

Abstract
This review covers three different chemical explanations that could account for the requirement of selenium in the form of selenocysteine in the active site of mammalian thioredoxin reductase. These views are the following: (1) the traditional view of selenocysteine as a superior nucleophile relative to cysteine, (2) the superior leaving group ability of a selenol relative to a thiol due to its significantly lower pKa and, (3) the superior ability of selenium to accept electrons (electrophilicity) relative to sulfur. We term these chemical explanations as the “chemico-enzymatic” function of selenium in an enzyme. We formally define the chemico-enzymatic function of selenium as its specific chemical property that allows a selenoenzyme to catalyze its individual reaction. However we, and others, question whether selenocysteine is chemically necessary to catalyze an enzymatic reaction since cysteine-homologs of selenocysteine-containing enzymes catalyze their specific enzymatic reactions with high catalytic efficiency. There must be a unique chemical reason for the presence of selenocysteine in enzymes that explains the biological pressure on the genome to maintain the complex selenocysteine-insertion machinery. We term this biological pressure the “chemico-biological” function of selenocysteine. We discuss evidence that this chemico-biological function is the ability of selenoenzymes to resist inactivation by irreversible oxidation. The way in which selenocysteine confers resistance to oxidation could be due to the superior ability of the oxidized form of selenocysteine (Sec-SeO2−, seleninic acid) to be recycled back to its parent form (Sec-SeH, selenocysteine) in comparison to the same cycling of cysteine-sulfinic acid to cysteine (Cys-SO2− to Cys-SH).
Keywords: Selenocysteine, Seleninic acid, Sulfinic acid, Nucleophile, Leaving group, Electrophile
Introduction
Selenium (Se), in the form of selenocysteine (Sec, U—the 21st amino acid) occurs in 25 proteins1 in the human proteome (Atkins and Gesteland 2000; Bock et al. 1991; Kryukov et al. 2003). Insertion of Sec into a protein is much more complicated than the other 20 amino acids because a UGA stop codon must be recoded as a sense codon for Sec as shown in Fig. 1 (Forchhammer et al. 1989; Heider et al. 1992; Leinfelder et al. 1988a; Low and Berry 1996). The complexity of this recoding process signifies that Sec must fulfill a chemical function that a conventional cysteine (Cys) residue cannot, and this chemical distinctiveness exerts biological pressure on the genome to maintain the Sec-insertion machinery. In this review, we term this chemical distinctiveness as the “chemico-biological” function of Se in enzymes, and we will provide a novel explanation (a chemico-biological rationale) for the use of Sec in enzymes. We will also review the chemical properties of Se that are thought to confer catalytic superiority relative to sulfur (S) in the form of a Cys residue, and we term these properties the “chemico-enzymatic” function of Se in enzymes. We formally define the chemico-enzymatic function of Se as its specific chemical property that allows a selenoenzyme to catalyze its individual reaction. For other reviews on this subject please see Odom (1983), Birringer et al. (2002), Jacob et al. (2003), and Wessjohann et al. (2007).
Fig. 1.
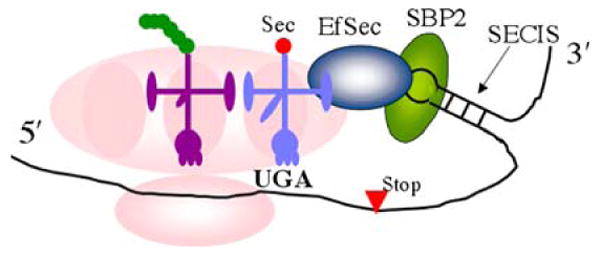
The Sec-insertion machinery. A special stem-loop structure called a selenocysteine insertion sequence (SECIS) element in the 3′ untranslated region of the mRNA (Krol 2002) binds a protein called SECIS binding protein-2 (SBP2), which then recruits a selenocysteine elongation factor (EfSec) that binds the specialized tRNA Sec (light blue). This complex is brought to the ribosomal A-site where the aminoacylated Sec residue is coupled to the growing polypeptide chain in the P-site (magenta tRNA). There are additional, required accessory factors such as selenophosphate synthetase that are not shown in the diagram (Squires and Berry 2008) (color figure online)
Selenocysteine: an ancient relic of the anaerobic world?
When the components of the Sec-insertion machinery were first being discovered and characterized, the prevailing view of Sec was that it was a relic of an earlier form of the genetic code when no (or very little) oxygen was present in the earth's atmosphere. The view expressed by Böck and co-workers was that:
“UGA was originally a sense codon for Sec in the anaerobic world, perhaps two to three billion years ago, and after introduction of oxygen into the biosphere this highly oxidizable amino acid could be maintained only in anaerobic organisms or in aerobic systems which evolved special protective mechanisms.”
The authors go on further to say that as oxygen was introduced into the atmosphere the UGA codon could have “acquired other functions such as its more familiar role in termination.” (Leinfelder et al 1988b). While Jukes thought that it was unlikely that there could have been an abrupt switch from UGA as a sense codon for Sec to that of a stop codon, he echoed their view of Sec as an ancient relic saying, “SeCys (sic) in enzymes is perhaps a vestigial reminder of the anaerobic world of 2 or 3 billion years ago.” (Jukes 1990). These statements show that a prevailing view of Sec at the time of its initial discovery was that it was so sensitive to oxygen that it must be a relic of a long ago era that has managed to persist to the present day. While we do not agree that Sec is a “relic”, we do agree that the selenium–oxygen connection is the key to understanding the presence of Sec in enzymes. We will return to the selenium–oxygen connection at the end of this review.
Selenocysteine: a sophisticated evolutionary innovation?
Another view of Sec in enzymes is that it is a “highly sophisticated innovation” that gives a Sec-containing enzyme a “catalytic advantage” over a Cys-containing enzyme (Osawa et al. 1992). While we cannot catalogue every instance in the literature of this view of Sec, a Google search of the World Wide Web with the phrase “catalytic advantage” and “selenocysteine” reveals 160 occurrences. This view is best characterized by a commentary called “Selenium speeds reactions” on the Sec-containing methionine sulfoxide reductase (MSRB1) (Robinson 2005). The view of Sec as a sophisticated innovation implies that for each occurrence of Sec in an enzyme there is a unique and specific reason for the use of Se to enhance the enzymatic reaction relative to that of S. This view also implies that since Se “speeds reactions” Sec should have widely substituted for Cys in enzymes, which clearly has not occurred. Specific reasons for the usage of Sec might include the enhanced nucleophilic character of Se relative to S, or another might be the much lower pKa of a selenol relative to that of a thiol. However, the chemico-physical differences that are thought to confer a catalytic superiority to Se relative to S are not as large as is generally assumed. For example, Se does have more nucleophilic character than S, but it is only in the range of six to tenfold (Pearson and Songstad 1967, 1968), which would only impart a rather modest rate enhancement relative to S given the very high energetic cost of maintaining the entire Sec-insertion machinery shown in Fig. 1. A recent review comparing the physicochemical properties of Se and S as related to their biochemical function (Wessjohann et al. 2007), notes their high similarity and states: “…a functional advantage of selenium over sulfur in enzyme reactions becomes not immediately evident.” The authors suggest a higher concentration of Cys-enzyme may compensate for the lower reactivity of Cys relative to Sec, especially given the very high cost of inserting a Sec residue into a protein in the first place. Despite this very insightful comment on the properties of Sec-enzymes relative to Cys-enzymes, researchers in the field continue to define the specific function of Se for every enzyme that contains Sec. We ourselves have pursued this hypothesis in our studies of the selenoenzyme thioredoxin reductase (TR) (Lacey et al. 2008). Instead of a specific explanation for the use of Sec in each selenoenzyme, we now believe that there may be a universal reason for the usage of Sec in enzymes. We will return to this idea when we discuss the chemico-biological function of Se in enzymes. We also note that the debate over whether the use of Sec is a relic or innovation has been previously discussed by Gladyshev and Kryukov (2001).
Different chemico-enzymatic views of Se in thioredoxin reductase
We first begin our discussion with the possible chemico-enzymatic functions that Sec might have in a selenoenzyme such as TR. These possible functions are broken down into three distinct views listed here: (1) the superior nucleophilicity of Se relative to S, (2) the superior leaving group ability of a selenolate compared to that of a thiolate (related to the higher acidity of RSeH relative to RSH), (3) the superior electrophilic character of Se relative to S. The higher electrophilicity of Se is also related to its greater ability to become hypervalent relative to S. The chemico-enzymatic function of Se in a selenoenzyme is predicated on the idea that Se is required in the rate-determining step of the enzymatic mechanism. We must therefore examine the mechanism of TR and review what is believed to be the rate-determining step of this enzyme.
Structure and mechanism of thioredoxin reductase
There are two classes of TRs and they differ by size and the number of redox centers. The low Mr TRs from bacteria have two redox centers (FADH and a disulfide redox center), while high Mr TRs from higher eukaryotes contain three redox centers (FADH and two disulfide redox centers) (Williams et al. 2000). High Mr TRs can be divided into two types: type I TRs contain a C-terminal disulfide redox center with the sequence Xaa-Cys1-Cys2-Xaa (Cys-TRs), while the type II TRs contain a C-terminal disulfide redox center with the sequence Gly-Cys1-Gly-Gly-Gly-Lys-Cys2-Gly. Mammalian type I TRs are further differentiated since they use Sec in the Cys2 position (Sec-TRs). High Mr TRs are homodimers and are similar in structure and mechanism to glutathione reductase (GR) and lipoamide dehydrogenase (LipDH) (Arscott et al. 1997). Sec-TR works by using a hydride from NADPH to reduce a bound flavin. The reduced flavin on one subunit reduces the conserved N-terminal disulfide redox center (CICVNVGCCT), which in turn reduces the 8-membered selenosulfide ring of the Cys1-Sec2 dyad that is attached to the enzyme through a flexible peptide linker on the opposite subunit (Fig. 2a). The 8-membered selenosulfide ring acts as an internal substrate in TR, and once it is reduced it passes its electrons to the disulfide bond of thioredoxin (Trx). The overall flow of electrons from NAPDH to the substrate is shown schematically in Fig. 2b. GR and LipDH work similarly, but are essentially truncated forms of TR that do not contain the flexible C-terminal tail. In place of the C-terminal redox center, GR and LipDH reduce external substrates GSSG (oxidized glutathione) and lipoic acid, respectively.
Fig. 2.
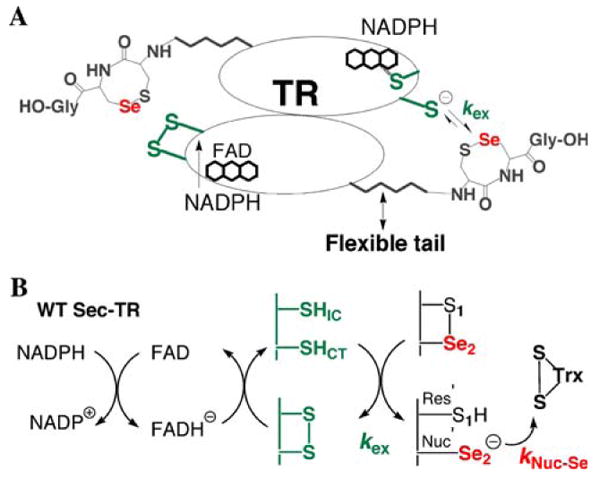
a Position of the C-terminal selenosulfide ring relative to the N-terminal redox center. The arrow shows the direction of electrons from the N-terminus on one subunit to the C-terminus on the opposite subunit. b Flow of electrons from NADPH to the substrate Trx. The N-terminal redox center consists of the conserved sequence CICVNVGCCT. CysIC is termed the interchange Cys because it reacts with the C-terminal disulfide and CysCT is termed the charge transfer Cys because it forms a charge transfer complex with FAD. The C-terminal redox center consists of a conserved Cys1-Sec2 dyad. The rate constant kNuc-Se represents the nucleophilic attack step (attack of the selenolate onto the disulfide bond of Trx), while rate constant kex represents the electron transfer step between N- and C-terminal redox centers
A unique feature of the TR mechanism is that a resolving Cys residue (Cys1) attacks the mixed selenosulfide bond of the TR-Trx complex, resulting in the formation of a rare 8-membered ring structure. While an 8-membered ring is the smallest ring size that can form from a disulfide bond in a protein, as is the case when a disulfide forms between adjacent Cys residues (Carugo et al. 2003; Hudaky et al. 2004), in Sec-TR the result is an 8-membered selenosulfide ring. This ring structure is reduced (and opened) by attack of the thiolate from CysIC. This thiolate can either attack S1 (path 1) or Se2 (path 2) as shown in Fig. 3. Figure 3 also shows the different possible ways in which Se is involved in the reaction mechanism of TR, which are discussed in the next section.
