

Abstract
Background and purpose:
Although microsomal prostaglandin E synthase (mPGES)-1 is known to contribute to stroke injury, the underlying mechanisms remain poorly understood. This study examines the hypothesis that EP3 receptors contribute to stroke injury as downstream effectors of mPGES-1 neurotoxicity through Rho kinase activation.
Experimental approach:
We used a glutamate-induced excitotoxicity model in cultured rat and mouse hippocampal slices and a mouse middle cerebral artery occlusion–reperfusion model. Effects of an EP3 receptor antagonist on neuronal damage in mPGES-1 knockout (KO) mice was compared with that in wild-type (WT) mice.
Key results:
In cultures of rat hippocampal slices, the mRNAs of EP1–4 receptors were constitutively expressed and only the EP3 receptor antagonist ONO-AE3-240 attenuated and only the EP3 receptor agonist ONO-AE-248 augmented glutamate-induced excitotoxicity in CA1 neurons. Hippocampal slices from mPGES-1 KO mice showed less excitotoxicity than those from WT mice and the EP3 receptor antagonist did not attenuate the excitotoxicity. In transient focal ischaemia models, injection (i.p.) of an EP3 antagonist reduced infarction, oedema and neurological dysfunction in WT mice, but not in mPGES-1 KO mice, which showed less injury than WT mice. EP3 receptor agonist-induced augmentation of excitotoxicity in vitro was ameliorated by the Rho kinase inhibitor Y-27632 and Pertussis toxin. The Rho kinase inhibitor HA-1077 also ameliorated stroke injury in vivo.
Conclusion and implications:
Activity of mPGES-1 exacerbated stroke injury through EP3 receptors and activation of Rho kinase and/or Gi. Thus, mPGES-1 and EP3 receptors may be valuable therapeutic targets for treatment of human stroke.
This article is commented on by Andreasson, pp. 844–846 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2010.00715.x
Keywords: ischaemia, prostaglandin E2, prostaglandin E synthase, mPGES-1, EP3 receptors, Rho kinase, excitotoxicity
Introduction
Stroke remains a major cause of death and neuronal disability worldwide. In the early stages of cerebral ischaemia, activation of glutamate receptors initiates the ischaemic cascade that causes most of the cerebral damage (Butcher et al., 1990; Lee et al., 1999). At later times after ischaemia, inflammation is a major factor in the progression of the injury (Barone and Feuerstein, 1999; Dirnagl et al., 1999).
Prostaglandin E2 (PGE2), one of the most likely candidates for propagation of inflammation, is known to be produced and to accumulate at the lesion sites of the ischaemic brain (Kempski et al., 1987; Iadecola et al., 2001; Ikeda-Matsuo et al., 2006). PGE2 is sequentially synthesized from arachidonic acid in two enzymic steps: cyclooxygenase (COX) and PGE2 synthase (PGES). Among the two COX isoforms, COX-1 and COX-2, COX-2 is the more highly inducible form (Kaufmann et al., 1997) and is involved in the pathogenic events occurring in cerebral ischaemia (Nogawa et al., 1997; Iadecola et al., 2001; Sasaki et al., 2004). Similarly, of the three PGES isozymes, cytosolic PGES, microsomal PGES (mPGES)-1 and mPGES-2, mPGES-1 is the only inducible form (Jakobsson et al., 1999; Murakami et al., 2002; Ikeda-Matsuo et al., 2005). Recently, using a model of focal cerebral ischaemia in mPGES-1 knockout (KO) mice, we demonstrated that induction of mPGES-1 contributed to the exacerbation of stroke injury through ischaemic PGE2 production (Ikeda-Matsuo et al., 2006). More recently, we have found that mPGES-1 activity is required for COX-2 to exert neuronal damage in ischaemic injury (Ikeda-Matsuo et al., 2010).
How, then, is the toxicity of post-ischaemic PGE2 mediated? PGE2 acts on four G protein-coupled receptors (EP1–EP4; nomenclature follows Alexander et al., 2009) that have very distinct and potentially antagonistic signalling cascades. EP1 receptors couple to Gq, and activation of this receptor results in increased intracellular Ca2+ concentrations (Kawano et al., 2006); EP2 and EP4 receptors couple to Gs to increase cyclic AMP (cAMP) formation, whereas most EP3 receptor isoforms, which are generated by alternative splicing, couple to Gi to decrease cAMP (Narumiya et al., 1999). EP3 receptors can also be coupled to the G12/13, resulting in activation of the small G protein Rho followed by activation of Rho kinase, an effector of RhoA (Katoh et al., 1996; Hatae et al., 2002; Shum et al., 2003; Macias-Perez et al., 2008). Thus, there is great potential for variability in the response of target cells to PGE2 based on the receptors activated (Narumiya et al., 1999). The various roles of the PGE2 receptors in neuronal death induced by excitotoxicity and ischaemic stroke have been clarified by genetic deletion and selective inhibition of each EP receptor. Deletion and inhibition of the EP1 receptors partially reduce the neuronal damage caused by excitotoxicity and ischaemic stroke (Ahmad et al., 2006; Kawano et al., 2006). Deletion of the EP2 receptors increases the infarct volume in mice after ischaemic stroke (McCullough et al., 2004). In contrast, a selective agonist of the EP2 receptor induces caspase-dependent apoptosis (Takadera et al., 2004) and a EP4 receptor agonist has been shown to reduce excitotoxic brain injury (Ahmad et al., 2005). Interestingly, a recent study showed that genetic deletion of the EP3 receptor reduces the neuronal damage caused by oxygen/glucose deprivation (OGD) or ischaemic stroke (Saleem et al., 2009), while the EP3 receptor agonist exacerbates acute excitotoxic or ischaemic-induced brain injury (Ahmad et al., 2007). We have also confirmed the amelioration of excitotoxicity and stroke injuries by inhibition and deletion of EP3 receptors through anti-inflammatory and anti-apoptotic mechanisms (under submission).
Thus, EP3 receptors could be one of the important effectors for the neurotoxicity of PGE2. However, several points remain to be elucidated. First, is the EP3 receptor a downstream effector of mPGES-1 neurotoxicity? Second, can an EP3 receptor antagonist protect against excitotoxicity? And finally, what is the mechanism underlying EP3 receptor-mediated neurotoxicity? In order to address these issues, we examined the hypothesis that EP3 receptors contribute to the exacerbation of stroke injury as downstream effectors of mPGES-1 neurotoxicity and that the activation of the EP3 receptors exerted its neurotoxic effect through Rho kinase and/or Gi activation. We performed both an in vitro study using hippocampal slices exposed to glutamate and an in vivo study employing transient focal ischaemia models in mPGES-1 KO and wild-type (WT) mice. The results demonstrated that an EP3 receptor antagonist conferred protection against neurotoxicity in vitro and in vivo in WT mice, but not in mPGES-1 KO mice, and that Rho kinase was involved in EP3 receptor-mediated neurotoxicity and ischaemic stroke.
