

Abstract
Loss-of-function mutations of ABCC6 cause pseudoxanthoma elasticum (PXE). This Mendelian disorder is characterized by elastic calcification leading to dermal, ocular, and cardiovascular symptoms like coronary artery disease (CAD) and stroke. Although PXE is a recessive disease, microscopic dermal lesions, serum alterations, and higher anecdotal incidence of stroke or CAD among carriers were reported. Here we investigated the association of the c.3421C>T loss-of-function mutation of ABCC6 and CAD and stroke. A previous study demonstrated the association of the c.3421C>T mutation with CAD; however, the frequency found in the control population was unexpectedly high, contradicting, thus, the prevalence of PXE. In the present study, genomic DNA from 749 healthy blood donors was used as control, while 363 and 361 patients suffering from stroke and CAD were investigated, respectively. One carrier was found in our control group, which is in accordance with the reported prevalence of this mutation. No significant association was found between carrier status and stroke in our cohort. In contrast, a significant association of carrier status and CAD was observed (5/361 carriers: p = 0.016, odds ratio [OR] = 10.5). We propose that carriers of ABCC6 loss-of-function mutations benefit from CAD prevention therapy.
Introduction
It has been demonstrated that recessive mutations in the ABCC6 gene are responsible for the development of pseudoxanthoma elasticum (PXE; OMIM 264800; www.pxe.org) (Bergen et al., 2000; Le Saux et al., 2000; Ringpfeil et al., 2000), a rare Mendelian disorder. To date, approximately 180 PXE-associated mutations have been published (Plomp et al., 2008). These mutations were identified mainly in the coding region; although they can be found throughout the gene, it has been observed that they cluster in various functionally important parts of the protein (Fulop et al., 2009) or in genetically fragile regions of the gene. The most frequent mutation, accounting for 20–30% of all mutations, is the c.3421C>T nucleotide change leading to the R1141X nonsense mutation (Pfendner et al., 2008). A large deletion between exons 23 and 29 is the second most frequent mutation with 5–20% prevalence in various reports. Finally, the 5′ end of the gene is also genetically fragile due to the various potential gene conversion events between the 99% identical and expressed nonprocessed pseudogenes and the functional ABCC6 gene (Aranyi et al., 2005; Symmons et al., 2008).
ABCC6 codes for MRP6, an adenosine triphosphate (ATP)-binding cassette transporter protein that is most probably involved in transmembrane transport of presently unknown low molecular weight organic anion(s), while the pathomechanism of the disease remains elusive. At the microscopic level, PXE leads to the calcification and fragmentation of elastic fibers (Lebwohl et al., 1994; Struk et al., 1997). Patients suffer from symptoms affecting the skin (predominantly yellow papules at the flexural area, which may coalesce over time), the eye, and the cardiovascular system (Goodman et al., 1963) like hypertonia, intermittent claudication, ischemic stroke, and myocardial infarction [for recent reviews, see Pfendner et al. (2008) and Li et al. (2009)]. Although heterozygote carriers only rarely present the symptoms of PXE, some subclinical signs due to the loss of one functional ABCC6 allele can be often observed (Bergen, 2006; Ringpfeil et al., 2006; Pfendner et al., 2008). For instance, at the microscopic level, some foci of abnormal elastic mineralization can be detected (Martin et al., 2007; Pfendner et al., 2008). Further, two important factors of ectopic calcification, namely, matrix Gla protein and fetuin-A, are present at a significantly lower level in serum in both PXE patients and carriers, than in unaffected wild-type individuals (Hendig et al., 2006, 2008). The existence of these subclinical symptoms among carriers may explain the anecdotal reports on the higher incidence of ischemic stroke and coronary artery disease (CAD) in this population.