Fig. 3.
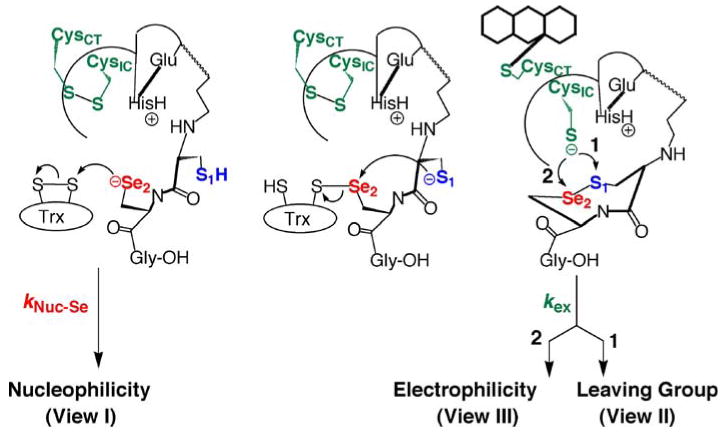
Reduction of macromolecular substrates by TR. The mechanism shows the different ways Sec could be involved in the rate-determining step of the reaction mechanism. If the nucleophilic attack of the selenolate onto the disulfide bond of Trx (represented by the rate constant kNuc-Se) is the rate-determining step, then the chemical property of Se that is most important is its nucleophilicity. The Se atom is also involved in accepting electrons from the N-terminal redox center (represented by the rate constant kex). If kex is the rate-determining step, then the chemical property of Se that is most important depends upon the mechanism by which the unique 8-membered ring between Cys1 and Sec2 is reduced and opened. The ring could be opened by attack on S (path 1) or Se (path 2). Attack at Se would mean that Se is needed due to its high electrophilic character. Attack at S would mean that Se is needed due to its superior leaving group ability relative to S
The rate-determining step in the reaction mechanism
Se could be involved in the rate-determining step in the reaction mechanism by any of the three chemico-enzymatic views listed above. The most common belief about the role of Se in the mechanism of TR is that it is needed as a strong nucleophile (view I) such that attack of the selenolate onto the disulfide bond of Trx is the rate-limiting step (represented in Figs. 2b, 3 by the rate constant kNuc-Se). The Se atom is also involved in the thiol/selenosulfide exchange step between N- and C-terminal redox centers, and therefore kex could be the rate-limiting step (depicted in Fig. 2a, b). The way in which Se is involved in this step depends on the mechanism by which the 8-membered ring is opened (Fig. 3). If the ring is opened by using path 1 (Eckenroth et al 2007b; Lacey et al 2008), then Se is needed as a good leaving group due to the low pKa of a selenol (view II). On the other hand if the ring is opened through path 2 (Bauer et al. 2003; Biterova et al. 2005; Cheng et al. 2009), Se is needed due to its superior electrophilic character (view III). These three views are discussed separately below.
Selenium as a superior nucleophile: view I
The work of Zhong and Holmgren (2000) has made it clear that Se facilitates the flow of electrons from the N-terminal redox center to the substrate Trx. This was shown by their observation that a thiolate–flavin charger transfer complex is formed in the Cys-mutant of rat TR1. This study showed that the reductive half-reaction occurs in the absence of Se. In other words, electrons were able to flow from NADPH to the N-terminal redox center efficiently, but the transfer of electrons from the N-terminal redox center to the substrate was somehow inefficient in the Cys-mutant. The conventional view of how Se facilitates the flow of electrons to Trx is through its superior nucleophilicity (chemico-enzymatic view I—implying kNuc-Se is the rate-limiting step). Many prominent scientists in the field have explicitly stated this position. Some of these statements are listed here, such as this one by Stadtman and Rhee (Lee et al. 2000):
“The large rate enhancement exerted by a selenolate anion in dithiol-disulfide interchange reactions can explain the requirement of the C-terminal Sec residue for efficient reduction of the TrxS2 substrate.”
Or this statement by Mugesh and Sing (2000):
“Although selenium shares many chemical properties with its neighboring homologue sulfur, selenium differs from sulfur in a number of ways, the most significant being that selenol is a more powerful nucleophile than thiol. Hence, in several enzymes, the incorporation of a chemically more active selenol confers a dramatic catalytic advantage.”
Operating under this view of the role of Sec, kNuc-s ≪ kNuc-Se when Sec is mutated to Cys because the impairment of nucleophilic character in the mutant is such that a great loss in rate occurs. It is indeed true that the enzyme loses 175–550-fold Trx-reductase activity in kcat when Sec is mutated to Cys. While it is tempting to believe that kNuc-Se is the rate-limiting step, there is no direct experimental evidence to substantiate this belief because this individual rate constant has never been measured. Further, the difference in nucleophilicities between Se and S, while not insignificant, is modest. Pearson and co-workers measured the nucleophilic reactivity constants of a wide range of nucleophiles (Y) toward methyl iodide in methanol (Pearson and Songstad 1967, 1968). This constant, n, is the log of the ratio of kY/kS. The rate constant kY is the rate constant for the formation of CH3Y and kS is the second order rate constant for methanolysis. Formally: nMeI = log (kY/kS). For benzeneselenolate (C6H5Se−), n is 10.7 and for benzenethiolate (C6H5S−), n is 9.92. Thus a comparison of the fully ionized species shows that there is a ∼6-fold difference in nucleophilicities between Se and S. If we compare the neutral atoms in dimethylsulfide and dimethylselenide, the values of n are 5.34 and 6.32, respectively—a ∼10-fold difference in nucleophilicities. This modest difference in nucleophilicities between the two atoms means that at most a rate enhancement of 1 kcal/mol is achieved by using Se instead of S (only considering nucleophilicity). It is hard to imagine how this small difference in free energy could account for the large bioenergenic expenditure needed for the biosynthesis of all of the components of the Sec-insertion machinery. The small difference in nucleophilicities also does not explain the large difference in kcat between the wild type (WT) Sec-TR and the mutant Cys-TR.
The difference in nucleophilicities between S and Se does increase significantly as the ionization state of the two atoms diverge. For example at pH 7.4, a Sec residue with a pKa of ∼5.5 will be significantly more nucleophilic than a Cys residue with a pKa of ∼8. However, there are many examples of low pKa Cys residues in enzymes such as: Cys164 of chalcone synthase (pKa = 5.5), Cys25 of papain (pKa ∼ 3–4), and Cys30 of DsbA (pKa = 3.5) (Jez and Noel 2000; Johnson et al. 1981; Lewis et al. 1981; Nelson and Creighton 1994). To the authors of this manuscript, it seems unlikely that the Sec-insertion system evolved so that an enzyme could utilize a chalcogen amino acid with a low pKa.
Selenium as a superior leaving group: view II
In contrast to the view that Se is needed in the nucleophilic attack step, we have taken the position that the rate-limiting step is kex, the transfer of electrons from the N-terminal redox center to the C-terminal selenosulfide motif (shown in Figs. 2a, 3). This idea was developed by studying a truncated form of the enzyme missing the last 8 amino acids (TRΔ8) and using a synthetic selenosulfide-containing peptide (corresponding to the missing 8 amino acids) as an external substrate for the truncated enzyme as shown in Fig. 4 (Flemer et al. 2008; Lacey et al. 2008). The truncated TR/peptide substrate system is a type of protein semisynthesis that has allowed us to isolate the exchange step from the overall reaction (Eckenroth et al. 2006). This system also enables us to overcome the high barriers of producing a recombinant selenoenzyme because the Sec residue is in a synthetic peptide (Tormay and Böck 1997). Selenoproteins isolated from natural sources or produced by recombinant methods can be missing the Sec residue due to inefficient translation of the UGA codon as Sec; this problem is avoided in our synthetic peptide substrate system.
Fig. 4.
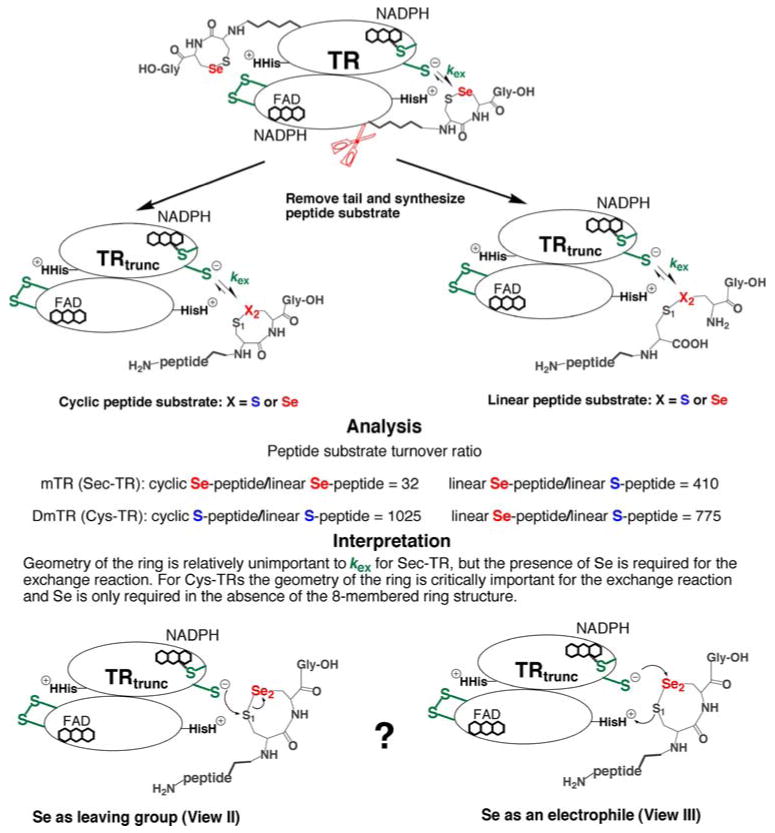
The importance of Se to kex. Using the truncated TR/peptide substrate system illustrated above, we demonstrated that Se is critically important to the exchange step between N- and C-terminal redox centers (Lacey et al. 2008). Mammalian TR (mTR) can reduce a selenosulfide (S1–Se2) bond whether it is in the context of a cyclic 8-membered ring (rate = 260 min−1·mM−1), or a linear, interchain selenosulfide (rate = 8.2 min−1·mM−1). However, both cyclic and linear disulfide (S1–S2) peptides were very poor substrates (rate = 0.02 min−1·mM−1). This shows that for mTR Se is important in accelerating the transfer of electrons from CysIC to the S1–Se2 bond. We originally interpreted this rate acceleration as being due to the superior leaving group ability of a selenolate compared to a thiolate (bottom left), but now favor the role of Se as a superior electrophile (bottom right and see discussion in the text). In the case of DmTR (a Cys-TR that uses Cys2 in place of Sec2), the truncated enzyme could only reduce a disulfide (S1–S2) when it was part of an 8-membered ring (rate = 41 min−1·mM−1) and not a linear disulfide (rate = 0.04 min−1·mM−1). This shows that unlike the Sec-TR, geometry was critically important for electron transfer to occur from CysIC to the S1–S2 bond. The Cys-TR could reduce a linear peptide containing a S1–Se2 bond (rate = 31 min−1·mM−1), demonstrating that Se accelerates the ability of CysIC to transfer electrons in the absence of proper catalytic geometry
In the case of the mammalian Sec-TR, the results of these studies showed that when Se was substituted with S, the peptide substrate was turned over very slowly. This was true for both cyclic (8-membered ring) and linear peptides (interchain peptides connected by either a disulfide or selenosulfide linkage as shown in Fig. 4). The cyclic, Sec-containing peptide (mimicking the WT enzyme) was a moderately better substrate than the linear Sec-containing peptide (by a factor of 32), showing that for the mammalian enzyme, geometry and ring strain were far less important factors for substrate utilization than the presence of a Se atom. These data show that electron transfer between the N-terminal redox center and the Sec-peptide substrate is disrupted when Se is missing from the peptide. This demonstrates that when the peptide is linked to the enzyme, as is the case in the holoenzyme, Se is needed in the reaction mechanism in the step before nucleophilic attack on TrxS2 (the exchange step governed by rate constant kex). Our view is also consistent with an intact reductive half-reaction as noted by Zhong and Holmgren (2000), except our interpretation is that the disruption in the flow of electrons is not due to an impairment in nucleophilicity that results from S for Se substitution, but is instead due to an inability of the C-terminal disulfide (in the Cys-mutant) to accept electrons in comparison to a C-terminal selenosulfide (wild type TR).
