

The enediyne natural products are potent antitumor antibiotics.1 They share a common unprecedented structural feature, an enediyne core containing two acetylenic groups conjugated by a double bond or incipient double bond (Figure 1A), that impart cytotoxicity via unique mechanisms of DNA damage.2 Recent progress in elucidating enediyne biosynthesis3 has enabled rational engineering of new analogues with altered activities.4 For example, manipulations of the C-1027 (1, Figure 1A) biosynthetic pathway produced analogues with DNA interstrand cross-linking activity, a property that could be exploited to target hypoxic tumors.4
Figure 1.

Selected enediynes and biosynthesis of the aromatic acid moieties: (A) the enediyne chromophores of C-1027 (1), NCS (2), and MDP (3) with the aromatic acid moieties highlighted in red; (B) biosynthesis of the naphthoic acid moiety of 2; and (C) early and (D) revised biosynthesis of the salicylyl moiety of 3.
The successful design of C-1027 analogues inspires parallel efforts to manipulate the neocarzinostatin (NCS, 2, Figure 1A) biosynthetic pathway for novel analogues. The naphthoic acid moiety of 2 is proposed to arise by the activities of the NcsB iterative type I polyketide synthase (PKS),5 the NcsB3 hydroxylase, and the NcsB1 O-methyltransferase.6 The NcsB2 CoA ligase finally activates the resultant naphthoic acid as a CoA thioester,7 setting the stage for its coupling to the enediyne core, catalyzed by the NcsB4 acyltransferase5,7 (Figure 1B). Significantly, both NcsB1 and NcsB2 exhibit broad substrate promiscuity, a property that can be exploited to engineer NCS analogues.6,7
Maduropeptin (MDP, 3, Figure 1A) also contains an aromatic acid moiety, a 3,6-dimethylsalicylic acid (4) whose construction might be targeted to engineer analogues. We recently cloned and characterized the MDP biosynthetic gene cluster.8 Parallel to that of the NCS naphthoic acid,6,7 the biosynthesis of 4 was proposed to be catalyzed by the MdpB iterative type I PKS and the MdpB1 C-methyl transferase; the MdpB2 CoA ligase then activated 4 as a CoA thioester (5) prior to its coupling to the enediyne core intermediate, catalyzed by the MdpB3 acyltransferase (Figure 1C).8 We were therefore inspired to study MdpB1 and MdpB2 with the hope that, like NcsB1 and NcsB2, similar substrate promiscuity may be uncovered and exploited to engineer MDP analogues. Here we report the surprising finding that MdpB2 acts before MdpB1, leading to a revised pathway for the biosynthesis of the salicylyl moiety of 3 (Figure 1D). Moreover, we demonstrate that MdpB2 is a promiscuous CoA ligase capable of activating a variety of salicylic acids.
We first attempted to characterize MdpB1 as a C-methyltransferase with 6-methylsalicylic acid (6) as a substrate in vitro.9 MdpB1 was overproduced in E. coli and purified to homogeneity (Figure S1), and 6 was synthesized.9 Unexpectedly, MdpB1, under all conditions examined, was unable to methylate 6 in the presence of S-adenosyl-L-methionine (SAM) to yield the predicted product 4 (Figure 1C), suggesting that the proposed pathway should be revised.
We next overproduced MdpB2 in E. coli and purified it to homogeneity (Figure S1) to examine the predicted CoA ligase activity in vitro. Upon incubation of 6 with MdpB2 in the presence of ATP and CoA, a new product was detected by HPLC analysis (Figure 2A) and identified as the corresponding CoA thioester 7.9 This suggested that MdpB2 may act, before MdpB1, on 6 as depicted in Figure 1D.
Figure 2.

HPLC analysis of MdpB2- and MdpB1-catalyzed sequential conversion of 6 (○) to 5 (◆): (A) MdpB2-catalyzed conversion of 6 (○) to 7 (●) (I) and control without enzyme (II) and (B) authentic standards of 7 (●) and 4 (▼) (I) and time course of MdpB1-catalyzed conversion of 7 (●) to 5 (◆) and its degradation product 4 (▼) upon 1.5 h (II) and 4.5 h (III) incubation.