Methods
Animals
All animal care and experimental procedures complied with the guidelines given by the Japanese Pharmacological Society. mPGES-1 KO mice and WT mice (C57BL/6J × 129/SvJ background) back-crossed to C57BL/6J mice for >8 generations to avoid artefactual differences caused by genetic background were used (Uematsu et al., 2002). In spite of the reported gender differences in infarct size after middle cerebral artery (MCA) occlusion and reperfusion (Hayashi et al., 2005), our preliminary data showed no significant gender differences in not only infarct volume, but also degree of oedema, neurological score and ischaemic PGE2 production 24 h after ischaemia (Ikeda-Matsuo et al., 2006). Therefore, both male and female mice were studied at a weight of 25–30 g and data from both sexes were pooled. Hayashi et al. (2005) used age-matched younger animals and a 90 min occlusion model, while we used adult animals weighing 24–30 g and a 120 min occlusion model to ensure adequacy of occlusion. The reason we could not observe gender differences may be connected to the severity of our protocol compared with that of Hayashi et al. (2005). Wistar rat pups were purchased from SLC (Shizuoka, Japan).
Organotypic hippocampal culture
Brains were rapidly removed from 7-day-old Wistar rat, mPGES-1 KO or WT mouse pups, and 300-µm-thick horizontal entorhino-hippocampal slices were placed on transparent membranes (Millicell-CM, Millipore, Bedford, MA, USA) in 6-well culture plate and cultured with 700 µl of culture medium consisting of 50% minimal essential medium, 25% horse serum and 25% Hanks' balanced salt solution supplemented with 3 mg·mL−1 glucose, 2 mM l-glutamine, 100 U·mL−1 penicillin G and 125 µg·mL−1 streptomycin in a humidified incubator at 37°C in 5% CO2. After 7 days in vitro, slice cultures were exposed to 1 mM glutamate for 15 min for rats and 30 min for mice with or without EP receptor antagonists, EP receptor agonists and/or NS-398. Then medium was changed to normal culture medium containing 5 µg·mL−1 propidium iodide (PI) with or without kinase inhibitors, EP receptor antagonists, EP receptor agonists and/or NS-398 and cultured for 24 h. After that, all cells were killed by 24 h incubation with 0.3% Triton X-100 at a low temperature (4°C). PI fluorescence images were obtained with the confocal laser scanning system (LSM510) on an Axiovert200M inverted microscope (Carl Zeiss, Germany). The fluorescence intensity at CA1 pyramidale was obtained by measuring averaged grey-scale values of the desired area using graphics software (Photoshop ver. 7.0, Adobe Systems, San Jose, CA, USA).
mRNA analysis
RNA extraction and the semiquantitative reverse transcriptase polymerase chain reaction were performed as described (Ikeda et al., 2000). PCR cycles were titrated to establish amplification conditions for each primer, to document linearity, and to permit quantitative analyses of signal strength; the conditions were as follows: denaturation at 94°C for 30 s, annealing at T°C, which is described below for each set of primers, for 30 s, and extension at 72°C for 45 s (X cycles). The cDNA fragments for EP1, EP2, EP3 and EP4 receptors was amplified with specific primers for mice (EP1: sense 5′-TTAACCTGAGCCTAGCGGATG-3′ and antisense 5′-CGCTGAGCGTATTGCACACTA-3′, 665 bp, T = 56°C, X = 30 cycles; EP2: sense 5′-AGGACTTCGATGGCAGAGGAGAC-3′ and antisense 5′-CAGCCCCTTACACTTCTCCAATG-3′, 402 bp, T = 60°C, X = 36 cycles; EP3α: sense 5′-TGACCTTTGCCTGCAACCTG-3′ and antisense 5′-AGCTGGAAGCATAGTTGGTG-3′, 377 bp, T = 56°C, X = 33 cycles; EP3β: sense 5′-CTAATTGCAGTTCGCCTGGCT-3′ and antisense 5′-CGTCTCAAGTGCAGAGTCTTC-3′, 320 bp, T = 58°C, X = 34 cycles; EP3γ: sense 5′-TGACCTTTGCCTGCAACCTG-3′ and antisense 5′-AGACAATGAGATGGCCTGCC-3′, 409 bp, T = 56°C, X = 33 cycles; EP4: sense 5′-CATTCCGCTCGTGGTGC-3′ and antisense 5′-AGGTGGTGTCTGCTTGGGT-3′, 425 bp, T = 56°C, X = 36 cycles) and rats (EP1: sense 5′-TTAACCTGAGCCTAGTGGATG-3′ and antisense 5′-CGCTGAGCGTATTACACACTA-3′, 665 bp, T = 56°C, X = 33 cycles; EP2: sense 5′-AGGACTTCTATGGCGGAGGAGAC-3′ and antisense 5′-CGGCCCTTTACGTTCCTCCAACG-3′, 402 bp, T = 60°C, X = 36 cycles; EP3: sense 5′-CCCGGCACGTGGTGCTTCAT-3′ and antisense 5′-AGCTGGAAGCATAGTTGGTG-3′, 437 bp, T = 56°C, X = 36 cycles; EP4: the same as mice). The quality of RNA samples was evaluated using GAPDH-specific primers (5′-AGA-CAG-CCG-CAT-CTT-CTT-GT-3′, 5′-CCA-CAG-TCT-TCT-GAG-TGG-CA-3′, T = 56°C, X = 20 cycles). PCR products were analysed on 2% agarose gels and visualized by ethidium bromide staining.
PGE2 assay
The concentration of the PGE2 in dissected brain tissues or culture medium was determined using an enzyme immunoassay kit (Cayman Chemical, Ann Arbor, MI, USA) as described previously (Ikeda-Matsuo et al., 2005). The brain tissues were quickly frozen in liquid nitrogen and weighed to determine the wet weight. Prostanoids were extracted by homogenization of the tissues in 70% methanol solution containing 10 µM indomethacin and centrifugation at 15 000×g for 20 min at 4°C. The supernatant was evaporated and dissolved and diluted with the assay buffer. The hippocampal culture medium was also diluted with the assay buffer. The PGE2 concentration was determined according to the instructions provided with the kit.
Induction of transient focal ischaemia
MCA occlusion was carried out under halothane anaesthesia as described previously (Ikeda-Matsuo et al., 2006). The right common carotid artery was exposed through a midline incision, and occlusion of the MCA was achieved by inserting a 6-0 nylon monofilament with a heat-blunted tip coated with silicon thread through the proximal external carotid artery into the internal carotid artery and up to the MCA (9 mm from the internal carotid/pterygopalatine artery bifurcation). The occlusion of the MCA was maintained for 2 h, followed by reperfusion for 24 h. The incision wound was treated with povidone iodine to avoid infection. A rectal thermometer was used to measure body temperature, which was maintained at 37°C by using a warm pad during the operation. The mortality associated with the technique was 6.4%. In sham-operated animals, an incision was made and the external carotid artery was tied off but the monofilament was not inserted, thereby avoiding MCA ischaemia. The EP3 receptor antagonist ONO-AE3-240 was suspended in 1% sodium carboxymethylcellulose (CMC) in physiological saline, and the vehicle consisted of 1% CMC saline alone. The EP3 receptor antagonist (3 mg·kg−1; i.p.) or vehicle was administered 2, 8 or 14 h after MCA occlusion. HA-1077 was diluted in saline and administered i.p. at 10 mg·kg−1 per day for 2 days before MCA occlusion. These mice were killed 24 h after MCA occlusion.