What is the manner in which Se accelerates the rate of electron transfer from CysIC to the C-terminal selenosulfide? Our original interpretation of the data using external peptide substrates for the truncated TR was that the superior leaving group ability of a selenol (due to its low pKa) enabled the exchange reaction to occur because the selenolate need not be protonated as the S1–Se2 bond is being broken as shown at the bottom left of Fig. 4. This represents view II of the chemico-enzymatic function of Se. We believed that this low pKa leaving group was necessary because there was a physical barrier (distance) between the enzymic general acid (HisH+ shown in Fig. 4) and the selenol. As we stated (Lacey et al. 2008):
“From a mechanistic viewpoint, this physical separation explains the need for the utilization of Sec by the mammalian enzyme because of the need for a low pKa leaving group in the ring opening step.”
Leaving group ability as a driving force for the exchange reaction was supported by the fact that the truncated TR could also reduce DTNB (pKa of thiol = 4.75) (Danehy et al. 1971), but other unactivated disulfide substrates such as cystine were turned over slowly.
The reason that a selenol is a superior leaving group relative to a thiol is because the acidity of a selenol is ∼ 1,000× greater than that of a typical thiol. The relevant biochemical comparison is between the selenol of selenocysteine (pKa 5.2) versus the thiol of cysteine (pKa ∼ 8.3) (Huber and Criddle 1967; Danehy and Noel 1960). Thus even at pH 4, the fraction of ionized selenolate is 10%, enabling reactions to occur at much lower pH compared to a thiol. The exceptional reactivity of a selenol was demonstrated by Noren and co-workers, who showed that the selenol of Sec could be biotinylated with iodoacetyl-LC-biotin at pH 2.5 (Sandman et al. 2000). The thiol of Cys was completely unreactive at this pH. The difference in reactivities at acidic pH was also quantified by Raines and co-workers who showed that the rate of attack of a selenol on a thioester was 1,000-fold faster than that of a thiol at pH 5 (Hondal et al. 2001). In a similar vein, Maeda et al. (2006) developed a fluorescent probe that would react with thiol nucleophiles, but is pH selective. This fluorophore, 3-(2,4-dinitrobenzenesulfonyl)-2,7-dimethylfluorescein (BESThio), reacted poorly with reduced glutathione at pH values <7, but would do so to a much greater extent at pH 8.5. They used this chemoselectivity to specifically label the selenol of Sec at pH 5.8 compared to the thiol of Cys. They further showed that BESThio was selective for the Sec residues of glutathione peroxidase and TR. Thus, due to the superior acidity of Se relative to S, we placed Se in the leaving group position in the “ring opening” step in Fig. 3 (path 1) and placed S1 as the site of attack by CysIC.
At the time of our publication of the data in Lacey et al. (2008), we did not consider the alternative way in which Se could accelerate the exchange reaction, which was to act as a superior electrophile (shown at the bottom right of Fig. 4). The electrophilic character of Se and the data supporting the electrophilicity of Se in accelerating the exchange reaction is discussed below.
Selenium as a superior electrophile: view III
Recently, we discovered that a diverse array of small molecule substrates could be turned over by the N-terminal redox center of truncated TR as shown in Fig. 5 (Lothrop et al. 2009). For comparison the activities of the full-length and truncated mammalian TR toward some of the substrates shown in Fig. 5 are summarized in Table 1. Our model of substrate utilization based on leaving group ability did not explain the ability of the N-terminal redox center to reduce substrates such as lipoic acid (pKa of thiol = 10.7) (Gascoigne and Radda 1967), or substrates that lacked a low pKa leaving group such as selenite (SeO3−2) (Kumar et al. 1992). The shared chemical property of all of the molecules shown in Fig. 5 is high electrophilicity (view III of the chemico-enzymatic function of Se). The S–S bond of lipoic acid is electrophilic due to the high degree of strain in the disulfide bond caused by the 35° C–S–S–C dihedral angle, instead of 90°–100°, for unstrained disulfides (Singh and Whitesides 1990; Burns and Whitesides 1990). The S–S bond of DTNB is electrophilic due to the strong polarization of the bond conferred to it by symmetrical 2-nitrobenzoate groups. Arnér has reported that various quinone compounds (already known substrates of TR such as vitamin K), are substrates for the N-terminal redox center (Anestål et al. 2008). A quinone is electrophilic due to the presence of di-α,β-unsaturated carbonyl groups. We also recently discovered that SeO3−2 was a substrate for the N-terminal redox center, but the corresponding S-analog, sulfite (SO3−2), was not (Lothrop et al. 2009). The only difference between these two small molecule substrates is the electrophilic character of the central atom. Thus, selenite must be electrophilic due to the electrophilic nature of Se itself. Applying this logic to our truncated TR/peptide substrate system argues for the much stronger electrophilic character of Se in a selenosulfide bond compared to the electrophilic character of S in a disulfide bond. This rationale supports the mechanism shown at the bottom right of Fig. 4. This must mean that the Se atom is attacked in the thiol/selenosulfide exchange reaction between N- and C-termini and explains why kex is very slow in the Cys-mutant of TR and accounts for the overall loss in kcat (175—550-fold) when Sec is mutated to Cys with Trx as the substrate. While we did not originally consider the electrophilic character of Se as being important to the reaction mechanism, it is clear from the pattern of substrate usage shown in Fig. 5, that the high electrophilic character of Se must be the driving force in the exchange reaction. The electrophilic nature of Se has long been recognized in the chemical literature (Back 1987), but this property of Se has been thus far unrecognized in the biochemical literature. The electrophilic nature of Se is discussed in more detail below.
Fig. 5.
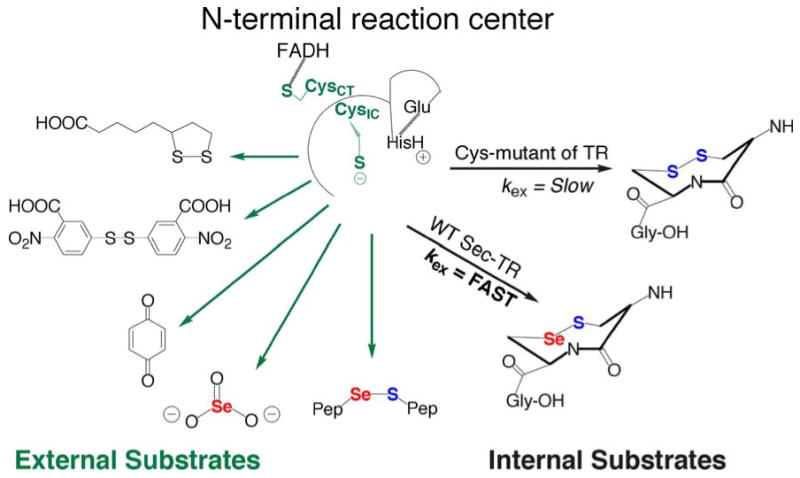
External and internal substrates for the N-terminal redox center of TR. We discovered that for the mitochondrial TR, the truncated enzyme containing only the N-terminal redox center could reduced a variety of small molecule substrates (named from top left and continuing counter clockwise) such as lipoic acid, DTNB, quinones (reported by Anestål et al. 2008), selenite, and Sec-containing peptides. All of these small molecules are good electrophiles. Our hypothesis is that kex is the rate-limiting step and Se is needed due to its superior electrophilicity. This would explain the decrease in kcat in the Cys-mutant because kex would be slow
Table 1.
Comparison of substrate activities with full-length and truncated mammalian TR
| Substrate | Activity (min−1) | |
|---|---|---|
| mTR-GCUG | mTRΔ8 | |
| 5 mM lipoic acida,b | 38.2 | 87.7 |
| 1 mM selenitea,b | 308 | 49 |
| DTNBb,c,d | 1,251 | 2,905 |
| Juglone (a quinone)d,e | 360f | 360g |
The TR assayed is mouse mitochondrial TR
From Lacey et al. (2008)
This is the kcat value under steady-state conditions
The TR assayed is full-length rat cytosolic TR
The TR assayed is truncated rat cytosolic TR made by deleting the last two amino acids (Sec-Gly)
We would first, however, like to make a special point with respect to the reduction of selenite by TR. The reduction of selenite by cytosolic TR was first reported by Holmgren and co-workers (Kumar et al. 1992). They noted a distinct lag phase at low concentrations of selenite. We also observed the same phenomenon with the mitochondrial enzyme (Lothrop et al. 2009). We believe that the reason for this lag phase is that the active substrate species is not selenite, but is in fact selenium dioxide (SeO2). In a study of the reaction of selenite with thiols, Kice et al. (1980) found that selenite (SeO3−2) reacts much faster with thiols than does benzeneseleninic acid (1,000-fold faster). Kice suggested the reason for the much higher reactivity of selenite is because selenite can dehydrate to SeO2 and this dehydration does not involve a net reduction as the Se atom is in the +4 oxidation state in both cases (Kice et al. 1980). As Kice points out, SeO2 is expected to be much more reactive toward a thiol than selenite (as a result of resonance) and we believe that the reason for the lag phase is that TR is actually reducing the small amount of SeO2 in solution, which is in equilibrium with SeO3−2 (see Scheme 1). This hypothesis could be verified by 77Se NMR experiments with TR.
Scheme 1.
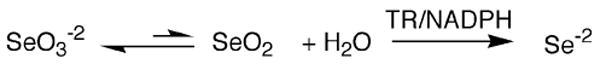
Proposed mechanism for the reduction of selenite by TR. We propose that the reactive species in the enzymatic reaction is selenium dioxide
Electrophilic character of selenium
The high electrophilic character of Se is evidenced by its high reactivity toward butyllithium (n-BuLi) in comparison to S. As shown in Scheme 2, n-BuLi readily attacks the Se atom of bis(phenylseleno)methane (PhSeCH2SePh), while in the case of the sulfur homolog (bis(phenylthio)methane), the butylate anion acts as a base to abstract a proton from the carbon acid so that lithium exchanges with a hydrogen atom. In this reaction, exchange with S occurs only to a small extent such that attack at Se is ∼104 times faster than attack at S (Reich et al. 2002). In an experiment to show that attack of the butylate anion occurs preferentially at Se, (Seebach et al. (1977) used phenylseleno(phenylthio)methane (PhSeCH2SPh) for the exchange reaction and found that the selenoether was the product (Scheme 3). These experiments demonstrate the much greater electrophilic character of Se compared to S.
