, followed by Ag-catalyzed fluorination of the resulting strychnine-derived ammonium salt proceeded in 75% yield. Subsequent hydrogenolysis yielded previously unknown fluoro-strychnine (30) in 60% overall yield from 29 by late-stage fluorination.
Scheme 1.

Late-stage fluorination of ezetimibe and strychnine. a: i) PhNTf2, Et3N, DMAP, CH2Cl2, 23 °C, 95%; ii) LiCl, 5 mol% Pd(PPh3)4, (n-Bu3Sn)2, dioxane, 100 °C, 50%; b: Ag2O (5 mol%), NaHCO3, NaOTf, F-TEDA-PF6, acetone, 65 °C, 90%; c: Et3N, 5 mol% Pd(PPh3)4, (n-Bu3Sn)2, dioxane, 100 °C, 34%; d: BnBr, acetone; AgOTf; Ag2O (5 mol%), NaHCO3, NaOTf, F-TEDA-PF6, 65 °C; e: 1,4-cyclohexadiene, Pd/C, MeOH, 40 °C, 60% for two steps.
Mechanistically, silver redox catalysis is much less understood than, for example, palladium redox catalysis, in part due to its scarcity.11 Cross-coupling catalysis most commonly relies on mononuclear complexes derived from transition metals like palladium, which typically undergoes predictable two-electron redox pathways during reductive elimination, while silver undergoes one-electron redox chemistry.
We propose the catalytic cycle shown in Scheme 2 for silver-catalyzed fluorination: Aryl transmetallation from tin to Ag(I) can afford arylsilver species such as I,12a,17 possibly aggregated with additional Ag(I) under conditions of catalysis. We suggest that subsequent Ag-based fluorination affords a high-valent arylsilver fluoride complex such as II. A multinuclear silver complex, such as dinuclear complex II, may be responsible for carbon–fluorine bond formation because two-electron redox processes can be facilitated by metal-metal cooperation, in which more than one metal contributes to the redox process.18 Carbon–fluorine reductive elimination from potential intermediate II provides aryl fluoride and regenerates the Ag(I) catalyst.
Scheme 2.

Proposed mechanism for silver-catalyzed fluorination.
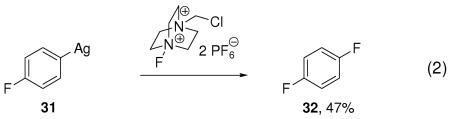
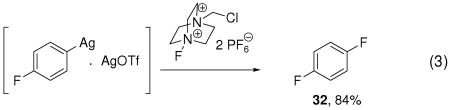
Indirect evidence for the presence of a multinuclear arylsilver complex was obtained by fluorination of complex 31 with and without additional Ag(I) (eqs 2, 3). Fluorination of the mononuclear arylsilver complex 31 afforded aryl fluoride 32 in 47% yield, while addition of one additional equivalent of AgOTf to 31 afforded 32 in 84% yield. We were unable to isolate a high-valent Ag complex, but our results imply that a direct cleavage of the carbon–silver bond of the aryl Ag(I) complex I by carbon fluorination does not occur: Addition of 25 equivalents of water to the fluorination reaction of tributylarylstannane afforded phenols as byproduct, as shown in the dashed box in Scheme 2. The formation of phenols is consistent with fluoride-hydroxide exchange at a high-valent silver complex, followed by carbon–oxygen reductive elimination. Addition of the radical inhibitor butylated hydroxytoluene (BHT) had no effect on the yield of fluorination, which is also consistent with a high-valent silver intermediate.
 |
 |
Conclusion
We present a silver-catalyzed fluorination reaction, which can be applied to complex small molecules. The functional group tolerance and substrate scope presented here has not been demonstrated for any other fluorination reaction to date. A current disadvantage of the method is the need for the preparation of arylstannanes as starting materials. In addition, the fluorination reaction reported by Buchwald can afford aryl fluorides from triflates directly, while our fluorination reaction requires an additional synthetic step from triflate or halide to stannane. On the other hand, advantages of our fluorination reaction compared to the Buchwald method are milder reaction conditions, stereospecificity, and tolerance toward protic functional groups, which make our fluorination reaction particularly useful for late stage fluorination of complex small molecules, which may not be suitable for other fluorination reactions.