Quantification of infarct volume
The animals were killed and the brain tissue was removed. Brains were sectioned coronally into five 2 mm sections and incubated with 2% triphenyltetrazolium chloride (TTC) in saline for 10 min at 37°C. The area of infarct, identified by the lack of TTC staining, was measured on the rostral and caudal surfaces of each slice using Scion image software (Scion Corp., Frederick, MD, USA) and numerically integrated across the thickness of the slice to obtain an estimate of the infarct volume in each slice. The volumes from all slices were summed to calculate the total infarct volume over the entire infarcted hemisphere. The infarct volume was measured separately in the cerebral cortex, striatum and hemisphere, and corrected for swelling by comparing the volume of the neocortex in the infarcted hemisphere with that in the non-infarcted hemisphere. The infarct volumes of cortex or striatum was expressed as percentage of the whole brain volume [infarct volume = (infarct volume of cortex or striatum/volume of whole brain) × 100]. The degree of oedema was calculated as follows [oedema per cent = (volume of post-ischaemic hemisphere − volume of contralateral hemisphere)/volume of contralateral hemisphere × 100].
Behavioural experiment
All mice used for infarct volume estimation and Western blot analysis for Rho kinase activation were evaluated for post-ischaemic neurological deficits on a 5-point scale at 1 day of reperfusion by an investigator unaware of the treatment and genotypic conditions, as follows: 0, no deficit; 1, flexion of the torso and contralateral forelimb when lifted by the tail; 2, contralateral forelimb weakness upon application of pressure to the side of the body; 3, circling to the affected side; and 4, no spontaneous locomotor activity.
Western blotting
For measurement of Rho kinase activity, Western blot analysis was performed as previously described (Ikeda-Matsuo et al., 2005; Yamaguchi et al., 2009) with slight modifications. The cultured slices were treated with drug(s), and the reactions were terminated with ice-cold 5% trichloroacetic acid. The slices were sonicated and precipitated by centrifugation, and then solubilized in SDS sample buffer. Proteins were separated on SDS-polyacrylamide gels, and then transferred to PVDF membranes (Immobilon-P; Millipore). After blocking with 5% skim milk, the membranes were incubated with the appropriate primary antibody solution against anti-pT850 myosin-binding subunit (MBS) (1:1000) and anti-β-actin (1:20 000) in Solution 1 of the Can-get-Signal enhancer solutions for 1.5 h. Horseradish peroxidase (HRP)-conjugated anti-rabbit or mouse IgG antibody (1:20 000) was used as a secondary antibody and incubated in Solution 2 of the Can-get-Signal enhancer solutions for 1.5 h. Immunoreactive proteins were detected using LumiGLO, and the images were captured using a CCD camera (Light Capture; ATTO, Tokyo, Japan) and then analysed using ‘CS analyzer’ software (ATTO).
Statistical analysis
Results are expressed as the mean ± standard error. Statistical significance was evaluated with one-way analysis of variance followed by Tukey's method. For behavioural experiment, Steel-Dwass' method was used to analyse the data of neurological score. Values of P < 0.05 were considered to indicate statistical significance.
Materials
Selective agonists for EP1 (ONO-DI-004), EP2 (ONO-AE1-259), EP3 (ONO-AE-248) and EP4 (ONO-AE1-329) receptors, and selective antagonists for EP1 (ONO-8713), EP3 (ONO-AE3-240) and EP4 (ONO-AE3-208) receptors were gifts from Ono Pharmaceutical (Osaka, Japan). Each agonist and antagonist is highly selective for each receptor, and the Ki values were as follows: ONO-DI-004 (150 nM for the EP1 receptor and more than 10 µM for the other receptors); ONO-AE1-259 (3 nM for the EP2 receptor and more than 6 µM for the other receptors); ONO-AE-248 (7.5 nM for the EP3 receptor and more than 3 µM for the other receptors); ONO-AE1-329 (9.7 nM for the EP4 receptor and more than 1 µM for the other receptors); ONO-8713 (0.3 nM for the EP1 receptor and more than 1 µM for the other receptors); ONO-AE3-240 (590, 0.23 and 58 nM for the EP1, EP3 and EP4 receptors respectively, and more than 10 mM for the EP2 receptor); ONO-AE3-208 (30 and 1.3 nM for the EP3 and EP4 receptors respectively, and more than 10 µM for the other receptors) (Suzawa et al., 2000; Watanabe et al., 2000; Kabashima et al., 2002; Amano et al., 2003). NS-398, TTC and anti-β-actin monoclonal antibody were from Sigma (St Louis, MO, USA). H-89, Ro-31-8220 and Pertussis toxin (PTX) were from Calbiochem (Darmstadt, Germany). Other materials and their sources were as follows: anti-pT805 myosin-binding protein (MBP; Upstate, Charlottesville, VA, USA); HRP-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA); Y-27632 (Tocris, Ellisville, MO, USA); fasudil (HA-1077; Asahi Chemical Ind, Tokyo, Japan); LumiGLO Western blot detection reagent (Cell Signalling, Danvers, MA); Can-Get-Signal enhancer solution (Toyobo, Osaka, Japan). Other reagents were obtained from Wako Pure Chemical Industries (Osaka, Japan).
Results
Involvement of EP receptors in neuronal damage after transient ischaemic and excitotoxic injury
Before starting the in vitro study of ischaemic neurotoxicity, we first examined whether or not the production of PGE2 and the expression of EP receptors in cultured hippocampal slices exposed to glutamate showed tendencies similar to those in ischaemic cortices in vivo. We investigated the PGE2 content and EP mRNA levels in the mouse brain after 2 h of focal ischaemia followed by 24 h of reperfusion. In our experimental stroke model, the PGE2 production in the ipsilateral cortex of MCA-occluded mice was about threefold higher than that in the contralateral cortex (Figure 1A). Expression of the mRNAs of each of the EP1, EP2, EP3α, EP3β, EP3γ and EP4 receptors was observed in both the contralateral and ipsilateral cortex, and the mRNA levels of these receptors in the ipsilateral cortex were almost the same as those in the contralateral cortex (Figure 1B). We next investigated the production of PGE2 and expression of EP receptors in an in vitro ischaemia model. Rat hippocampal slices were stimulated with 1 mM glutamate for 15 min and then cultured with normal medium for 24 h. In hippocampal slice cultures, glutamate increased the PGE2 levels up to 2.5-fold higher than the control level (Figure 1C). All of the EP receptors were constitutively expressed in the hippocampal slices with or without glutamate exposure (Figure 1D).
Figure 1.