Scheme 2.
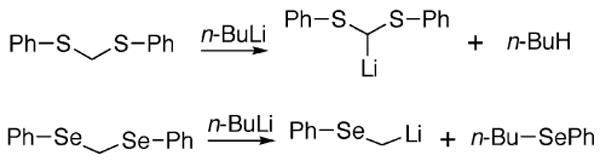
Butyllithium exchange reaction with bis(phenylthio) methane (top) and bis(phenylseleno)methane (bottom). With the S-compound, butylate acts as a base to abstract hydrogen, whereas with the Se-compound it acts as a nucleophile to attack Se
Scheme 3.

Preferential attack in a butyllithium exchange reaction using phenylseleno(phenylthio)methane. When the butylate anion is faced with the choice of either attacking S or Se, attack at Se is preferred
This greater electrophilic character of Se is related to its ability to become “hypervalent”, e.g. the ability to have more than two covalent bonds (Hellwinkel 1972; Musher 1972). Both S and Se have the ability to form multiple bonds, for example, SF6 for sulfur. The periodic trend is for elements to have greater hypervalent character as the atom becomes larger (descending down group 16 of the periodic table). Thus the ability of the chalcogens to form multiple bonds is in the order Te ≫ Se > S. So as a nucleophile (Nu:) attacks R–Se–Y to form R–Se–Nu, the transition state involves a hypervalent selenium atom (3 bonds). The larger size of Se enables more hypervalent character and stabilizes the transition state, making the reaction of the compound containing Se faster relative to the reaction of the compound containing S. The reaction is undoubtedly enhanced due to the weaker Se–Y bond compared to a S–Y bond (note that a Se–S bond is ∼20 kcal/mol weaker than a S–S bond). Using this logic, a thiol (R′SH) should attack the Se atom of a selenosulfide (RS–SeR″) to form R′S– SeR″, the opposite conclusion that we reached in Lacey et al. (2008). To our knowledge, there has never been a study of a thiol/selenosulfide exchange reaction of the type R′SH/RS–SeR″ that demonstrated which atom (Se or S) is attacked in the exchange reaction (Scheme 4).
Scheme 4.
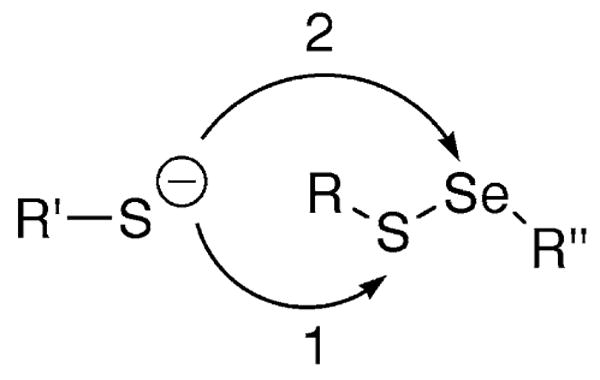
Potential routes of attack of a thiolate on a selenosulfide bond
There have been a few studies of thiol/bis(alkylthio)selenide reactions of the type RSH/R′S–Se–SR′. One study by Kice and Sleblocka-Tilk (1982) found that attack at Se or S depended on the nature of the R group. When R′ was t-butyl, attack at Se was preferred, when R′ was i-propyl, both S and Se could be attacked to a nearly equal degree, when R′ was n-butyl, attack at S was preferred strongly. The only other study that we are aware of is one done by Rabenstein et al. (1991) who used d-penicillamine and bis(d-penicillamine)selenide. The results of this study showed that attack at Se was faster, in agreement with the work by Kice showing attack at Se is preferred with bulky groups on S. Bachrach and co-workers used computational methods to study the attack of HS− and CH3S− on selenosulfides HS–SeH, CH3S–SeH, and HS–SeCH3 in the gas-phase. The result of this study showed that attack at Se was both kinetically and thermodynamically favored (Bachrach et al. 2004). We note, however, that extrapolating results from the gas-phase to the solution-phase is difficult.
An enzyme active site is quite different from reactions that take place free in solution and enzymes are able to modulate the reaction pathways to gain catalytic advantage. One factor that selenoenzymes may use to control the Se/S selectivity ratio in a thiol/selenosulfide exchange reaction is non-bonded interactions. Both S and Se can participate in weakly covalent interactions that are called “non-bonding”. There are two types of these non-bonding interactions. The first type involves donation of a lone pair on Se to a neighboring atom that acts as a weak electrophile. The second type usually involves donation of a lone pair from O or N to the antibonding σ* orbital of a Se–R fragment (Coles 2006). Mugesh has proposed that a proximal His residue in the TR active site is involved in such an interaction. In this model a lone pair on N from His would donate electron density to the S1 atom of the Gly-Cys1-Sec2-Gly motif. Such an interaction would have the effect of elongating and weakening the S1–Se2 bond, thereby enhancing the electron density around Se2 and making S1 more electropositive (Sarma and Mugesh 2006). This interaction would facilitate attack of CysIC onto S1 so that Se2 would be in the leaving group position. Mugesh argues that this would regenerate the catalytic selenolate more quickly. However, the free selenolate would be available to be active in the back reaction (attack on the mixed disulfide between CysIC and Cys1). Whichever atom of the Cys1-Sec2 dyad is in the leaving group position, TR must have a mechanism to suppress this back reaction. While Mugesh has presented very strong evidence for these non-bonded interactions in model systems (Sarma and Mugesh 2005), this type of interaction is difficult to demonstrate definitively for the enzyme.
Electrophilicity as a unifying concept for Cys- and Sec-TRs
Based on our finding that substrate utilization by the N-terminal redox center of mammalian TR is governed by electrophilic character, we must conclude that path 2 (attack at Se—last panel of Fig. 3) is utilized in the critical exchange reaction between N- and C-terminal redox centers. Thus Se2 is attacked and S1 receives a proton from HisH+ in this model.
This rationale can also explain how Cys-TRs (that use Cys2 in place of Sec2), such as the one from Drosophila melanogaster (DmTR), can function in the absence of Sec. Instead of increasing the nucleophilic character of S to compensate for a lack of Se (as is commonly assumed), Cys-TRs could increase the electrophilic character of S2 through the use of a strong S1:::+H–His interaction as shown in Fig. 6. This would polarize the S1–S2 bond, rendering S2 more electrophilic. This hypothesis explains the experimental finding that the two S atoms of the C-terminal Cys1-Cys2 dyad in DmTR are non-equivalent. For example, using our truncated TR/peptide substrate system with DmTR, we were able to chemically distinguish S1 from S2 by replacing either atom with Se in the peptide substrate as shown in Fig. 7 (Lacey et al 2008). When Se replaced S1 in the dyad, the turnover rate of the peptide substrate decreased tenfold. When Se replaced S2 in the dyad, the turnover rate of the peptide substrate increased 12-fold. We originally explained the increase in activity with Se in the S2 position as being due to superior leaving group ability of a selenolate relative to a thiolate. We explained the decrease in rate when Se was in the S1 position as being due to attack of CysIC onto the “incorrect” atom. However, in light of our recent findings that the N-terminal redox center can only utilize highly electrophilic substrates, the increase in rate with Se in the S2 position is due to the superior intrinsic electrophilicity of Se relative to S. Similarly, the decrease in activity observed with Se in the S1 position is most likely due to a “seleno effect”, analogous to the well known thio effect observed in phosphodiesterases (Kravchuk et al. 2001; Herschlag 1994). If there is a strong S1:::+H–His interaction as we posit, then replacing S1 with Se would slow down the reaction because a selenolate is much less basic than a thiolate (i.e. Se has lower ability to accept a proton relative to S), and this is what is experimentally observed (Lacey et al. 2008).
Fig. 6.
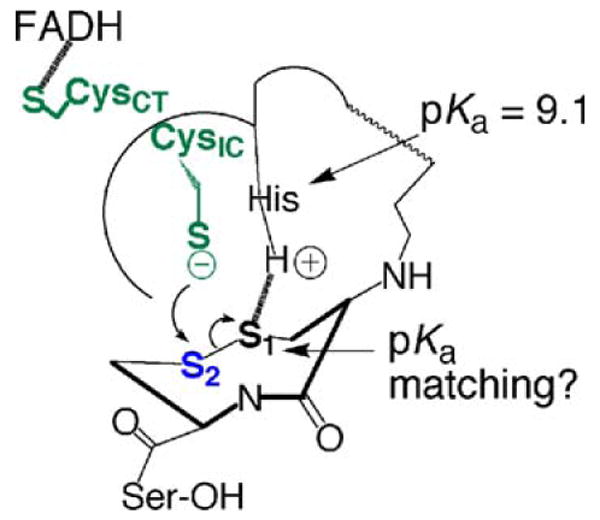
Proposed electrophilic activation mechanism in DmTR. DmTR uses S2 in place of Se2 and this enzyme catalyzes the reduction of its cognate Trx substrate efficiently. If Se is needed in mTR as an efficient electrophile, than a Cys-TR such as DmTR must increase the electrophilicity of the S1–S2 bond in order for efficient electron transfer to occur from CysIC. This could be accomplished through a S1:::+H–His interaction, which would increase the electrophilic character of S2. We, and the Williams group previously determined the pKa of HisH+ in DmTR to be ∼9.1 (Lacey et al. 2008; Huang et al 2008); this is very close to the pKa values of typical thiols in proteins. In the transition state the pKa of the thiol of Cys1 could be closely matched to the imidazolium group of HisH+. If this is the case, it would make for a very strong S1:::+H–His interaction, greatly increase the electrophilicity of S2, and facilitate electron transfer from CysIC
Fig. 7.
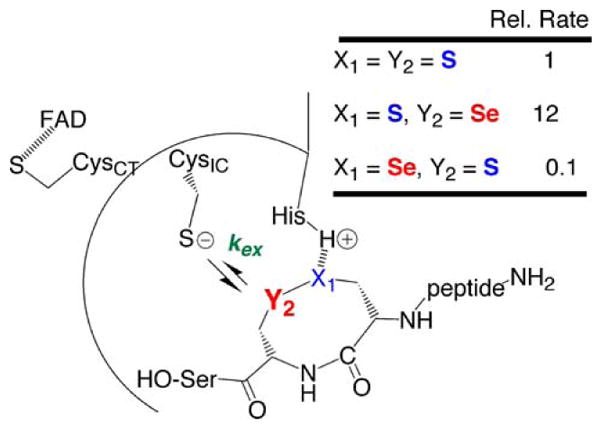
Positional effects of Se placement on kex. Using our truncated TR/peptide substrate system with DmTR we were able to measure the rate of peptide turnover when Se is either placed in the Cys1 position or the Cys2 position in the C-terminal dyad in DmTR. The results show that the two S atoms of the dyad are non-equivalent as described in the text. A strong interaction between X1 and HisH+ (H-bond or protonation) can explain the decrease in rate when X1 = Se because of the decreased basicity of a selenolate relative to a thiolate. Note that in this figure, X1 is equivalent to the S1 position and Y2 is equivalent to the S2 position
Our explanation of a strong S1:::+H–His interaction in DmTR can also explain why ring geometry is so important to its catalytic cycle, while ring geometry is relatively unimportant for the Sec-TR as shown by the data in Fig. 4. For example, DmTR could utilize an 8-membered ring disulfide substrate (the natural situation), but the linear disulfide substrate was turned over very slowly. In contrast, while the Sec-TR preferred the 8-membered ring selenosulfide substrate to the linear peptide by a factor of 32, the linear selenosulfide peptide substrate could still be turned over at a significant rate. This empirical finding fits well with our explanation that high intrinsic electrophilicity of Se is the driving force in the exchange reaction in mammalian TR. Thus the S1–Se2 bond need not be held in a rigid ring in order for reduction of this bond to occur and the S1:::+H–His interaction is relatively unimportant in the mammalian TR. In contrast, a special geometry is required for the corresponding S1–S2 bond to be reduced in DmTR because the S1–S2 bond must be activated (made more electrophilic) due to the proposed S1:::+H–His interaction. Therefore the exact position of S1 and S2 relative to CysIC and HisH+ in the tetrapeptide active site is critical for the Cys-TR, while this same positioning of active site groups in the Sec-TR is less important as long as a Se atom is present to accept electrons (as shown by the data in Fig. 4).