Although the cardiovascular symptoms are not well documented at the epidemiologic level due to the high interindividual variability of the PXE phenotype and the rarity of the disease, several mouse models also show that the cardiovascular system is affected by the loss-of-function mutations of ABCC6. One of them is the Dyscalc I mouse, which shows dystrophic aortic and cardiac calcification contributing to myocardial calcification. This phenotype was shown to be due to mutations in the Abcc6 gene (Meng et al., 2007; Aherrahrou et al., 2008). Two Abcc6 knock-out strains were generated, both characterized by general soft tissue and medium/small arterial calcification phenotype (Gorgels et al., 2005; Klement et al., 2005).
This compelling evidence strongly suggests that ABCC6 is a good candidate to be a risk factor for CAD. Indeed, a strong correlation between a rare sequence variant of the ABCC6 gene (c.3421C>T leading to the R1141X nonsense mutation) and CAD has been demonstrated in a Dutch cohort (Trip et al., 2002). This result has been published several years ago, but no confirmation was reported since then although this risk factor might have important clinical relevance. Further, a surprisingly high frequency of the mutant allele was observed in the control population, raising doubts about the validity of this unique study on the association of ABCC6 mutation carrier status and CAD. While the prevalence of PXE has been reported to be between 1/25,000 and 1/100,000 (Struk et al., 1997; Gotting et al., 2004; Chassaing et al., 2005), the data of Trip et al. (2002) suggest a prevalence of 1/4000.
Altogether, in light of these data we decided to study the association of ABCC6 loss-of-function mutation status and two cardiovascular disorders: CAD and stroke. We analyzed the allele frequency (AF) of the c.3421C > T (R1141X) mutation in healthy blood donors, patients with CAD, and patients suffering from ischemic stroke. We confirmed that the carrier status of the c.3421C > T mutation is a risk factor for CAD and demonstrated that it is not for stroke.
Materials and Methods
Patients and healthy blood donors
Genomic DNA isolated from anticoagulated peripheral blood of 749 healthy blood donors (453 men and 296 women, 38.89 ± 11.64 years old) was used as the control. Only apparently healthy (checked by physical examination) adults without clinically manifest cardiovascular diseases, not taking medicines for chronic diseases, were subjected to the study. Consecutive patients (n = 363, 217 men and 146 women, 59.66 ± 15.14 years old) with acute ischemic stroke were included in the present study. The diagnosis and therapy of stroke in the Stroke Center are in accordance with current stroke guidelines (Adams et al., 2003); however, none of the patients received thrombolytic therapy. All patients underwent cranial computed tomography examinations, and patients with intracerebral hemorrhage were excluded from the study. Unrelated Caucasian patients (n = 361, 277 men and 84 women, 59.57 ± 8.52 years old) with severe CAD were also included in the present study. The diagnosis was based on clinical signs of stable or unstable angina pectoris, typical electrocardiogram changes, and signs of severe coronary stenosis confirmed by coronary angiography using Judkins' technique. The study was approved by the local ethics committee, and each individual signed an informed consent.
Detection of c.3421C>T mutation
The c.3421C>T mutation was investigated in the genomic DNA samples obtained from patients and healthy blood donors. We used the method described by Gotting et al. (2004). Briefly, we amplified a 307 bp DNA fragment of the ABCC6 gene containing the c3421 nucleotide. The primers used were as follows: LCU forward CTCCCATCCATCCTTCT and LCL reverse CCTCGCTACCATACAATATGA. The aTaq DNA Polymerase (Promega, Mannheim, Germany) was used for the amplification reaction carried out in a LightCycler Instrument (Roche, Mannheim, Germany). Anchor-labeled (GGCAGCACAGTGGTCCGG-6-Fam) and detector-labeled (Cy5.5-ATTCCGAACCCAGGCCC-P) oligonucleotides were used to generate a fluorescence resonance energy transfer (FRET) signal for Tm determination. To enhance the signal, the concentration ratio of the LCL/LCU primer was 3:1 (Szilvasi et al., 2005). The Tm of the wild type was 64°C, while that of the c.3421C>T mutated was 55°C. Negative (wild-type) and positive (heterozygote c3421C>T mutation carrier) controls were systematically included in the screening.