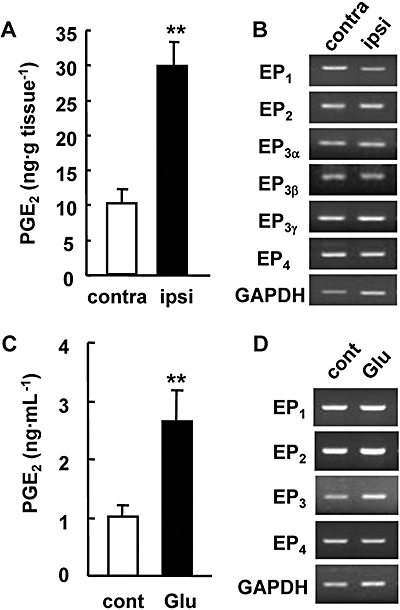
Production of prostaglandin E2 (PGE2) and the expression of EP receptors in the ischaemic cortex of the mice or in cultured rat hippocampal slices exposed to glutamate. (A) The production of PGE2 in the ipsilateral (ipsi) and contralateral (contra) cortex of mice 24 h after middle cerebral artery occlusion. n= 8 mice; **P < 0.01. (B) Reverse transcriptase polymerase chain reaction (RT-PCR) analysis for EP receptor mRNAs in the cortex 24 h after ischaemia. Representative data from three separate experiments are presented. GAPDH signals were used as loading controls. (C) The accumulation of PGE2 in the culture medium 24 h after 1 mM glutamate exposure for 15 min. n= 4; **P < 0.01. (D) RT-PCR analysis for EP receptor mRNAs in the cultured hippocampal slices 24 h after glutamate exposure. Representative data from three separate experiments are presented.
To elucidate the roles of EP receptors in the excitotoxicity induced by glutamate, cellular damage in the hippocampal slices was assessed by fluorescent image analysis of PI uptake (Figure 2A–D). The exposure of slices to glutamate resulted in neuronal death, which was detected as an increase in the uptake of PI in the CA1 region of the hippocampus (Figure 2C). The increase in PI uptake induced by glutamate exposure was inhibited significantly by the EP3 receptor antagonist ONO-AE3-240 in a concentration-dependent manner (0.1–10 µM), but not by other EP receptor antagonists even at a concentration of 10 µM (Figure 2E and F). Thus, we next investigated the effect of the EP3 receptor agonist ONO-AE-248 on glutamate-induced excitotoxicity. However, we could not detect any enhancement of glutamate-induced excitotoxicity even at the concentration of 1 µM (data not shown). Because we observed significant PGE2 production after glutamate exposure (Figure 1A) and a protective effect of the EP3 receptor antagonist on glutamate-induced excitotoxicity (Figure 2F), it was suggested that the extent of the contribution by endogenous PGE2 does not leave room for an additive effect on glutamate-induced excitotoxicity by EP receptor activation in our model. Therefore, to investigate the effect of EP receptor agonists, endogenous PGE2 production was inhibited by the COX-2 inhibitor NS-398 (Table 1). As expected, NS-398 attenuated approximately 50% of the glutamate-induced excitotoxicity (Figure 3A and B). The EP3 receptor agonist ONO-AE-248 exaggerated the excitotoxicity, while agonists for EP2 (ONO-AE1-259) and EP4 receptors (ONO-AE1-329) had no effects at the concentration of 1 µM (Figure 3D and E). Because the Ki values of ONO-AE-248, ONO-AE1-259 and ONO-AE1-329 for the EP2, EP3 and EP4 receptors, respectively, are similar, while that of the EP1 agonist (ONO-DI-004) for the EP1 receptor is 10–100 times lower (see the Methods section), we examined the effect of ONO-DI-004 at higher concentration. ONO-DI-004 did not affect glutamate-induced excitotoxicity even at the concentration of 10 µM (Figure 3C and E). ONO-AE-248 exacerbated the glutamate-induced excitotoxicity in a concentration-dependent manner (0.01–1 µM, Figure 3F), while it alone had no effect on the survival of CA1 neurons even at the concentration of 1 µM (data not shown).
Figure 2.
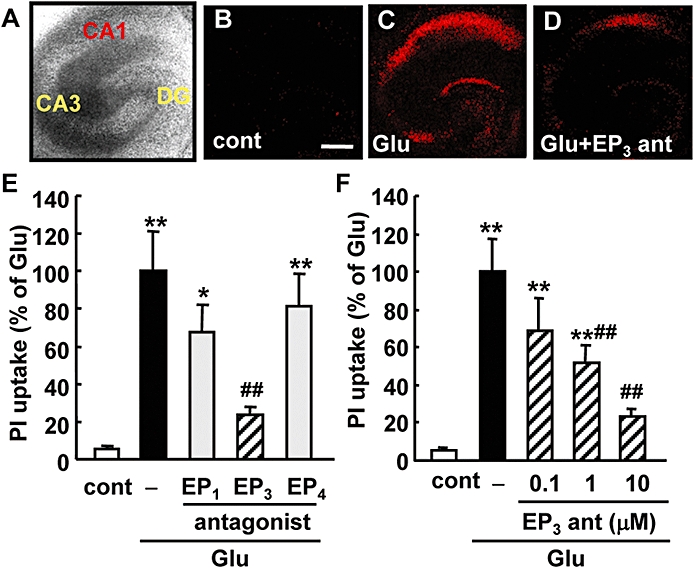
Effects of EP receptor antagonists on the glutamate-induced excitotoxicity in a rat hippocampal slice culture. EP receptor antagonists were added during and after the 1 mM glutamate exposure for 15 min. Twenty-four hours later, propidium iodide (PI) uptake was analysed. A representative differential interference microscopic image of a cultured hippocampal slice (A) and confocal images of PI fluorescence of a control slice (cont, B), a slice that received glutamate exposure (Glu, C) and a slice that received glutamate exposure with 10 µM of an EP3 receptor antagonist, ONO-AE3-240 (Glu + EP3 ant, D) are shown (scale bar: 400 µm). (E) Quantitative data from PI uptake analysis in the CA1 region with or without glutamate (Glu) and/or the EP1 receptor antagonist ONO-8713 (10 µM), EP3 receptor antagonist ONO-AE3-240 (10 µM) or EP4 receptor antagonist ONO-AE3-208 (10 µM) were scaled to a percentage of the response of glutamate alone. n= 5–14 slices per group. (F) Concentration-dependent protective effect of ONO-AE3-240 on glutamate-induced PI uptake in CA1. n= 5–12 slices per group. **P < 0.01, *P < 0.05 versus control, ##P < 0.05 versus glutamate alone, $$P < 0.01 versus glutamate with NS-398.
Table 1.
Production of PGE2 in the culture medium of hippocampal slices
| Rat | mPGES-1+/+ mice | mPGES-1−/− mice | |
|---|---|---|---|
| CTL | 1.01 ± 0.19 | 0.182 ± 0.020 | 0.183 ± 0.031 |
| GLU | 2.66 ± 0.52** | 0.273 ± 0.024* | 0.188 ± 0.028# |
| GLU + NS-398 | 0.29 ± 0.03## | – | – |
The amount of PGE2 (ng·mL−1) in the culture medium of slices from rats, mPGES-1 knockout (−/−) and wild-type (+/+) mice was measured 24 h after 1 mM glutamate (GLU) exposure and in control hippocampal slices (CTL). NS-398 (1 µM) were applied during and after the glutamate exposure (n= 3).
P < 0.01,
P < 0.05 versus control,
P < 0.01,
P < 0.05 versus glutamate.
mPGES, microsomal prostaglandin E synthase; PGE2, prostaglandin E2.
Figure 3.