Further support for this “electrophilic activation mechanism” comes from conventional mutagenesis experiments performed by our group. We found that when one or two Ala residues are inserted in between Cys1 and Cys2 in DmTR, kcat decreases by 150- and 300-fold, respectively (Eckenroth et al. 2007b). When the same Ala insertion experiment was performed for the mammalian Sec-TR, kcat only decreased four to sixfold (Eckenroth et al. 2007a). Our electrophilic activation hypothesis can explain these results. In the case of the Cys-TR (DmTR) the resulting change in ring geometry due to Ala insertion would disrupt the S1:::+H–His interaction, which would greatly affect the electrophilic character of S2, which in turn would decrease kex and thus cause a decrease in kcat. However, since the mammalian Sec-TR does not greatly depend on the S1:::+H–His interaction to activate the S1–Se2 bond, kex and kcat are not greatly effected by the disruption in ring geometry caused by Ala insertion.
cis and trans ring conformers revisited
An interesting feature of type I TRs is that they use a rare vicinal (between adjacent residues) disulfide bond during the catalytic cycle. A vicinal disulfide bond forms a strained 8-membered ring and has only been found in a very small percentage of the structures deposited in the PDB (Hudaky et al. 2004). The reason that nature has chosen this rare ring structure for type I TRs is unknown, but we have previously explained the difference between Cys- and Sec-TRs in terms of differences in cis/trans amide ring geometry.
We noted that the sulfur atoms of a vicinal disulfide ring could be superposed onto the sulfur atoms of oxidized glutathione (see Figure 1 of Lothrop et al. 2009 and Figure 4 of Eckenroth et al 2007b) only when the amide geometry of the ring was cis. This superposition and the structural and evolutionary relationship between GR and TR led us to hypothesize that Cys-TRs would utilize cis amide ring geometry to catalyze the reaction. Due to these relationships we placed S2 of the Cys1-Cys2 dyad in the leaving group position in the exchange reaction of DmTR. We reasoned that the situation for Sec-TRs would be somewhat different due to the presence of the Se atom in the Cys1-Sec2 dyad. An 8-membered selenosulfide ring is about 15% larger than the corresponding disulfide ring (Gromer et al. 1998). This size difference is due to longer C–Se and S–Se bonds compared to the corresponding C–S and S–S bonds. We therefore proposed that Sec-TRs would use trans amide geometry in the exchange reaction and we placed Se2 in the leaving group position because it occupied the analogous position to S2 in DmTR.
However, due to the evidence that we discussed at length above regarding the electrophilicity of Se and our proposed electrophilic activation mechanism for Cys-TRs, we must now rotate our placement of the oxidized tetrapeptides by 180° in the tetrapeptide binding pocket of the respected enzymes. This alignment now places Se2 (mTR) and S2 (DmTR) in the correct position to be attacked in the exchange reaction (path 2 of Fig. 3). This 180° rotation has caused us to rethink whether the oxidized Cys1-Cys2 dyad of DmTR utilizes cis amide ring geometry. If the ring in DmTR uses trans geometry, then the overall reaction mechanism of this Cys-TR would be nearly identical to that of the mammalian enzyme except for the use of our proposed “electrophilic activation” mechanism to compensate for the lack of an electrophilic Se atom. The use of trans amide ring geometry in DmTR could also help to make the S1–S2 bond more electrophilic because the trans conformer should be more strained than the cis conformer. We note that in protein structures all of the vicinal disulfide ring structures characterized to date are found in a strained trans conformation (Hudaky et al. 2004). The relief of ring strain could be an additional factor in the Cys-TR that helps to electrophilically activate the S1–S2 bond and thus compensate for the lack of Sec.
Is Sec needed to catalyze specific enzymatic reactions?
We have described three different chemical (chemicoenzymatic) views of why Se might be needed for enzymatic catalysis in TR. However, an important question to ask is “Is Se really needed at all to catalyze a specific enzymatic reaction?” Comparison of the proteomes of higher vertebrates with other complex eukaryotes shows that Cys can substitute for Sec in the same enzyme without significant loss in activity. For example, the TR from D. melanogaster (DmTR) can reduce its cognate substrate (DmTrx) with high catalytic efficiency (1.9 × 108 min−1·M−1), which is only slightly lower than the human Sec-containing TR (7.6 × 108 min−1·M−1). As the authors state: “These data speak against the proposition that Sec in the C-terminal Cys-X sequence of high Mr TRs is a chemical necessity.” (Kanzok et al. 2001). In addition, the genome of C. elegans has maintained the entire Secinsertion machinery to decode a single UGA codon for the Sec-containing cytosolic TR (Taskov et al 2005). However, C. elegans also has a mitochondrial TR that contains Cys (Gladyshev et al. 1999; Lacey and Hondal 2006). Both enzymes ostensibly catalyze the same enzymatic reaction—reduction of the disulfide bond of Trx. It is unlikely that dissimilarity of Trx molecules between different cellular compartments account for the use of Sec and Cys in the same organism. Therefore, if Sec is not needed to catalyze a specific enzymatic reaction, then there must be a completely different reason for the use of Sec in enzymes that explains the biological pressure to maintain the Secinsertion machinery. Despite this knowledge, researchers continue to ask “Why selenoproteins?” (Gromer et al. 2003) and offer a “niche” rationale (sophisticated innovation) that Sec-enzymes can catalyze some reactions better than the Cys-enzyme. The niche rationale that has been offered for the role of Sec in TR is that the presence of Se explains the broad substrate specificity of the enzyme and that the low pKa of the selenol gives it very high activity. An example of this view is stated by Nalvarte et al. (2004):
“The combination of a low pKa of Sec (typically 5.25) and an easily accessible C-terminal localization of the Sec-containing active site should explain why reduced TrxRs are highly reactive at physiological pH and display a broad substrate specificity.”
We have documented similar statements in the Supporting Information of our most recent paper (Lothrop et al. 2009). In this same paper, we showed that Sec was not required for many of the activities of mitochondrial TR, refuting the claim that Sec conferred broad substrate specificity. Subsequent to this paper we have developed a new hypothesis that explains the biological pressure to maintain the UGA recoding apparatus for Sec. This biological pressure is based upon the superior chemical property of Se (relative to S) to confer resistance to irreversible oxidation and we thus name it the “chemico-biological” rationale for the presence of Sec in enzymes. This hypothesis with supporting evidence is discussed in the next section.
A new rationale for the occurrence of Se in enzymes: the selenium paradox
Gladyshev (2006) recently presented evidence that the trait for Sec insertion was present in the last universal common ancestor (LUCA) and that the number of selenoproteins increased as the amount of oxygen in the earth's atmosphere increased. He especially noted that selenoprotein P, the only known eukaryotic protein with multiple Sec residues (10) (Burk and Hill 2005), appeared at a time in evolution when the amount of oxygen in the atmosphere was increasing dramatically. This strongly argues against Sec insertion as being an anaerobic relic, but rather for the use of Sec as an adaptation to the increased presence of oxygen. However, it is well known that Se is much more sensitive to oxidation than is S. Thus if one introduces Se into an enzyme it can be more easily oxidized to selenenic (RSeOH) acid or seleninic acid (RSeO2−), which would inactivate the enzyme. This situation is seemingly paradoxical, as it would first appear that S is better able to resist the dangers of oxidation because it is slower to oxidize relative to Se.
This condition is shown in Scheme 5 as the rate constant for oxidation of RSeH to the RSeOH form (k1) being greater than the corresponding oxidation of RSH to RSOH (rate constant k4). Both the selenenic and sulfenic (RSOH) acid forms can be converted back to the selenol and thiol, respectively, by another thiol (represented by rate constants kPxSe and kPxS with kPxSe > kPxS). Further oxidation can occur to the seleninic and sulfinic (RSO2−) forms. For Cys-enzymes oxidation to the Cys-SO2− form is referred to as being “over-oxidized” (Rhee et al. 2005) because it is extremely difficult to reduce the sulfinic acid form back to a thiol. We suggest that an extremely large chemical advantage occurs for the Sec-enzyme under conditions where the Sec-enzyme and Cys-enzyme are over-oxidized to their Sec-SeO2− and Cys-SO2− forms, respectively, due to the superior ability of Sec-SeO2− to be recycled back to Sec-SeH relative to Cys-SO2− cycling to Cys-SH.
Scheme 5.
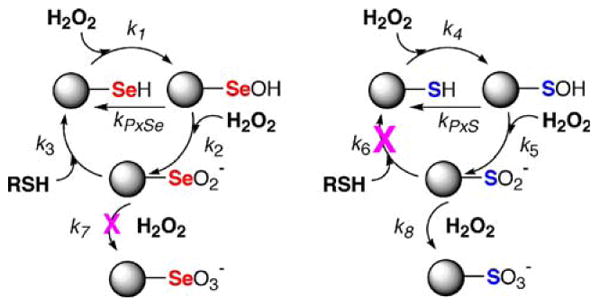
The chemico-biological rationale for the presence of Se in an enzyme. The answer to the selenium paradox is that while Se is more easily oxidized than S (k1 > k4), RSeO2− is much more easily recycled back to RSeH compared to RSO2− cycling to RSH (k3 ≫ k6). The scheme shows that the chemistry of Se is such that it allows for reaction with oxygen, but the reaction is reversible so that cycling to the original form (RSeH) occurs readily at either the selenenic acid (RSeOH) stage or the seleninic acid (RSeO2−) stage. The scheme shows that S also reacts with oxygen, but conversion back to the original form (RSH) is only possible at the sulfenic acid (RSOH) stage. When sulfenic acid reacts with another equivalent of oxygen it is irreversibly oxidized to RSO2− and RSO3−
A biologically important example of over-oxidation is the oxidation of the peroxidatic Cys residue of 2-Cys peroxiredoxins (Prxs). When Prxs are converted to the Cys-SO2− form, they become inactivated and require the action of another enzyme, sulfiredoxin, to restore function (Biteau et al. 2003). This is the only known example of reduction of Cys-SO2− to Cys-SH in mammalian proteins. Moreover, it is known in the chemical literature that it is very difficult to convert a sulfinic acid back to a thiol, and this is accomplished only under very harsh conditions (Finlayson et al. 1979; Oae et al. 1980). In contrast, seleninic acid can be converted back to a selenol by another thiol under milder conditions (Kice and Lee 1978). Unfortunately, the paucity of the literature is such that a direct comparison of identical Se- and S-compounds does not exist. We suggest that the ratio k3/k6 is >106 and we term this the “chemico-biological” function of Se in enzymes. The solution to the paradox is that while Se is more sensitive to oxygen than is S, it is much more easily restored back to its original form by another thiol. This chemical fact makes proteins containing Sec more resistant to oxygen because they can cycle through higher oxidation states much faster.