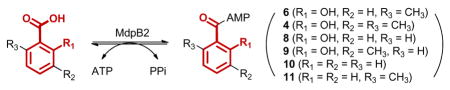
We then chemically synthesized 49 and compared the steady-state kinetic parameters of MdpB2 towards 4 (Figure 1C) and 6 (Figure 1D) to differentiate the two pathways. CoA ligase catalysis occurs in two steps: (i) ATP-dependent activation of carboxylic acids as acyl-AMPs and (ii) addition of CoA to the acyl-AMP to generate the acyl-CoA product. Although the rate-limiting step of the MdpB2 reaction is unknown, we probed adenylation specificity using the ATP-[32P]pyrophosphate exchange assay, which has been widely used for characterizing the substrate specificities of adenylating enzymes.10 Although 4 was also converted to its CoA thioester 5, whose identity was verified by HPLC analysis (Figure S3A) and high resolution mass spectrometry (HRMS),9 MdpB2 apparently prefers 6 (kcat of 2.3 min−1 and KM of 0.041 μM) over 4 (kcat of 0.59 min−1 and KM of 2.1 μM) (Table 1 and Figure S2). The 200-fold higher specificity (kcat/KM) of MdpB2 for 6 over 4 supports the proposed pathway in Figure 1D, implying that MdpB1 C-methylates the CoA activated 7 rather than the free acid 6 as a substrate.
Table 1.
Kinetic parameters of MdpB2 towards aromatic substrates featuring 1,2-hydroxy acid functionality (highlighted in red).
 | |||
|---|---|---|---|
| Substrate | KM (μM) | kcat (min−1) | rel kcat/KM |
| y | 0.041 ± 0.005 | 2.32 ± 0.06 | 1 |
| 4 | 2.1 ± 0.3 | 0.59 ± 0.02 | 0.0049 |
| 8 | 0.23 ± 0.05 | 0.90 ± 0.03 | 0.069 |
| 9 | 1.2 ± 0.2 | 0.86 ± 0.04 | 0.013 |
| 10 | ≥0.0042a | ||
| 11 | ≥0.0013a | ||
Values represent specific activities at 100 μM substrate concentration.
The kinetic insight from MdpB2 prompted re-evaluation of MdpB1 in light of the pathway proposed in Figure 1D. Incubation of MdpB1 with 7 and SAM yielded two products in a time-dependent fashion upon HPLC analysis (Figure 2B). One was the expected product 59, and the other product was identified as 4, indicating that 5 was not stable under the assay conditions. These results corroborate the revised pathway of Figure 1D and highlight its unusual biosynthetic logic of CoA-activation one step earlier than chemically necessary and demonstrate that MdpB1 is a C-methyltransferase specific for a CoA-tethered aromatic substrate.
Finally we characterized MdpB2 substrate promiscuity and observed a requirement for the ortho-hydroxy and carboxylate groups of the aromatic acid substrate. As summarized in Table 1, MdpB2 can also utilize salicylic acid (8) and its 3-methyl derivative (9), in addition to 4, as a substrate, albeit with 15-, 80-, and 200-fold lower specificities than 6, respectively (Figure S2). In contrast, MdpB2 barely recognized benzoic acid (10) and its methyl derivative (11) as substrates, exhibiting specific activities of only 0.25 h−1 and 0.08 h−1, respectively, at substrate concentrations of 100 μM. Although the kinetic analyses were based on the ATP-[32P]pyrophosphate exchange assays9, formation of the corresponding CoA thioester product was confirmed in all cases by HPLC (Figure S3) and HRMS.9 In contrast to other aryl-CoA ligases such as NcsB27 or SsfL113, MdpB2 is apparently more specific for the 1,2-hydroxy acid functionality, reminiscent of that reported for the NcsB1 O-methyltransferase.6
In summary, this study characterizes a novel C-methyltransferase that acts on a CoA-tethered aromatic substrate in natural product biosynthesis. Relatively few C-methyltransferases act on aromatic substrates in natural product biosynthesis.11 Notably, polyketomycin (POK) also possesses the 4 moiety (Figure S4A) and the POK biosynthetic gene cluster predicts proteins with high sequence identities (65–73%) to MdpB (PokM1), MdpB1 (PokMT1), and MdpB2 (PokM3) (Figure S4B).11a Indeed, a ΔpokMT1 strain produced a 6-containing POK analogue, confirming PokMT1 as a C-methyltransferase (Figure S4A). Our present in vitro work using purified MdpB1 sets the stage to further characterize this unusual enzyme capable of C-methylating a CoA-tethered aromatic substrate. To our knowledge, only an O-methyltransferase acting on caffeoyl-CoA has been described to date.12 Although the significance of the unusual biosynthetic logic is unclear, MdpB1 may represent an emerging class of C-methyltransferases exploitable for tailoring CoA thioester-tethered intermediates in natural product biosynthetic pathways.
Supplementary Material
Acknowledgments
We thank the Analytical Instrumentation Center of the School of Pharmacy, University of Wisconsin-Madison for support in obtaining NMR and mass spectrometric data. This work was supported in part by NIH grants CA78747 and CA113297. J.L. is the recipient of a China Scholarship Council fellowship, G. P. H. is the recipient of an NSERC (Canada) postdoctoral fellowship, and Y.L. is the recipient of a Visiting Scholar Fellowship from the Chinese Academy of Sciences.