Statistical analysis
AFs (%) are presented with 95% confidence intervals (95% CI). To compare the frequencies of ABCC6 genotypes between the blood donor and patient groups, Fisher's exact test was used. OR and 95% CI were also calculated.
Calculation of the frequency of PXE
Based on data from literature, the frequency of c3421C>T mutation (AF) was considered to account for 25% of all PXE-causing mutations. Therefore, the total frequency of PXE-causing mutations in the control population is fourfold higher than that determined for the c3421C>T mutation. The following equation was used to calculate the prevalence of PXE (P) in the control population:
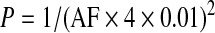 |
(1) |
The maximal and minimal prevalence (Pm) were calculated by taking into account the 95% CI of AF determined in the various studies:
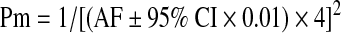 |
(2) |
Results and Discussion
Determination of the frequency of R1141X in the control population
Before investigating the eventual association of the R1141X loss-of-function mutation of ABCC6 with CAD or stroke, we determined the frequency of this allele in our control population consisting of 749 healthy blood donors. This seemed to be important, since the two existing studies on the AF of the R1141X mutation in healthy Caucasians reported significantly different results (p = 0.028; Table 1) (Trip et al., 2002; Gotting et al., 2004). While the prevalence calculated from the German cohort (Gotting et al., 2004) confirms the generally accepted prevalence of PXE, the prevalence of the disease calculated from the Dutch cohort serving as control group for the first association study with CAD (Trip et al., 2002) seems to be extremely high (1:4000). We found only 1 heterozygous out of 749 blood donors in our control cohort (Table 1). The corresponding frequency of the R1141X allele is almost identical to the German cohort (Gotting et al., 2004) (p = 0.495). We have also evaluated the prevalence and maximal prevalence values of PXE in the Hungarian population based on these data, which turned out to be 1:128000 and 1:15600, respectively. These data led us to the conclusion that the prevalence of PXE in our control group is in accordance with other reports and it can be used for further investigations.
Table 1.
Frequency of R1141X Heterozygotes in the Control Groups of the Three Different Studies
| Population | Dutch | German | Hungarian |
|---|---|---|---|
| n | 1057 | 910 | 749 |
| Carriers | 8 | 1 | 1 |
| AF 95% CI | 0.38 ± 0.27% | 0.06 ± 0.11% | 0.07 ± 0.13% |
n, cohort size; AF, allele frequency; CI, confidence interval.
Determination of the frequency of R1141X in patients with ischemic stroke
Various anecdotal reports exist about a higher incidence of stroke in PXE patients than in unaffected individuals. Further, Vanakker et al. (2008) reported recently the high incidence of stroke in a small group of carriers of loss-of-function ABCC6 mutations. This led us to investigate a cohort of stroke patients to determine whether the most frequent PXE-causing mutation, accounting for approximately 25% of all mutations, is a risk factor for this disease. A patient group composed of 363 individuals suffering from stroke (diagnosed by computed tomography) was compiled. Only one heterozygous individual was found in the patient group (Table 2). This frequency of heterozygotes among stroke patients is not significantly different from controls (p = 0.44); thus, the carrier status of R1141X mutation of the ABCC6 gene is not a risk factor for stroke. However, we should take into consideration the size of our cohort of stroke patients and the frequency of the mutated allele in the control population (see above) while evaluating these results. According to these parameters, the detection limit of a significant association is at least equal to an 8.25-fold increase of the frequency of carriers of the mutated allele among patients versus controls. Our data indicate that the frequency of carriers is only two times higher in patients, which is under that detection limit.
Table 2.