Another important, and unrecognized way in which the use of Se in an enzyme may help to resist oxidation is that it is very difficult for RSeO2− to be further oxidized to RSeO3−, while RSO2− can be easily oxidized to RSO3− (k7 ≪ k8 in Scheme 5). In biochemical terms this means that Sec-SeO2− cannot be oxidized to the selenonic acid form (Sec-SeO3−), while Cys-SO2− can be oxidized to the sulfonic acid form (Cys-SO3−–—termed “hyper-oxidized”). Biologically there is no known method of reducing Cys-SO3− to Cys-SH. In chemical terms, this means that it is more difficult for Se in the +4 oxidation state to be oxidized to the +6 oxidation state, while this same +4 to +6 transition occurs with relative ease for S.2 Experimental support for this position is shown in Fig. 8 which shows the progress of the oxidations of phenyl methyl selenide and phenyl methyl sulfide. The plot shows that PhSeMe is more easily oxidized to the selenoxide than the corresponding oxidation of the sulfide to the sulfoxide, but it also shows the difficulty in oxidizing the selenoxide to the selenone (+4 to +6 oxidation state) compared to the oxidation of the sulfoxide to the sulfone. The author of this study (Reich 1978) has suggested that the reason for the difficulty in oxidizing the selenoxide is that it is much more polar than the sulfoxide (greater Se–O single bond character compared to S–O). Thus Se of R2Se=O or Sec-SeO2− is more positively charged than the S analogs, and hence the lone pair on Se is harder to donate to an oxidant. Thus Scheme 5 shows the chemical basis for selenium's peroxidatic and antioxidant properties. On the basis of the above analyses we suggest that Se will only occur in an enzyme when this enzyme needs to be very resistant to irreversible oxidation and thus resistant to inactivation.
Fig. 8.
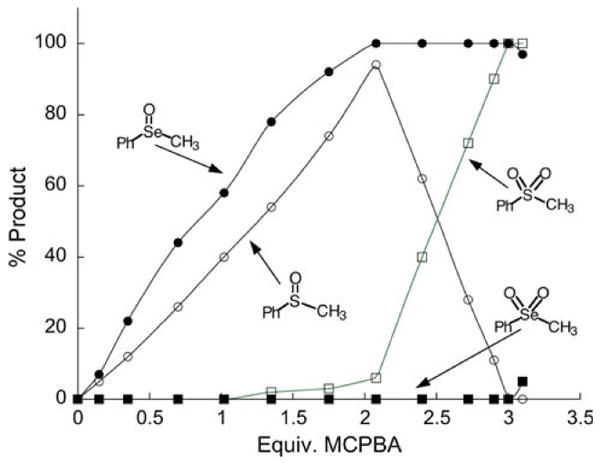
Progress of the oxidation of PhSeMe and PhSMe to the +4 and +6 oxidation states by meta-chloroperoxybenzoic acid (MCPBA). The results of this study show that the selenide is oxidized to the selenoxide more easily compared to the oxidation of the sulfide to the sulfoxide. However as shown by the plot, the selenoxide resists further oxidation while the sulfoxide can be readily oxidized to the sulfone (reproduced from Reich 1978)
Scheme 5 clearly illustrates the specific area where the chemical property of Se is far superior to that of S—the interaction with oxygen. Another clear example of this is in the comparison of the reduction of methionine selenoxide and methionine sulfoxide by another thiol. Methionine selenoxide is easily reduced to selenomethionine by glutathione, cysteine, and ascorbate, while methionine sulfoxide is not reduced back to methionine under the same conditions (Krause and Elfarra 2009; Assmann et al. 1998). This ease of reduction is directly related to the weaker and longer Se=O bond compared to the S=O bond. For example, the bond dissociation energy (BDE) of the Se=O bond is 101.5 kcal/mol (Smoes and Drowart 1984), while the BDE of the S=O bond is 124.7 kcal/mol (Benson 1978). The bond length of the Se–O bond in selenite is 1.7 Å, while the S–O bond length in sulfite is 1.5 Å (Larsson and Kierkegaard 1969; Wickleder 2002). The weakness of the Se–O bond is reflected by the fact that racemization of a selenoxide in water occurs on a time scale of minutes, while racemization of sulfoxides occurs on a time scale of days or longer (Shimizu et al 1988). Thus when methionine is oxidized to methionine sulfoxide, two isomers result (R, S) and these two stereoisomers are stereochemically frozen on a biological time scale. This is obviously why two different methionine sulfoxide reductases, each specific for either the R or S isomer, have evolved.
The ease of reduction and racemization of the selenoxide compared to the sulfoxide is a direct consequence of the greater electrophilic and hypervalent character of Se in comparison to S (as we have discussed above). This is because the reduction of the selenoxide involves attack of a thiolate on the Se atom of R2Se=O, and racemization involves attack of the oxygen atom of water on the Se atom of the selenoxide. The racemization of the selenoxide is facilitated by its oxygen atom first becoming protonated (Shimizu et al 1988), and this is also likely to be true in the case of the reduction of the selenoxide.
Our hypothesis that the Se atom in TR confers high resistance to oxidation is supported by our recent discovery that methaneseleninate (CH3SeO2−) is a substrate for the truncated TR. The rate of reduction of 200 μM CH3SeO2− is 12 mol NADPH/min/mol TR at pH 7.4, but is much faster at pH 6.1 (300 mol NADPH/min/mol TR) (Grout, Snider, and Hondal, unpublished data). Previously, it was known that methaneseleninate was a substrate for TR, but it was not determined which redox center of the enzyme is responsible for this activity (Gromer and Gross 2002). Our studies show that the full-length TR (containing Sec) reduces methaneseleninate, but only fourfold faster than the truncated enzyme containing only the N-terminal redox center. Seleninic acids are known to be good electrophiles and this example again illustrates the far superior electrophilicity of Se compared to S since the corresponding S-compound, hydroxymethanesulfinate (HOCH2SO2−) was not turned over by the truncated TR. This suggests to us that it would be extremely difficult to “overoxidize” TR, because as shown in Fig. 9, the Se atom would be oxidized to the seleninic acid form, but this could then be an internal substrate for the N-terminal reaction center. The N-terminal reaction center would then be a type of “internal rescue system” that would restore the function of the enzyme. If this is true, then TR might be at the top of a “redox pyramid” and as bursts of peroxide signals move through a cell, the function of TR would then be to restore the cell back to redox homeostasis. Experiments are underway in my laboratory to further substantiate this hypothesis.
Fig. 9.
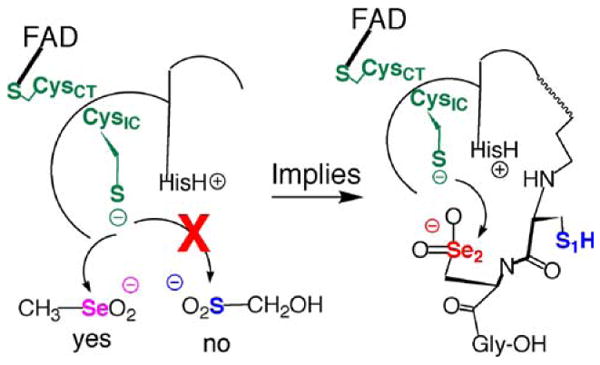
Proposed internal rescue system of TR for reducing an oxidized seleninic acid residue in the C-terminal reaction center by the N-terminal reaction center. Our results show that the truncated enzyme will reduce methaneseleninic acid as an external substrate. This result strongly implies that when the Sec residue in the C-terminal redox center becomes oxidized it can be quickly reduced back to Sec-SeH or Sec-Se–S-R, restoring the activity of the enzyme and rendering TR resistant to inactivation
Evidence for our hypothesis from the study of selenosubtilisin
Selenosubtilisin was the first semisynthetic selenoenzyme produced and studied. Selenosubtilisin is made by chemically converting the active site Ser residue of subtilisin to Sec (Wu and Hilvert 1989). The resulting selenoenzyme has peroxidase activity (Wu and Hilvert 1990). The crystal structure of selenosubtilisin shows that it is in the seleninic acid form (Enz-SeO2−) (Syed et al. 1993). NMR studies of selenosubtilisin show that it also exists in the selenenic acid (Enz-SeOH) and selenosulfide (Enz-Se-SR) forms (House et al. 1993). If our hypothesis is incorrect, then the Enz-SeO2− form should be inactive, however, all three forms of selenosubtilisin have peroxidase activity (Bell et al. 1993). The data shows that the Enz-SeO2− form of selenosubtilisin is not irreversibly inactivated as would have occurred had this been a Cys-containing enzyme, but rather the Enz-SeO2− form is converted back to an active form after treatment of two equivalents of thiol (Bell et al. 1993 and see Scheme I of Syed et al. 1993).
Is the chemico-enzymatic function of Se in TR related to the chemico-biological function of Se in TR?
As shown in Fig. 5, the N-terminal redox center will only react with strong electrophiles. The reason for this must be that the thiolate of CysIC is a weak nucleophile. We suggest the reason for the weak nucleophilicity of CysIC is that this is an evolved mechanism that protects CysIC from being converted to CysIC-SOH. The weak nucleophilicity of CysIC thus makes it resistant to oxidation and inactivation, and this resistance to oxidation would enable CysIC to be in the active form so that it can reduce the oxidized seleninic acid in the C-terminal redox center of TR. In initial experiments, we could incubate the truncated TR with up to 50 mM H2O2 without inactivating its DTNB reductase activity (Snider and Hondal, unpublished data). If the thiolate of CysIC of TR evolved into a weak nucleophile as a protective mechanism, this would then necessitate the enzyme to increase the electrophilicity of the C-terminal redox center so it could more easily accept electrons from the N-terminal reaction center. A selenosulfide bond was one way of accomplishing this increased reactivity. So for TR, the issues of the chemico-enzymatic function of Se (high electrophilicity) and chemico-biological function of Se (seleninic acid form converted easily back to the selenol) are related if this hypothesis is correct.
Conclusion
The specific chemical function (chemico-enzymatic function) of Se in TR has historically been described in terms of its superior nucleophilicity. While Se is a stronger nucleophile than S, the six to tenfold difference between these two atoms does not account for the overall loss in Trxreductase activity when Sec is mutated to Cys, nor does this difference account for the high expense of maintaining the components of the Sec-insertion machinery. Our group had previously argued for the requirement of Se in the active site of TR in terms of superior leaving group ability, but this property does not explain the broad substrate specificity of the N-terminal redox center. We now believe that Se is needed due to its superior electrophilicity because this property is shared with all of the small molecule substrates that are reduced by the N-terminal redox center. This property, however, does not account for the biological pressure on the genome that is needed to maintain the presence of the Sec-insertion machinery because Cys-TRs such as DmTR have evolved to catalyze the reduction of their cognate Trx molecules with high efficiency. We name the biological pressure to maintain the Sec-insertion machinery the chemico-biological function of Se. We believe that this chemico-biological function is the superior ability of Se to be highly resistant to irreversible oxidation, conferring Se-containing enzymes resistance to inactivation by oxidation. Unlike the other physicochemical properties of Se and S, which are very similar, the chemical rate of recycling of the oxidized Sec residue back to its parent form is very large compared to the same cycling of an oxidized Cys residue (Cys-SO2− to Cys-SH). This ability to resist irreversible oxidation can explain the biological pressure to maintain the Sec-insertion machinery.
Finally, while we have presented a novel hypothesis for the chemico-biological function of Se in enzymes (resistance to inactivation by resisting irreversible oxidation), we cannot, however, at the present time rule out that the use of Sec in some enzymes is for a unique and specific purpose (niche rationale). We hope the hypothesis presented here will spur more research to uncover the reason for the rare, but wide spread use of Sec in enzymes.
Acknowledgments
The authors would like to express deep gratitude to Hans J. Reich for the many hours of conversation spent on this subject and for his help in guiding our hypotheses. This work was supported by National Institutes of Health Grant GM070742 to RJH.
Footnotes
There are in fact 25 genes for selenocysteine-containing proteins in the human genome. The actual number of proteins is somewhat larger due to alternatively spliced transcripts.
There is considerable confusion in the literature in calculating the oxidation number of sulfur and selenium compounds. For the compounds shown in Fig. 8, we have chosen to base the oxidation number on that of sulfuric acid (S = +6). Using this as a reference compound, the S atom in methyl phenyl sulfoxide has an oxidation number of +4 and the S atom in methyl phenyl sulfone has an oxidation number of +6. This convention has the benefit of having the valence number equal to the oxidation number.