The Ag-catalyzed fluorination reaction likely proceeds through a mechanism distinct from traditional cross-coupling mechanisms. Noteworthy is the C–F bond formation under comparatively mild reaction conditions, by reductive elimination from a putative multinuclear high-valent silver fluoride complex. Silver catalysis, with potential redox synergy between two or more metals, is a new avenue for carbon–heteroatom cross-coupling chemistry.
Supplementary Material
Acknowledgments
This work was supported by NSF (CHE-0952753) and NIH-NIGMS (GM088237). We thank Air Products and Chemicals, Inc. for a generous donation of F-TEDA-BF4.
Footnotes
Supporting Information Available: Experimental procedures and spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Yamazaki T, Taguchi T, Ojima I. In: Fluorine in Medicinal Chemistry and Chemical Biology. Ojima I, editor. Wiley-Blackwell; United Kingdom: 2009. [Google Scholar]; (b) Jeschke P. Chembiochem. 2004;5:570. doi: 10.1002/cbic.200300833. [DOI] [PubMed] [Google Scholar]; (c) Babudri F, Farinola GM, Naso F, Ragni R. Chem Commun. 2007:1003. doi: 10.1039/b611336b. [DOI] [PubMed] [Google Scholar]
- 2.Müller K, Faeh C, Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 3.(a) Wilkinson JA. Chem Rev. 1992;92:505. [Google Scholar]; (b) O'Hagan D.Chem Soc Rev 20083730818197347 [Google Scholar]; (c) Furuya T, Klein JEMN, Ritter T. Synthesis. 2010;11:1804. doi: 10.1055/s-0029-1218742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For selected fluorination reactions, see:; (a) Balz G, Schiemann G. Ber Deutsch Chem Ges. 1927;60:1186. [Google Scholar]; (b) Adams DJ, Clark JH. Chem Soc Rev. 1999;28:225. [Google Scholar]; (c) Sun H, DiMagno SG. Angew Chem Int Ed. 2006;45:2720. doi: 10.1002/anie.200504555. [DOI] [PubMed] [Google Scholar]; (d) Sandford G. J Fluorine Chem. 2007;128:90. [Google Scholar]; (e) Yamada S, Gavryushin A, Knochel P. Angew Chem Int Ed. 2010;49:2215. doi: 10.1002/anie.200905052. [DOI] [PubMed] [Google Scholar]; (f) Anbarasan P, Neumann H, Beller M. Angew Chem Int Ed. 2010;49:2219. doi: 10.1002/anie.200905855. [DOI] [PubMed] [Google Scholar]
- 5.Baudoux J, Cahard D. Org React. 2007;69:347. [Google Scholar]
- 6.Furuya T, Kuttruff CA, Ritter T. Curr Opin Drug Disc Dev. 2008;11:803. [PubMed] [Google Scholar]
- 7.For stoichiometric and catalytic transition metal-mediated C–F bond formation reactions, see:; (a) Tius MA, Kawakami JK. Synth Commun. 1992;22:1461. [Google Scholar]; (b) Tius MA, Kawakami JK. Synlett. 1993:207. [Google Scholar]; (c) Tius MA, Kawakami JK. Tetrahedron. 1995;51:3997. [Google Scholar]; (d) Akana JA, Bhattacharyya KX, Muller P, Sadighi JP. J Am Chem Soc. 2007;129:7736. doi: 10.1021/ja0723784. [DOI] [PubMed] [Google Scholar]; (e) Kaspi AW, Yahav-Levi A, Goldberg I, Vigalok A. Inorg Chem. 2008;47:5. doi: 10.1021/ic701722f. [DOI] [PubMed] [Google Scholar]; (f) Furuya T, Kaiser HM, Ritter T. Angew Chem, Int Ed. 2008;47:5993. doi: 10.1002/anie.200802164. [DOI] [PubMed] [Google Scholar]; (g) Gorske BC, Mbofana CT, Miller SJ. Org Lett. 2009;11:4318. doi: 10.1021/ol9016782. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Wu T, Yin G, Liu G. J Am Chem Soc. 2009;131:16355. doi: 10.1021/ja9076588. [DOI] [PubMed] [Google Scholar]