Footnotes
Supporting Information Available: Full experimental details, SDS-PAGE gels, kinetic data, HPLC chromatograms, and comparison between MDP and POK biosynthesis. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Maeda H. Adv Drug Deliv Rev. 2001;46:169–185. doi: 10.1016/s0169-409x(00)00134-4. [DOI] [PubMed] [Google Scholar]; (b) Sievers EL, Linenberger M. Curr Opin Oncol. 2001;13:522–527. doi: 10.1097/00001622-200111000-00016. [DOI] [PubMed] [Google Scholar]
- 2.(a) Nicolaou KC, Dai WM. Angew Chem, Int Ed Engl. 1991;30:1387–1416. [Google Scholar]; (b) Galm U, Hager MH, Van Lanen SG, Ju J, Thorson JS, Shen B. Chem Rev. 2005;105:739–758. doi: 10.1021/cr030117g. [DOI] [PubMed] [Google Scholar]
- 3.Van Lanen SG, Shen B. Curr Top Med Chem. 2008;8:448–459. doi: 10.2174/156802608783955656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Kennedy DR, Gawron LS, Ju JH, Liu W, Shen B, Beerman TA. Cancer Res. 2007;67:773–781. doi: 10.1158/0008-5472.CAN-06-2893. [DOI] [PubMed] [Google Scholar]; (b) Kennedy DR, Ju J, Shen B, Beerman TA. Proc Natl Acad Sci USA. 2007;104:17632–17637. doi: 10.1073/pnas.0708274104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu W, Nonaka K, Nie LP, Zhang J, Christenson SD, Bae J, Van Lanen SG, Zazopoulos E, Farnet CM, Yang CF, Shen B. Chem Biol. 2005;12:293–302. doi: 10.1016/j.chembiol.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 6.(a) Luo Y, Lin S, Zhang J, Cooke HA, Bruner SD, Shen B. J Biol Chem. 2008;283:14694–14702. doi: 10.1074/jbc.M802206200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cooke HA, Guenther EL, Luo Y, Shen B, Bruner SD. Biochemistry. 2009;48:9590–9598. doi: 10.1021/bi901257q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooke HA, Zhang J, Griffin MA, Nonaka K, Van Lanen SG, Shen B, Bruner SD. J Am Chem Soc. 2007;129:7728–7729. doi: 10.1021/ja071886a. [DOI] [PubMed] [Google Scholar]
- 8.Van Lanen SG, Oh TJ, Liu W, Wendt-Pienkowski E, Shen B. J Am Chem Soc. 2007;129:13082–13094. doi: 10.1021/ja073275o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.See Supporting Information for full experimental details.
- 10.Linne U, Marahiel MA. Methods Enzymol. 2004;388:293–315. doi: 10.1016/S0076-6879(04)88024-8. [DOI] [PubMed] [Google Scholar]
- 11.(a) Daum M, Peintner I, Linnenbrink A, Frerich A, Weber M, Paululat T, Bechthold A. ChemBioChem. 2009;10:1073–1083. doi: 10.1002/cbic.200800823. [DOI] [PubMed] [Google Scholar]; (b) Lozano MJ, Remsing LL, Quiros LM, Brana AF, Fernandez E, Sanchez C, Mendez C, Rohr J, Salas JA. J Biol Chem. 2000;275:3065–3074. doi: 10.1074/jbc.275.5.3065. [DOI] [PubMed] [Google Scholar]; (c) Zhang W, Watanabe K, Wang CCC, Tang Y. J Biol Chem. 2007;282:25717–25725. doi: 10.1074/jbc.M703437200. [DOI] [PubMed] [Google Scholar]; (d) Ishida K, Fritzsche K, Hertweck C. J Am Chem Soc. 2007;129:12648–12649. doi: 10.1021/ja075524e. [DOI] [PubMed] [Google Scholar]; (e) Pacholec M, Tao J, Walsh CT. Biochemistry. 2009;44:14969–14976. doi: 10.1021/bi051599o. [DOI] [PubMed] [Google Scholar]
- 12.Ibdah M, Zhang XH, Schmidt J, Vogt T. J Biol Chem. 2003;278:43961–43972. doi: 10.1074/jbc.M304932200. [DOI] [PubMed] [Google Scholar]
- 13.Pickens LB, Kim W, Wang P, Zhou H, Watanabe K, Gomi S, Tang YJ. Am Chem Soc. 2009;131:17677–17689. doi: 10.1021/ja907852c. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.