References
- Anestål K, Prast-Nielsen S, Cenas N, Arnér ES. Cell death by SecTRAPs: thioredoxin reductase as a prooxidant killer of cells. PLoS One. 2008;3:e1846. doi: 10.1371/journal.pone.0001846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arscott LD, Gromer S, Schirmer RH, Becker K, Williams CH., Jr The mechanism of thioredoxin reductase from human placenta is similar to the mechanisms of lipoamide dehydrogenase and glutathione reductase and is distinct from the mechanism of thioredoxin reductase from Escherichia coli. Proc Natl Acad Sci USA. 1997;94:3621–3626. doi: 10.1073/pnas.94.8.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assmann A, Briviba K, Sies H. Reduction of methionine selenoxide to selenomethionine by glutathione. Arch Biochem Biophys. 1998;349:201–203. doi: 10.1006/abbi.1997.0462. [DOI] [PubMed] [Google Scholar]
- Atkins JF, Gesteland RF. The twenty-first amino acid. Nature. 2000;407:463, 465. doi: 10.1038/35035189. [DOI] [PubMed] [Google Scholar]
- Bachrach SM, Demoin DW, Luk M, Miller JV., Jr Nucleophilic attack at selenium in diselenides and selenosulfides. A computational study. J Phys Chem A. 2004;108:4040–4046. [Google Scholar]
- Back TG. Electrophilic selenium reactions. In: Liotta D, editor. Organoselenium chemistry. Wiley-Interscience; New York: 1987. pp. 1–125. [Google Scholar]
- Bauer H, Massey V, Arscott LD, Schirmer RH, Ballou DP, Williams CH., Jr The mechanism of high Mr thioredoxin reductase from Drosophila melanogaster. J Biol Chem. 2003;278(3):3020–3028. doi: 10.1074/jbc.M303762200. [DOI] [PubMed] [Google Scholar]
- Bell IM, Fisher ML, Wu ZP, Hilvert D. Kinetic studies on the peroxidase activity of selenosubtilisin. Biochemistry. 1993;32:3754–3762. doi: 10.1021/bi00065a030. [DOI] [PubMed] [Google Scholar]
- Benson SW. Thermochemistry and kinetics of sulfur-containing molecules and radicals. Chem Rev. 1978;78:23–35. [Google Scholar]
- Birringer M, Pilawa P, Flohe F. Trends in selenium biochemistry. Nat Prod Rep. 2002;19:693–718. doi: 10.1039/b205802m. [DOI] [PubMed] [Google Scholar]
- Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- Biterova EI, Turanov AA, Gladyshev VN, Barycki JJ. Crystal structures of oxidized and reduced mitochondrial thioredoxin reductase provide molecular details of the reaction mechanism. Proc Natl Acad Sci USA. 2005;102:15018–15023. doi: 10.1073/pnas.0504218102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock A, Forchhammer K, Heider J, Leinfelder W, Sawers G, Veprek B, Zinoni F. Selenocysteine: the 21st amino acid. Mol Microbiol. 1991;5:515–520. doi: 10.1111/j.1365-2958.1991.tb00722.x. [DOI] [PubMed] [Google Scholar]
- Burk RF, Hill KE. Selenoprotein P: an extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu Rev Nutr. 2005;25:215–235. doi: 10.1146/annurev.nutr.24.012003.132120. [DOI] [PubMed] [Google Scholar]
- Burns JA, Whitesides GM. Predicting the stability of cyclic disulfides by molecular modeling: “effective concentrations” in thiol-disulfide interchange and the design of strongly reducing dithiols. J Am Chem Soc. 1990;112:6296–6303. [Google Scholar]
- Carugo O, Cemazar M, Zahariev S, Hudáky I, Gáspári Z, Perczel A, Pongor S. Vicinal disulfide turns. Protein Eng. 2003;16:637–639. doi: 10.1093/protein/gzg088. [DOI] [PubMed] [Google Scholar]
- Cheng Q, Sandalova T, Lindqvist Y, Arnér ES. Crystal structure and catalysis of the selenoprotein thioredoxin reductase-1. J Biol Chem. 2009;284:3998–4008. doi: 10.1074/jbc.M807068200. [DOI] [PubMed] [Google Scholar]
- Coles MP. Utilization of nonbonded interactions involving organoselenium compounds. Curr Org Chem. 2006;10:1993–2005. [Google Scholar]
- Danehy JP, Noel CJ. The relative nucleophilic character of several mercaptans toward ethylene oxide. J Am Chem Soc. 1960;82:2511–2515. [Google Scholar]
- Danehy JP, Elia VJ, Lavelle CJ. The alkaline decomposition of organic disulfides. IV. A limitation on the use of Ellman's reagent, 2,2′-dinitro-5,5′ -dithiodibenzoic acid. J Org Chem. 1971;36:1003–1005. [Google Scholar]
- Eckenroth B, Harris K, Turanov AA, Gladyshev VN, Raines RT, Hondal RJ. Semisynthesis and characterization of mammalian thioredoxin reductase. Biochemistry. 2006;45:5158–5170. doi: 10.1021/bi0517887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckenroth BE, Lacey BM, Lothrop AP, Harris KM, Hondal RJ. Investigation of the C-terminal redox center of high Mr thioredoxin reductases by protein engineering and semisynthesis. Biochemistry. 2007a;46:9472–9483. doi: 10.1021/bi7004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckenroth BE, Rould MA, Hondal RJ, Everse SJ. Structural and biochemical studies reveal differences in the catalytic mechanisms of mammalian and Drosophila melanogaster thioredoxin reductases. Biochemistry. 2007b;46:4694–4705. doi: 10.1021/bi602394p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlayson AJ, MacKenzie SL, Finley JW. Reaction of alanine-3-sulfinic acid with 2-mercaptoethanol. Can J Chem. 1979;57:2073–2077. [Google Scholar]
- Flemer S, Jr, Lacey BM, Hondal RJ. Synthesis of peptide substrates for mammalian thioredoxin reductase. J Pept Sci. 2008;14:637–647. doi: 10.1002/psc.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forchhammer K, Leinfelder W, Bock A. Identification of a novel translation factor necessary for the incorporation of selenocysteine into protein. Nature. 1989;342:453–456. doi: 10.1038/342453a0. [DOI] [PubMed] [Google Scholar]
- Gascoigne IM, Radda GK. The chemistry of flavins and flavoproteins. 3. The reaction of dihydrolipoic acid with flavins. Biochim Biophys Acta. 1967;131:498–507. doi: 10.1016/0005-2728(67)90009-6. [DOI] [PubMed] [Google Scholar]
- Gladyshev VN. Plenary lecture. 8th International symposium on selenium in biology and medicine; Madison, WI, USA. 2006. [Google Scholar]
- Gladyshev VN, Kryukov GV. Evolution of selenocysteine-containing proteins: significance of identification and functional characterization of selenoproteins. Biofactors. 2001;14:87–92. doi: 10.1002/biof.5520140112. [DOI] [PubMed] [Google Scholar]
- Gladyshev VN, Krause M, Xu XM, Korotkov KV, Kryukov GV, Sun QA, Lee BJ, Wootton JC, Hatfield DL. Selenocysteine-containing thioredoxin reductase in C. elegans. Biochem Biophys Res Commun. 1999;259:244–249. doi: 10.1006/bbrc.1999.0765. [DOI] [PubMed] [Google Scholar]
- Gromer S, Gross JH. Methylseleninate is a substrate rather than an inhibitor of mammalian thioredoxin reductase. Implications for the antitumor effects of selenium. J Biol Chem. 2002;277:9701–9706. doi: 10.1074/jbc.M109234200. [DOI] [PubMed] [Google Scholar]
- Gromer S, Wissing J, Behne D, Ashmans D, Schirmer RH, Flohe L, Becker K. A hypothesis on the catalytic mechanism of the selenoenzyme thioredoxin reductase. Biochem J. 1998;332:591–592. doi: 10.1042/bj3320591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromer S, Johansson L, Bauer H, Arscott LD, Rauch S, Ballou DP, Williams CH, Jr, Schirmer RH, Arnér ES. Active sites of thioredoxin reductases: why selenoproteins? Proc Natl Acad Sci USA. 2003;100:12618–12623. doi: 10.1073/pnas.2134510100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heider J, Baron C, Bock A. Coding from a distance: dissection of the mRNA determinants required for the incorporation of selenocysteine into protein. EMBO J. 1992;11:3759–3766. doi: 10.1002/j.1460-2075.1992.tb05461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellwinkel D. Hypervalent organic derivatives of tellurium and selenium. Ann NY Acad Sci. 1972;192:158–166. doi: 10.1111/j.1749-6632.1972.tb52586.x. [DOI] [PubMed] [Google Scholar]
- Herschlag D. Ribonuclease revisited: catalysis via the classical general acid-base mechanism or a triester-like mechanism? J Am Chem Soc. 1994;116:11631–11635. [Google Scholar]
- Hondal RJ, Nilsson BL, Raines RT. Selenocysteine in native chemical ligation and expressed protein ligation. J Am Chem Soc. 2001;123:5140–5141. doi: 10.1021/ja005885t. [DOI] [PubMed] [Google Scholar]
- House KL, Garber AR, Dunlap RB, Odom JD, Hilvert D. 1H-NMR spectroscopic studies of selenosubtilisin. Biochemistry. 1993;32:3468–3473. doi: 10.1021/bi00064a034. [DOI] [PubMed] [Google Scholar]
- Huang HH, Arscott LD, Ballou DP, Williams CH., Jr Acid-base catalysis in the mechanism of thioredoxin reductase from Drosophila melanogaster. Biochemistry. 2008;47:1721–1731. doi: 10.1021/bi702040u. [DOI] [PubMed] [Google Scholar]
- Huber RE, Criddle RS. Comparison of the chemical properties of selenocysteine and selenocysteine and their sulfur analogs. Arch Biochem Biophys. 1967;122:164–173. doi: 10.1016/0003-9861(67)90136-1. [DOI] [PubMed] [Google Scholar]
- Hudaky I, Gaspari Z, Carugo O, Cemazar M, Pongor S, Perczel A. Vicinal disulfide bridge conformers by experimental methods and by ab initio and DFT molecular computations. Proteins. 2004;55:152–168. doi: 10.1002/prot.10581. [DOI] [PubMed] [Google Scholar]
- Jacob C, Giles GI, Giles NM, Sies H. Sulfur and selenium: the role of oxidation state in protein structure and function. Angew Chem Int Ed Engl. 2003;42:4742–4758. doi: 10.1002/anie.200300573. [DOI] [PubMed] [Google Scholar]
- Jez JM, Noel JP. Mechanism of chalcone synthase. pKa of the catalytic cysteine and the role of the conserved histidine in a plant polyketide synthase. J Biol Chem. 2000;275:39640–39646. doi: 10.1074/jbc.M008569200. [DOI] [PubMed] [Google Scholar]
- Johnson FA, Lewis SD, Shafer JA. Perturbations in the free energy and enthalpy of ionization of histidine-159 at the active site of papain as determined by fluorescence spectroscopy. Biochemistry. 1981;20:52–58. doi: 10.1021/bi00504a600. [DOI] [PubMed] [Google Scholar]
- Jukes TH. Genetic code 1990. Outlook. Experientia. 1990;46:1149–1157. doi: 10.1007/BF01936925. [DOI] [PubMed] [Google Scholar]
- Kanzok SM, Fechner A, Bauer H, Ulschmid JK, Muller HM, Botella-Munoz J, Schneuwly S, Schirmer R, Becker K. Substitution of the thioredoxin system for glutathione reductase in Drosophila melanogaster. Science. 2001;291:643–646. doi: 10.1126/science.291.5504.643. [DOI] [PubMed] [Google Scholar]