- 8.(a) Subramanian MA, Manzer LE. Science. 2002;297:1665. doi: 10.1126/science.1076397. [DOI] [PubMed] [Google Scholar]; (b) Hull KL, Anani WQ, Sanford MS. J Am Chem Soc. 2006;128:7134. doi: 10.1021/ja061943k. [DOI] [PubMed] [Google Scholar]; (c) Wang X, Mei TS, Yu JQ. J Am Chem Soc. 2009;131:7520. doi: 10.1021/ja901352k. [DOI] [PubMed] [Google Scholar]; (d) Watson DA, Su M, Teverovskiy G, Zhang Y, García-Fortanet J, Kinzel T, Buchwald SL. Science. 2009;325:1661. doi: 10.1126/science.1178239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) de Meijere A, Diederich F. Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH; Weinheim: 2004. [Google Scholar]; (b) Muci AR, Buchwald SL. In: Cross-Coupling Reactions. Miyaura N, editor. Springer; Berlin: 2002. [Google Scholar]; (c) Hartwig JF. Nature. 2008;455:314. doi: 10.1038/nature07369. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Furuya T, Ritter T. J Am Chem Soc. 2008;130:10060. doi: 10.1021/ja803187x. [DOI] [PubMed] [Google Scholar]; (e) Ball ND, Sanford MS. J Am Chem Soc. 2009;131:3796. doi: 10.1021/ja8054595. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Furuya T, Benitez D, Tkatchouk E, Strom AE, Tang P, Goddard WA, III, Ritter T. J Am Chem Soc. 2010;132:3793. doi: 10.1021/ja909371t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Linic S, Barteau MA. In: Handbook of Heterogeneous Catalysis. Ertl G, Knözinger H, Schüth F, Weitkamp J, editors. Wiley-VCH; Weinheim: 2008. [Google Scholar]
- 11.(a) Naodovic M, Yamamoto H. Chem Rev. 2008;108:3132. doi: 10.1021/cr068413r. [DOI] [PubMed] [Google Scholar]; (b) Weibel JM, Blanc A, Pale P. Chem Rev. 2008;108:3149. doi: 10.1021/cr078365q. [DOI] [PubMed] [Google Scholar]; (c) Álvarez-Corral M, Muñoz-Dorado M, Rodríguez-García I. Chem Rev. 2008;108:3174. doi: 10.1021/cr078361l. [DOI] [PubMed] [Google Scholar]; (d) Yamamoto Y. Chem Rev. 2008;108:3199. doi: 10.1021/cr078359u. [DOI] [PubMed] [Google Scholar]
- 12.(a) Furuya T, Strom AE, Ritter T. J Am Chem Soc. 2009;131:1662. doi: 10.1021/ja8086664. [DOI] [PubMed] [Google Scholar]; (b) Furuya T, Ritter T. Org Lett. 2009;11:2860. doi: 10.1021/ol901113t. [DOI] [PubMed] [Google Scholar]
- 13.Mehring M, Nolde C, Schürmann M. Inorg Chim Acta. 2009;362:745. [Google Scholar]
- 14.Davis AG, Gielen M, Pannell KH, Tiekink ERT. Tin Chemistry–Fundamentals, Frontiers and Applications. Wiley; United Kindom: 2008. [Google Scholar]
- 15.Sasikala CHVA, Reddy Padi P, Sunkara V, Ramayya P, Dubey PK, Bhaskar Rao Uppala V, Praveen C. Org Proc Res Dev. 2009;13:907. [Google Scholar]
- 16.Rosenmund P. Chem Ber. 1961;94:3342. [Google Scholar]
- 17.Krause E, Schmitz M. Ber Deutsch Chem Ges. 1919;52:2150. [Google Scholar]
- 18.(a) Powers DC, Ritter T. Nat Chem. 2009;1:302. doi: 10.1038/nchem.246. [DOI] [PubMed] [Google Scholar]; (b) Powers DC, Geibel MAL, Klein JEMN, Ritter T. J Am Chem Soc. 2009;131:17050. doi: 10.1021/ja906935c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.