Frequency of R1141X Heterozygotes in the Stroke, Control, and Coronary Artery Disease Groups
| Cohort | Stroke | Blood donors | Coronary artery disease |
|---|---|---|---|
| n | 363 | 749 | 361 |
| Carriers | 1 | 1 | 5 |
| AF 95% CI | 0.14 ± 0.28% | 0.07 ± 0.13% | 0.69 ± 0.62% |
Determination of the frequency of R1141X in patients with CAD
Trip et al. found that the carrier status for the R1141X mutation of the ABCC6 gene is a risk factor for the development of CAD. We decided to perform a similar study on a CAD cohort of 361 patients compiled in Hungary, since this would be the first confirmation of the findings of Trip et al. (2002).
We determined the number of heterozygotes among the CAD patients and found five individuals carrying the R1141X mutation (Table 2). The statistical analysis of these data showed that the frequency of the mutant allele within the CAD patient group is significantly higher (p = 0.016, OR = 10.5, 95% CI = 1.22–90.30) than in the control group, suggesting that carrying a mutant ABCC6 allele is a strong risk factor for the development of CAD.
We also performed further statistical analyses of the CAD patient group, but none of the tests showed significant enrichment of the carrier status within a subgroup of patients (male/female or age stratification). We should emphasize, however, once more that—due to the low frequency of the mutant allele—the studied sample size is too small to detect small differences. Nevertheless, it should be noted that all carriers suffered from angina pectoris (two of them had also concomitant myocardial infarction), supporting the idea that loss-of-function mutations in one ABCC6 allele may lead to arterial disease similar to those observed in PXE patients.
The data presented here are particularly important since this is the first confirmation of Trip et al.'s results on an independent patient/control cohort where the controls reflect well the generally accepted prevalence of PXE. We do not know the reason of the difference between the AFs found in Trip et al.'s and our study, but it may be due either to the different mutation detection system or to the selection of patients and controls. Indeed, the Dutch controls were selected from the Cardiovascular Disease Risk Factor Monitoring Project, whereas our controls were selected from healthy blood donors. The two groups might have some differences in the occurrence of risk factors. Accordingly, Trip et al. found a significantly higher R1141X AF in the CAD group than we did. This might be due to the fact that they analyzed younger patients than we did. Nevertheless, both this and the previous study demonstrated a very strong correlation between CAD and the haploinsufficiency of ABCC6 due to the R1141X mutation. Confirmation of these data by the analysis of another loss-of-function mutation of ABCC6 is unfortunately impossible in this sample size, as other PXE-causing mutations are much less frequent (Pfendner et al., 2008). Our present study was the first to investigate the eventual association between stroke and ABCC6 haploinsufficiency, but we failed to show such an association. However, it should be noted that our setup did not allow to demonstrate an OR lower than 8.25. In conclusion, we have identified a strong but rare risk factor of CAD. Based on the frequency of ABCC6 R1141X allele, approximately 0.5% of the control Caucasian population is a carrier of a loss-of-function mutation concerning several hundred thousands of individuals. Although general screening would not be appropriate, but as most of them are relatives of PXE patients they can easily be identified. Therefore, we propose that carriers of loss-of-function mutations should be systematically identified in PXE families and should benefit from CAD prevention therapy.
Acknowledgments
Positive (heterozygote c3421C>T mutation carrier) control genomic DNA was kindly provided by Prof. Nicolas Chassaing, Toulouse, France. This work has been funded by the following grants: National Research Fund of Hungary NI68950 (G.K., A.V., and T.A.), NF72689 (Z.P.), PF63953 (H.A. and A.T.), PD79183 (T.A.), and the NIH Grant K69102NIH R01 AR055225-01A2 (A.V.).
Disclosure Statement
No competing financial interests exist.