- Kice JL, Lee TWS. Oxidation-reduction reactions of organoselenium compounds. 1. Mechanism of the reaction between seleninic acids and thiols. J Am Chem Soc. 1978;100:5094–5102. [Google Scholar]
- Kice JL, Sleblocka-Tilk H. Reactivity of nucleophiles toward and the site of nucleophilic attack on bis(alkylthio) selenides. J Am Chem Soc. 1982;104:7123–7130. [Google Scholar]
- Kice JL, Lee TWS, Pan St. Mechanism of the reaction of thiols with selenite. J Am Chem Soc. 1980;102:4448–4455. [Google Scholar]
- Krause RJ, Elfarra AA. Reduction of l-methionine selenoxide to seleno-l-methionine by endogenous thiols, ascorbic acid, or methimazole. Biochem Pharmacol. 2009;77:134–140. doi: 10.1016/j.bcp.2008.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravchuk AV, Zhao L, Kubiak RJ, Bruzik KS, Tsai MD. Mechanism of phosphatidylinositol-specific phospholipase C: origin of unusually high nonbridging thio effects. Biochemistry. 2001;40:5433–5439. doi: 10.1021/bi002372q. [DOI] [PubMed] [Google Scholar]
- Krol A. Evolutionarily different RNA motifs and RNA-protein complexes to achieve selenoprotein synthesis. Biochimie. 2002;84:765–774. doi: 10.1016/s0300-9084(02)01405-0. [DOI] [PubMed] [Google Scholar]
- Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, Gladyshev VN. Characterization of mammalian selenoproteomes. Science. 2003;300:1439–1443. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- Kumar S, Bjornstedt M, Holmgren A. Selenite is a substrate for calf thymus thioredoxin reductase and thioredoxin and elicits a large non-stoichiometric oxidation of NADPH in the presence of oxygen. Eur J Biochem. 1992;207:435–439. doi: 10.1111/j.1432-1033.1992.tb17068.x. [DOI] [PubMed] [Google Scholar]
- Lacey BM, Hondal RJ. Characterization of mitochondrial thioredoxin reductase from C. elegans. Biochem Biophys Res Commun. 2006;346:629–636. doi: 10.1016/j.bbrc.2006.05.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey BM, Eckenroth BE, Flemer S, Hondal RJ. Selenium in thioredoxin reductase: a mechanistic perspective. Biochemistry. 2008;47:12810–12821. doi: 10.1021/bi800951f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson LO, Kierkegaard P. The crystal structure of sodium sulphite: a refinement allowing for the effect of crystal twinning. Acta Chem Scand. 1969;23:2253–2260. [Google Scholar]
- Lee SR, Bar-Noy S, Kwon J, Levine RL, Stadtman TC, Rhee SG. Mammalian thioredoxin reductase: oxidation of the C-terminal cysteine/selenocysteine active site forms a thioselenide, and replacement of selenium with sulfur markedly reduces catalytic activity. Proc Natl Acad Sci USA. 2000;97:2521–2526. doi: 10.1073/pnas.050579797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinfelder W, Forchhammer K, Zinoni F, Sawers G, Mandrand-Berthelot MA, Bock A. Escherichia coli genes whose products are involved in selenium metabolism. J Bacteriol. 1988a;170:540–546. doi: 10.1128/jb.170.2.540-546.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinfelder W, Zehelein E, Mandrand-Berthelot MA, Bock A. Gene for a novel tRNA species that accepts l-serine and cotranslationally inserts selenocysteine. Nature. 1988b;331:723–725. doi: 10.1038/331723a0. [DOI] [PubMed] [Google Scholar]
- Lewis SD, Johnson FA, Shafer JA. Effect of cysteine-25 on the ionization of histidine-159 in papain as determined by proton nuclear magnetic resonance spectroscopy. Evidence for a His-159–Cys-25 ion pair and its possible role in catalysis. Biochemistry. 1981;20:48–51. doi: 10.1021/bi00504a009. [DOI] [PubMed] [Google Scholar]
- Lothrop AP, Ruggles EL, Hondal RJ. No selenium required: Reactions catalyzed by mammalian thioredoxin reductase that are independent of a selenocysteine residue. Biochemistry. 2009;48:6213–6223. doi: 10.1021/bi802146w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low SC, Berry MJ. Knowing when not to stop: selenocysteine incorporation in eukaryotes. Trends Biochem Sci. 1996;21:203–208. [PubMed] [Google Scholar]
- Maeda H, Katayama K, Matsuno H, Uno T. 3′-(2,4-Dinitrobenzenesulfonyl)-2′, 7′-dimethylfluorescein as a fluorescent probe for selenols. Angew Chem Int Ed Engl. 2006;45:1810–1813. doi: 10.1002/anie.200504299. [DOI] [PubMed] [Google Scholar]
- Mugesh G, Sing HB. Synthetic organoselenium compounds as antioxidants: glutathione peroxidase activity. Chem Soc Rev. 2000;29:347–357. [Google Scholar]
- Musher JI. Some aspects of bonding and structure in hypervalent selenium and tellurium chemistry. Ann NY Acad Sci. 1972;192:52–59. doi: 10.1111/j.1749-6632.1972.tb52576.x. [DOI] [PubMed] [Google Scholar]
- Nalvarte I, Damdimopoulos AE, Nystöm C, Nordman T, Miranda-Vizuete A, Olsson JM, Eriksson L, Björnstedt M, Arnér ES, Spyrou G. Overexpression of enzymatically active human cytosolic and mitochondrial thioredoxin reductase in HEK-293 cells: effect on cell growth and differentiation. J Biol Chem. 2004;279:54510–54517. doi: 10.1074/jbc.M408494200. [DOI] [PubMed] [Google Scholar]
- Nelson JW, Creighton TE. Reactivity and ionization of the active site cysteine residues of DsbA, a protein required for disulfide bond formation in vivo. Biochemistry. 1994;33:5974–5983. doi: 10.1021/bi00185a039. [DOI] [PubMed] [Google Scholar]
- Oae S, Togo H, Numata T, Fujimori K. Facile reduction of sulfinic acid to disulfide with thiol and chlorotrimethylsilane. Chem Lett. 1980;9:1193–1196. [Google Scholar]
- Odom JD. Selenium biochemistry chemical and physical studies. Struct Bond. 1983;54:1–26. [Google Scholar]
- Osawa S, Jukes TH, Watanabe K, Muto A. Recent evidence for evolution of the genetic code. Microbiol Rev. 1992;56:229–264. doi: 10.1128/mr.56.1.229-264.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson RG, Songstad J. Application of the principle of hard and soft acids and bases to organic chemistry. J Am Chem Soc. 1967;89:1827–1836. [Google Scholar]
- Pearson RG, Songstad J. Nucleophilic reactivity constants toward methyl iodide and trans-[Pt(py)2Cl2] J Am Chem Soc. 1968;90:319–326. [Google Scholar]
- Rabenstein DR, Scott TM, Geo W. Nuclear magnetic resonance study of the kinetics of the penicillamine/bis(penicillamine) selenide symmetrical exchange reaction. J Org Chem. 1991;56:4176–4181. [Google Scholar]
- Reich HJ. Organoselenium oxidations. In: Trahanovsky WW, editor. Oxidation in organic chemistry, part C. Academic Press; New York: 1978. [Google Scholar]
- Reich HJ, Gudmundsson BO, Green DP, Bevan MJ, Reich IL. The role of ate complexes in the lithium-sulfur, lithium-selenium and lithium-tellurium exchange reactions. Helv Chim Acta. 2002;85:3748–3772. [Google Scholar]
- Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- Robinson R. Selenium speeds reactions. PLoS Biol. 2005;3:e419. [Google Scholar]
- Sandman KE, Benner JS, Noren CJ. Phage display of selenopeptides. J Am Chem Soc. 2000;122:960–961. [Google Scholar]
- Sarma BK, Mugesh G. Glutathione peroxidase (GPx)-like antioxidant activity of the organoselenium drug ebselen: unexpected complications with thiol exchange reactions. J Am Chem Soc. 2005;127:11477–11485. doi: 10.1021/ja052794t. [DOI] [PubMed] [Google Scholar]
- Sarma BK, Mugesh G. Biomimetic studies on selenoenzymes: modeling the role of proximal histidines in thioredoxin reductases. Inorg Chem. 2006;45:5307–5314. doi: 10.1021/ic052033r. [DOI] [PubMed] [Google Scholar]
- Seebach D, Meyer N, Beck AK. α-Heterosubstituted organyllithium compounds by selenium/lithium-exchange; directed coupling of carbonyl compounds. Justus Liebigs Ann Chem. 1977;5:846–858. [Google Scholar]
- Shimizu T, Kobayahi M, Kamigata N. Configurational stability of optically active selenoxides: racemization via achiral hydrate. Bull Chem Soc Jpn. 1988;61:3761–3763. [Google Scholar]
- Singh R, Whitesides GM. Degenerate intermolecular thiolatedisulfide interchange involving cyclic five-membered disulfides is faster by 103 than that involving six- or seven-membered disulfides. J Am Chem Soc. 1990;112:6304–6309. [Google Scholar]
- Smoes S, Drowart J. Determination of the dissociation energy of selenium monoxide by the mass-spectrometric Knudsen-cell method. J Chem Soc Faraday Trans 2. 1984;80:1171–1180. [Google Scholar]
- Squires JE, Berry MJ. Eukaryotic selenoprotein synthesis: mechanistic insight incorporating new factors and new functions for old factors. IUBMB Life. 2008;60:232–235. doi: 10.1002/iub.38. [DOI] [PubMed] [Google Scholar]
- Syed R, Wu ZP, Hogle JM, Hilvert D. Crystal structure of selenosubtilisin at 2.0-Å resolution. Biochemistry. 1993;32:6157–6164. doi: 10.1021/bi00075a007. [DOI] [PubMed] [Google Scholar]
- Taskov K, Chapple C, Kryukov GV, Castellano S, Lobanov AV, Korotkov KV, Guigó R, Gladyshev VN. Nematode selenoproteome: the use of the selenocysteine insertion system to decode one codon in an animal genome? Nucleic Acids Res. 2005;33:2227–2238. doi: 10.1093/nar/gki507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tormay P, Böck A. Barriers to heterologous expression of a selenoprotein gene in bacteria. J Bacteriol. 1997;179:576–582. doi: 10.1128/jb.179.3.576-582.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessjohann LA, Schneider A, Abbas M, Brandt W. Selenium in chemistry and biochemistry in comparison to sulfur. Biol Chem. 2007;388:997–1006. doi: 10.1515/BC.2007.138. [DOI] [PubMed] [Google Scholar]
- Wickleder MS. Sodium selenite, Na2SeO3. Acta Cryst. 2002;E58:i103–i104. [Google Scholar]
- Williams CH, Arscott LD, Muller S, Lennon BW, Ludwig ML, Wang PF, Veine DM, Becker K, Schirmer RH. Thioredoxin reductase: two modes of catalysis have evolved. Eur J Biochem. 2000;267:6110–6117. doi: 10.1046/j.1432-1327.2000.01702.x. [DOI] [PubMed] [Google Scholar]
- Wu ZP, Hilvert D. Conversion of a protease into an acyl transferase: selenosubtilisin. J Am Chem Soc. 1989;111:4513–4514. [Google Scholar]
- Wu ZP, Hilvert D. Selenosubtilisin as a glutathione peroxidase mimic. J Am Chem Soc. 1990;112:5647–5648. [Google Scholar]
- Zhong L, Holmgren A. Essential role of selenium in the catalytic activities of mammalian thioredoxin reductase revealed by characterization of recombinant enzymes with selenocysteine mutations. J Biol Chem. 2000;275:18121–18128. doi: 10.1074/jbc.M000690200. [DOI] [PubMed] [Google Scholar]