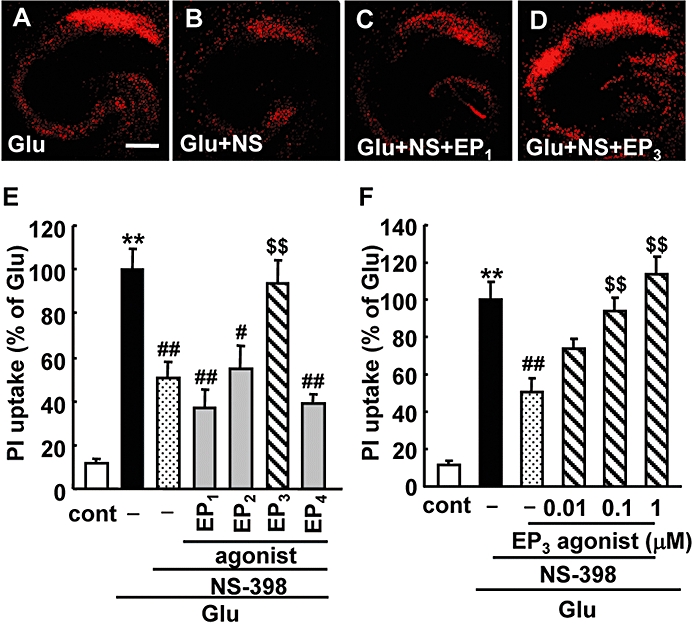
Effects of EP receptor agonists on the glutamate-induced excitotoxicity in a rat hippocampal slice culture. Representative confocal images of propidium iodide (PI) fluorescence of a slice that received 1 mM glutamate exposure for 15 min (Glu, A), a slice that received glutamate exposure with 1 µM NS-398 (Glu + NS, B), a slice that received glutamate exposure with NS-398 and 10 µM EP1 agonist ONO-DI-004 (Glu + NS + EP1, C) and a slice that received glutamate exposure with NS-398 and 0.1 µM EP3 agonist ONO-AE-248 (Glu + NS + EP3, D) are shown (scale bar: 400 µm). (E) Quantitated data from PI uptake analysis in the CA1 region with or without glutamate (Glu) and/or the EP1 agonist ONO-DI-004 (10 µM), EP2 agonist ONO-AE1-259 (1 µM), EP3 agonist ONO-AE-248 (0.1 µM), EP4 agonist ONO-AE1-329 (1 µM) and NS-398 (1 µM) were scaled to a percentage of the response of glutamate alone. n = 8–19 slices per group. (F) Concentration-dependent toxic effect of ONO-AE-248 on glutamate-induced PI uptake in CA1 in the presence of NS-398 (1 µM). n = 9–19 slices per group. **P < 0.01 versus control, ##P < 0.01, #P < 0.05 versus glutamate alone, $$P < 0.01 versus glutamate with NS-398.
EP3 receptors as downstream effectors of mPGES-1 neurotoxicity
We have previously reported that mPGES-1 enhanced neuronal death through massive PGE2 production in the ischaemic cortex (Ikeda-Matsuo et al., 2006). To address the question of whether or not the activation of EP3 receptors is involved in the mPGES-1-dependent neurotoxicity resulting from massive PGE2 production, we compared the effect of an EP3 receptor antagonist on the excitotoxicity in mPGES-1 KO and WT slices. ONO-AE3-240 significantly attenuated the glutamate-induced excitotoxicity in the slices obtained from WT mice (Figure 4A). In the mPGES-1 KO slices, the glutamate-induced PGE2 production observed in WT slices was almost completely abolished (Table 1). The glutamate-induced excitotoxicity was reduced, but not completely abolished, in the mPGES-1 KO slices; it was at almost the same level as observed in ONO-AE3-240-treated WT slices (Figure 4A and B). However, ONO-AE3-240 did not attenuate the excitotoxicity in mPGES-1 KO slices, while the EP3 receptor agonist ONO-AE-248 exacerbated it (Figure 4B and C).
Figure 4.
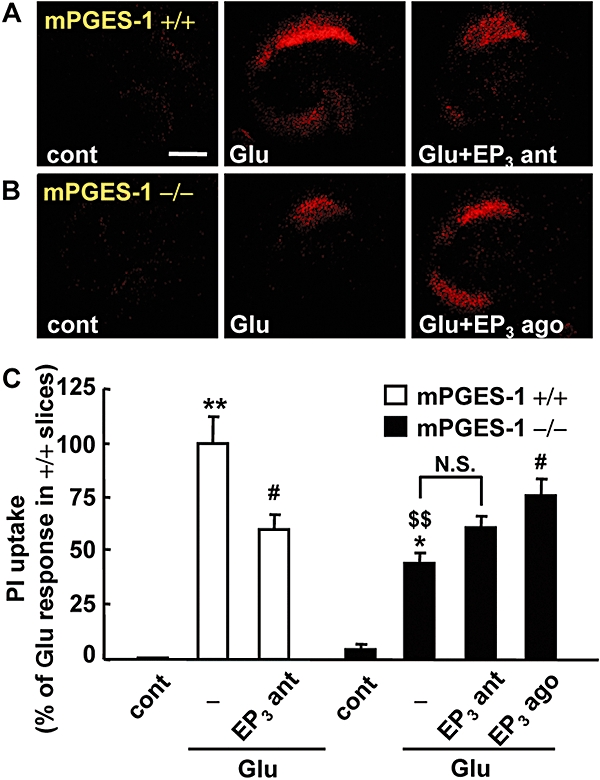
Effect of an EP3 receptor antagonist and agonist on the glutamate-induced excitotoxicity in a microsomal prostaglandin E synthase (mPGES-1) knockout (KO) (−/−) and a wild-type (WT) (+/+) mouse hippocampal slice culture. The EP3 receptor antagonist (ONO-AE3-240) and agonist (ONO-AE-248) were added during and after the 1 mM glutamate exposure for 30 min. Twenty-four hours later, propidium iodide (PI) uptake was analysed. Representative confocal images of PI fluorescence of a control slice (cont), a slice that received glutamate exposure (Glu) and slices that received glutamate exposure with 10 µM of an EP3 receptor antagonist (Glu + EP3 ant) or 0.1 µM of an EP3 receptor agonist (Glu + EP3 ago) in WT (+/+) mice (A) and mPGES-1 KO (−/−) mice (B) are shown (scale bar: 400 µm). (C) Quantitative data from PI uptake analysis in the CA1 region with or without glutamate (Glu) and EP3 receptor antagonist (EP3 ant) or agonist (EP3 ago) were scaled to a percentage of the glutamate response in slices from WT mice. n= 6–11 slices per group. **P < 0.01, *P < 0.05 versus the control slice from WT mice, #P < 0.05 versus glutamate alone in each genotype, $$P < 0.01 versus the control slice from mPGES-1 KO mice, N.S. (not significant).