References
- Adams HP., Jr. Adams RJ. Brott T. del Zoppo GJ, et al. Stroke Council of the American Stroke Association. Guidelines for the early management of patients with ischemic stroke: a scientific statement from the Stroke Council of the American Stroke Association. Stroke. 2003;34:1056–1083. doi: 10.1161/01.STR.0000064841.47697.22. [DOI] [PubMed] [Google Scholar]
- Aherrahrou Z. Doehring LC. Ehlers EM, et al. An alternative splice variant in Abcc6, the gene causing dystrophic calcification, leads to protein deficiency in C3H/He mice. J Biol Chem. 2008;283:7608–7615. doi: 10.1074/jbc.M708290200. [DOI] [PubMed] [Google Scholar]
- Aranyi T. Ratajewski M. Bardoczy V, et al. Identification of a DNA methylation-dependent activator sequence in the pseudoxanthoma elasticum gene, ABCC6. J Biol Chem. 2005;280:18643–18650. doi: 10.1074/jbc.M501139200. [DOI] [PubMed] [Google Scholar]
- Bergen AA. Pseudoxanthoma elasticum: the end of the autosomal dominant segregation myth. J Invest Dermatol. 2006;126:704–705. doi: 10.1038/sj.jid.5700129. [DOI] [PubMed] [Google Scholar]
- Bergen AA. Plomp AS. Schuurman EJ, et al. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat Genet. 2000;25:228–231. doi: 10.1038/76109. [DOI] [PubMed] [Google Scholar]
- Chassaing N. Martin L. Calvas P, et al. Pseudoxanthoma elasticum: a clinical, pathophysiological and genetic update including 11 novel ABCC6 mutations. J Med Genet. 2005;42:881–892. doi: 10.1136/jmg.2004.030171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulop K. Barna L. Symmons O, et al. Clustering of disease-causing mutations on the domain-domain interfaces of ABCC6. Biochem Biophys Res Commun. 2009;379:706–709. doi: 10.1016/j.bbrc.2008.12.142. [DOI] [PubMed] [Google Scholar]
- Goodman RM. Smith EW. Paton D, et al. Pseudoxanthoma elasticum: a clinical and histopathological study. Medicine (Baltim) 1963;42:297–334. [PubMed] [Google Scholar]
- Gorgels TG. Hu X. Scheffer GL, et al. Disruption of Abcc6 in the mouse: novel insight in the pathogenesis of pseudoxanthoma elasticum. Hum Mol Genet. 2005;14:1763–1773. doi: 10.1093/hmg/ddi183. [DOI] [PubMed] [Google Scholar]
- Gotting C. Schulz V. Hendig D, et al. Assessment of a rapid-cycle PCR assay for the identification of the recurrent c.3421C>T mutation in the ABCC6 gene in pseudoxanthoma elasticum patients. Lab Invest. 2004;84:122–130. doi: 10.1038/sj.labinvest.3700004. [DOI] [PubMed] [Google Scholar]
- Hendig D. Schulz V. Arndt M, et al. Role of serum fetuin-A, a major inhibitor of systemic calcification, in pseudoxanthoma elasticum. Clin Chem. 2006;52:227–234. doi: 10.1373/clinchem.2005.059253. [DOI] [PubMed] [Google Scholar]
- Hendig D. Zarbock R. Szliska C, et al. The local calcification inhibitor matrix Gla protein in pseudoxanthoma elasticum. Clin Biochem. 2008;41:407–412. doi: 10.1016/j.clinbiochem.2007.12.023. [DOI] [PubMed] [Google Scholar]
- Klement JF. Matsuzaki Y. Jiang QJ, et al. Targeted ablation of the abcc6 gene results in ectopic mineralization of connective tissues. Mol Cell Biol. 2005;25:8299–8310. doi: 10.1128/MCB.25.18.8299-8310.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Saux O. Urban Z. Tschuch C, et al. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat Genet. 2000;25:223–227. doi: 10.1038/76102. [DOI] [PubMed] [Google Scholar]