To extend these observations of slice culture to models of ischaemia in vivo, we used a mouse MCA occlusion–reperfusion model which showed an increase in PGE2 production in the post-ischaemic cortex (Figure 1A). ONO-AE3-240 (3 mg·kg−1) was injected i.p. three times, once each at 2, 8 and 14 h after MCA occlusion. ONO-AE3-240 reduced the infarct volume in the cortex of WT mice (Figure 5A); the infarct volume in the cortex of mice injected with ONO-AE3-240 was less than 40% that of vehicle-injected mice, while that in the striatum was not affected by the injection of ONO-AE3-240 (Figure 5B). As seen in our previous study (Ikeda-Matsuo et al., 2006), mPGES-1 KO mice in the present study were partially resistant to transient ischaemic injury; the infarct volume in the cortex of mPGES-1 KO mice was almost the same as that of ONO-AE3-240-injected WT mice. Interestingly, ONO-AE3-240 did not cause additional reduction of infarction in mPGES-1 KO mice. The degree of oedema was also reduced by ONO-AE3-240 in WT mice; the reduced level was almost identical to the level in mPGES-1 KO mice (Figure 5C). ONO-AE3-240 had no further protective effect on oedema in mPGES-1 KO mice. To determine the functional role of EP3 receptors in behavioural symptoms, we investigated the neurological dysfunction observed after transient ischaemia. ONO-AE3-240 ameliorated neurological deficits in WT mice to almost the same level as in mPGES-1 KO mice, while it did not ameliorate the neurological deficits of mPGES-1 KO mice (Figure 5D). ONO-AE3-240 did not alter mean arterial pressure (MAP), changes in cerebral blood flow (CBF) after reperfusion, or rectal temperature (data not shown). As we have reported previously, there were no significant differences between mPGES-1 KO and WT mice in MAP, changes in CBF before, during or after MCA occlusion, the anatomy of the circle of Willis or the origins of the cerebral arteries (Ikeda-Matsuo et al., 2006). Expression of the mRNAs of the EP1, EP2, EP3α, EP3β, EP3γ and EP4 receptors was observed in both the contralateral and ipsilateral cortex of mPGES-1 KO mice, and the mRNA levels of these receptors in the bilateral cortex of mPGES-1 KO mice were almost the same as those of WT mice (Figure 1B and other data not shown).
Figure 5.
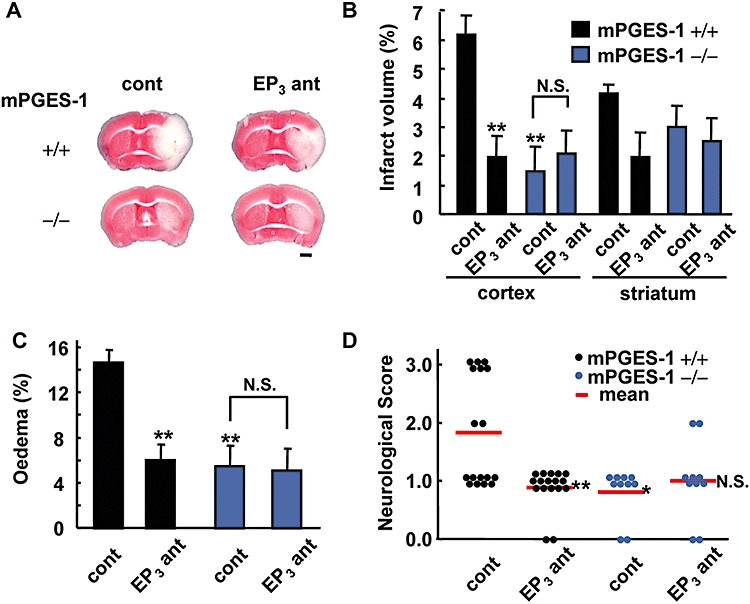
Protective effect of an EP3 receptor antagonist on post-ischaemic symptoms in wild-type (WT) mice but not in microsomal prostaglandin E synthase (mPGES-1) knockout (KO) mice. ONO-AE3-240 (3 mg·kg−1, i.p.) was injected three times at 2, 8 and 14 h after middle cerebral artery occlusion. (A) Representative triphenyltetrazolium chloride-stained coronal sections of the WT (+/+) mice and mPGES-1 KO (−/−) mice injected with vehicle (cont) or ONO-AE3-240 (EP3 ant) (scale bar: 1 mm). (B) The volume of the infarcted cortex and striatum 24 h after ischaemia was estimated and expressed as a percentage of the corrected tissue volume. n= 9 mice per group. (C) The corrected oedema percentage in the EP3 antagonist-injected WT and mPGES-1 KO mice. n= 9 mice per group. (D) Neurological dysfunction in the EP3 antagonist-injected WT and mPGES-1 KO mice 24 h after ischaemia. n= 10–17 mice per group, **P < 0.01 versus vehicle-treated WT mice (cont), N.S. (not significant) versus vehicle-treated mPGES-1 KO mice (cont).
Effect of Rho kinase inhibitor on glutamate-induced excitotoxicity and ischaemic injury
To further investigate the underlying mechanisms of EP3 receptor-mediated neurotoxicity, we examined the effects of protein kinase inhibitors on glutamate-induced excitotoxicity using rat hippocampal slices. EP3 receptors have been found to exert their effects through the small G protein Rho-involved pathway (Hatae et al., 2002; Macias-Perez et al., 2008). The glutamate-induced excitotoxicity was significantly inhibited by PTX, an inhibitor of Gi protein, and Y-27632, an inhibitor of Rho kinase, while H-89, a PKA inhibitor, and Ro-31-8220, a PKC inhibitor, had no protective effect (Figure 6A). In the presence of NS-398, in which the endogenous PGE2 production induced by glutamate was completely abolished (Table 1), the attenuated glutamate-induced excitotoxicity was not further inhibited by PTX and Y-27632, or by the EP3 receptor antagonist ONO-AE3-240 (Figure 6B). On the other hand, the attenuated glutamate-induced excitotoxicity in the presence of NS-398 was exacerbated by H-89 and Ro-31-8220. Exacerbation of glutamate-induced excitotoxicity induced by the EP3 receptor agonist ONO-AE-248, in the presence of NS-398 was completely inhibited by an EP3 receptor antagonist, ONO-AE3-240 (Figure 6C). PTX and Y-27632 significantly inhibited the ONO-AE-248-induced enhancement of the excitotoxicity, while H-89 and Ro-31-8220 had no protective effect (Figure 6C). Thus, PTX and Y-27632 completely inhibited the excitotoxicity related to EP3 receptor activation.
Figure 6.