- Lebwohl M. Neldner K. Pope FM, et al. Classification of pseudoxanthoma elasticum: report of a consensus conference. J Am Acad Dermatol. 1994;30:103–107. doi: 10.1016/s0190-9622(08)81894-4. [DOI] [PubMed] [Google Scholar]
- Li Q. Jiang Q. Pfendner E, et al. Pseudoxanthoma elasticum: clinical phenotypes, molecular genetics and putative pathomechanisms. Exp Dermatol. 2009;18:1–11. doi: 10.1111/j.1600-0625.2008.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L. Chassaing N. Delaite D, et al. Histological skin changes in heterozygote carriers of mutations in ABCC6, the gene causing pseudoxanthoma elasticum. J Eur Acad Dermatol Venereol. 2007;21:368–373. doi: 10.1111/j.1468-3083.2006.01940.x. [DOI] [PubMed] [Google Scholar]
- Meng H. Vera I. Che N, et al. Identification of Abcc6 as the major causal gene for dystrophic cardiac calcification in mice through integrative genomics. Proc Natl Acad Sci USA. 2007;104:4530–4535. doi: 10.1073/pnas.0607620104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfendner EG. Uitto J. Gerard GF. Terry SF. Pseudoxanthoma elasticum: genetic diagnostic markers. Expert Opin Med Diagn. 2008;2:63–79. doi: 10.1517/17530059.2.1.63. [DOI] [PubMed] [Google Scholar]
- Plomp AS. Florijn RJ. Ten Brink J, et al. ABCC6 mutations in pseudoxanthoma elasticum: an update including eight novel ones. Mol Vis. 2008;14:118–124. [PMC free article] [PubMed] [Google Scholar]
- Ringpfeil F. Lebwohl MG. Christiano AM. Uitto J. Pseudoxanthoma elasticum: mutations in the MRP6 gene encoding a transmembrane ATP-binding cassette (ABC) transporter. Proc Natl Acad Sci USA. 2000;97:6001–6006. doi: 10.1073/pnas.100041297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringpfeil F. McGuigan K. Fuchsel L, et al. Pseudoxanthoma elasticum is a recessive disease characterized by compound heterozygosity. J Invest Dermatol. 2006;126:782–786. doi: 10.1038/sj.jid.5700115. [DOI] [PubMed] [Google Scholar]
- Struk B. Neldner KH. Rao VS, et al. Mapping of both autosomal recessive and dominant variants of pseudoxanthoma elasticum to chromosome 16p13.1. Hum Mol Genet. 1997;6:1823–1828. doi: 10.1093/hmg/6.11.1823. [DOI] [PubMed] [Google Scholar]
- Symmons O. Varadi A. Aranyi T. How segmental duplications shape our genome: recent evolution of ABCC6 and PKD1 Mendelian disease genes. Mol Biol Evol. 2008;25:2601–2613. doi: 10.1093/molbev/msn202. [DOI] [PubMed] [Google Scholar]
- Szilvasi A. Andrikovics H. Kalmar L, et al. Asymmetric PCR increases efficiency of melting peak analysis on the LightCycler. Clin Biochem. 2005;38:727–730. doi: 10.1016/j.clinbiochem.2005.04.015. [DOI] [PubMed] [Google Scholar]
- Trip MD. Smulders YM. Wegman JJ, et al. Frequent mutation in the ABCC6 gene (R1141X) is associated with a strong increase in the prevalence of coronary artery disease. Circulation. 2002;106:773–775. doi: 10.1161/01.cir.0000028420.27813.c0. [DOI] [PubMed] [Google Scholar]
- Vanakker OM. Leroy BP. Coucke P, et al. Novel clinico-molecular insights in pseudoxanthoma elasticum provide an efficient molecular screening method and a comprehensive diagnostic flowchart. Hum Mutat. 2008;29:205. doi: 10.1002/humu.9514. [DOI] [PubMed] [Google Scholar]