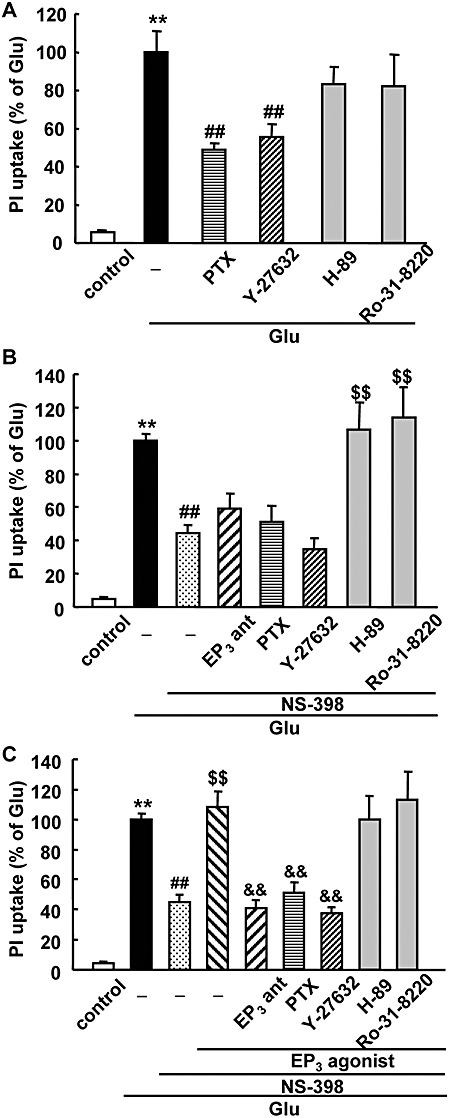
Effect of a Rho kinase inhibitor on glutamate-induced excitotoxicity and EP3-induced enhancement of excitotoxicity in a rat hippocampal slice culture. Quantitative data from propidium iodide (PI) uptake analysis in the CA1 region with or without glutamate and Pertussis toxin (PTX) and kinase inhibitors in the absence of NS-398 (A), in the presence of NS-398 (B) or in the presence of NS-398 and EP3 receptor agonist (C). PTX (1 µg·mL−1) was added from 2.5 h before 1 mM glutamate (Glu) exposure for 15 min. The EP3 receptor agonist (ONO-AE-248, 0.1 µM), EP3 receptor antagonist (EP3 ant, ONO-AE-240, 10 µM) and NS-398 (1 µM) were added during and after glutamate exposure. Y-27632 (10 µM), H-89 (1 µM) and Ro-31-8220 (1 µM) were added immediately after glutamate exposure. Twenty-four hours later, PI uptake was analysed. The data were scaled to a percentage of the glutamate response. n= 7–41 slices per group, **P < 0.01 versus control, ##P < 0.01 versus glutamate alone, $$P < 0.01 versus glutamate with NS-398, &&P < 0.01 versus glutamate with ONO-AE-248 and NS-398.
We next investigated the activation of Rho kinase by the EP3 receptor agonist ONO-AE-248 in rat hippocampal slices. Rho kinase activity, as measured by the Thr850 phosphorylation of MBS, was increased by more than twofold at 1 min after treatment of 0.1 µM ONO-AE-248 (Figure 7). This activation of Rho kinase was completely inhibited by treatment with 10 µM Y-27632. We also investigated the activation of Rho kinase 24 h after treatment with ONO-AE-248; however, we could not detect significant changes in Rho kinase activation at this time point (data not shown). In another experiment, we confirmed that the expression level of not only β-actin, but also pan-MBP, was not changed by treatment with ONO-AE-248 for 1 min (data not shown). In an in vivo ischaemic model, we also investigated the activation of Rho kinase in the mouse brain 24 h after transient ischaemia, but we could not detect the activation of Rho kinase in the ischaemic cortex (data not shown).
Figure 7.
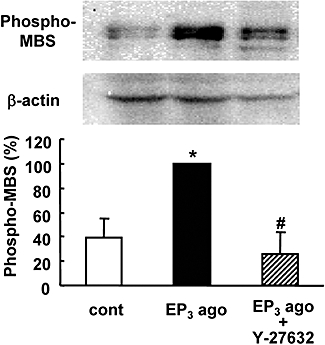
Effect of an EP3 receptor agonist on Rho kinase activity in rat hippocampal slices. The EP3 receptor agonist (EP3 ago; ONO-AE-248, 0.1 µM) was administered for 1 min with or without Y-27632 (10 µM). Rho kinase activity was determined by immunoblotting of slice lysates with antibodies against phosphorylated myosin-binding subunit (phospho-MBS) and β-actin. Rho kinase activity was normalized to β-actin and the data were scaled to a percentage of the EP3 agonist response. n= 3. *P < 0.05 versus control (cont), #P < 0.05 versus EP3 agonist alone.
To further elucidate the role of Rho kinase in the ischaemic brain in morbidity in vivo, we investigated the effect of HA-1077, another Rho kinase inhibitor clinically used for cerebrovascular disorder, on ischaemic brain injury. HA-1077 almost completely protected against infarction in both the cortex and striatum and against oedema formation after transient ischaemia (Figure 8A–C). Furthermore, HA-1077 significantly ameliorated the neurological dysfunction (Figure 8D).
Figure 8.
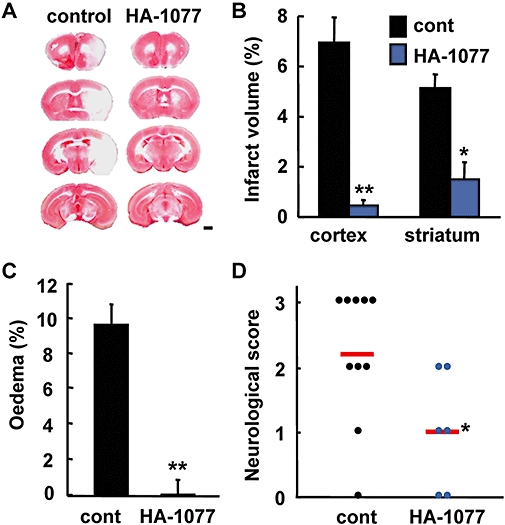
Effect of a Rho kinase inhibitor on stroke-reperfusion injury. HA-1077 (10 mg·kg−1, i.p.) or vehicle was injected twice at 24 and 48 h before middle cerebral artery occlusion. (A) Representative triphenyltetrazolium chloride-stained coronal sections of the vehicle (control) or HA-1077-injected mice (scale bar: 1 mm). (B) Volume of the infarcted cortex and striatum 24 h after ischaemia in the vehicle (cont)- or HA-1077-injected mice was estimated and expressed as a percentage of the corrected tissue volume. (C) The corrected oedema percentage in the vehicle- or HA-1077-injected mice. (D) Neurological dysfunction in the vehicle- or HA-1077-injected mice 24 h after ischaemia. n= 6–10 mice per group, **P < 0.01, *P < 0.05 versus control.
Discussion and conclusions
Here we have shown that mPGES-1 exacerbated ischaemic injury and glutamate-induced excitotoxicity through activation of EP3 receptors. Using mPGES-1 KO mice and antagonists of EP3 receptors, we provided unequivocal evidence that activity of mPGES-1 enhances the excitotoxicity and ischaemic insult through EP3 receptor activation both in vitro and in vivo. We have also shown that Rho kinase activation contributes to the neurotoxicity mediated by EP3 receptors.
We have previously demonstrated that up-regulation of mPGES-1 exacerbates the stroke injury observed after ischaemia and the excitotoxicity induced by glutamate in hippocampal slices, through extensive PGE2 production (Ikeda-Matsuo et al., 2006; 2010;). In this study, we show that EP3 receptors contribute to the excitotoxicity as downstream effectors of mPGES-1. This result is consistent with a recent report showing that OGD-induced cell death in hippocampal slice cultures of EP3 receptor KO mice is lower than in those of WT mice (Saleem et al., 2009). While we did not detect any effects of the EP1 receptor antagonist or agonist on glutamate-induced excitotoxicity, Kawano et al. (2006) showed that the EP1 receptor antagonist SC51089 attenuated the COX-2-dependent component of the injury produced by OGD in a hippocampal slice culture. The mechanisms of neuronal death induced by glutamate-induced excitotoxicity may differ from those of neuronal cell death induced by OGD; OGD-induced PGE2 may stimulate both EP1- and EP3 receptor-related pathways. Considering that an excess amount of extracellular glutamate is observed in the post-ischaemic cortex (Dávalos et al., 2000), EP3 receptors may contribute at least in part to post-ischaemic injury. In fact, in a recent study we demonstrated that pharmacological and genomic inhibition of EP3 receptors attenuated ischaemic injuries 24 h after transient focal ischaemia, suggesting that EP3 receptors have a toxic effect on the post-ischaemic brain (under submission). However, using similar methods, Li et al. (2008) showed that there were no differences in infarct size between WT and EP3 receptor KO mice. The differences in results between our study and that of Li et al. may be due to various technical factors. We used a siliconized flexible filament to minimize blood vessel damage such as endothelial injury and to reduce intracerebral haemorrhage and mortality (Shah et al., 2006). We also used halothane anaesthesia to minimize the pre- and post-conditioning effects derived from other anaesthetics, such as isoflurane (Lee et al., 2008). In fact, using similar methods (e.g. using a siliconized flexible filament and halothane anaesthesia), Saleem et al. (2009) also showed reduced infarct volume and neurological dysfunctions in EP3 receptor KO mice compared with WT mice. We used a glutamate-induced neurotoxicity model in rat and mouse hippocampal slice cultures and a mouse ischaemia-induced cerebral cortex and striatum infarction model. There might be region- and species-specific differences in the susceptibility to excitotoxicity and ischaemia and the role of EP3 receptors. In the present study, however, the protective effects of the EP3 receptor antagonist were observed in both rat and mouse hippocampal slices and also in the cerebral infarction model, suggesting that EP3 receptors play a role in common mechanisms of excitotoxic and ischaemic neuronal death in both species. Furthermore, the protective effects of the EP3 receptor antagonist observed in WT mice were completely abolished in mPGES-1 KO mice in both hippocampal slices exposed to glutamate and the focal cerebral ischaemia (Figures 4 and 5), suggesting that EP3 receptors are the most important effectors for the mPGES-1 neurotoxicity in these ischaemic models (Figure 9). In our transient ischaemia model, although significant ischaemic injury was observed at 24 h after ischaemia, it might not have reached the maximal level at this time point, and thus further experiments will be needed to elucidate the effect of EP3 receptor antagonists at later time points of re-perfusion and the effective time-window of EP3 receptor antagonist treatment.
Figure 9.
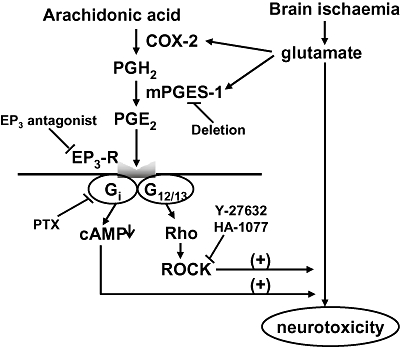
Pathway for EP3 receptor activation-induced exacerbation of stroke injury. The prostaglandin E2 (PGE2) produced by induction of COX-2 and microsomal prostaglandin E synthase (mPGES-1) during ischaemia exacerbates glutamate-induced neurotoxicity through activation of EP3 receptors (EP3-R) followed by activation of Rho kinase (Rho kinase) and/or Gi.
How, then, is the toxicity by EP3 receptor activation mediated? The EP3 receptor has been shown to play a role in the regulation of body temperature (Ushikubi et al., 1998; Oka et al., 2003). It has also been shown that the heightened temperatures induced by i.c.v. injection of PGE2 play a significant role in escalating the neural damage caused by global ischaemia (Thornhill and Asselin, 1999). Therefore, we measured rectal temperature before and after injection of a EP3 receptor antagonist in mice with MCA occlusion, and also in mPGES-1 KO mice. However, as we have reported recently, the rectal temperature in mPGES-1 KO mice did not differ from that of WT mice (Ikeda-Matsuo et al., 2006). Also, the rectal temperature after treatment with a EP3 receptor antagonist did not differ from those of mice given vehicle (unpublished experiments). With respect to vasoconstriction, it has been reported that stimulation of EP3 triggers vasoconstriction in porcine middle cerebral arteries (Hatae et al., 2002; Jadhav et al., 2004) and in the guinea-pig aorta (Shum et al., 2003). EP3 receptors also seem to contribute significantly to the PGE2-dependent regulation of arterial blood pressure in male mice (Audoly et al., 1999). However, under our experimental conditions, no significant difference was observed in CBF or MAP between the WT and mPGES-1 KO mice before, during or after reperfusion (Ikeda-Matsuo et al., 2006). Also, the EP3 receptor antagonist had no effect on MAP and the recovery of CBF after reperfusion (under submission). Therefore, the effect of EP3 receptors on the exacerbation of stroke injury may not be attributable either to the thermoregulatory effect or to the vasoregulatory and cerebral circulatory effect of EP3 receptors. As we are able to detect neuronal excitotoxicity after EP3 receptor activation even in the cultured hippocampal slice system, we speculate that some or all of the stroke injury observed in our in vivo system was the result of direct damage through the activation of EP3 receptors in neuronal cells at the parenchymal lesion site.
In fact, it has been demonstrated that EP3 receptors are constitutively and abundantly expressed throughout the brain (Nakamura et al., 2000). In particular, EP3 receptors are expressed exclusively in neurons and induced in glial cells after excitotoxic lesions by quinolinic acid (Slawik et al., 2004). In our mouse brain ischaemia models, all of the mouse EP3 receptor subtypes were constitutively expressed, and the expression level was not changed after transient ischaemia. In cultured hippocampal slices from rats, EP3 receptors were also constitutively expressed with or without glutamate stimulation. The enhancement of glutamate-induced excitotoxicity by an EP3 receptor agonist in the presence of NS-398, which inhibits endogenous PGE2 synthesis, was completely abolished by Rho kinase and Gi inhibitors, as well as by an EP3 receptor antagonist, while glutamate-induced toxicity in the presence of NS-398 but the absence of an EP3 receptor agonist was not attenuated by any of these inhibitors. This suggests that the neurotoxic effect of EP3 receptor activation is mediated through activation of Rho kinase and/or Gi (Figure 9). In an EP2 receptor-deficient mouse model, it has been shown that the neuroprotective function of the EP2 receptor in cerebral ischaemia is dependent on cAMP signalling (McCullough et al., 2004). Therefore, an EP3 receptor-related cAMP reduction through Gi activation may mediate the effects in a manner opposite to the mediation by EP2 receptors. EP3 receptors have also been shown to be linked to phospholipase C activation via Gi, and this activation leads to Ca2+ mobilization from internal stores and influx from the extracellular medium (Irie et al., 1994). Thus, much as in the case of the toxic effect of EP1 receptors (Kawano et al., 2006), EP3 receptors may contribute to neurotoxicity by augmenting the Ca2+ dysregulation underlying excitotoxic neuronal death.
We detected activation of Rho kinase by the EP3 receptor agonist in rat hippocampal slices, but not by focal ischaemia in the mouse brain. Because phosphorylation of MBS in hippocampal slices was observed at 1 min, but not 24 h, after treatment with the EP3 receptor agonist, the activation of Rho kinase in the ischaemic brain might have been detectable at a time point earlier than 24 h (Yamashita et al., 2007). In fact, we confirmed the involvement of Rho kinase not only in the glutamate-induced neurotoxicity in vitro, but also in the ischaemic infarction in mouse transient ischaemia models using the Rho kinase inhibitor. We used two types of Rho kinase inhibitors, Y-27632 for in vitro study and HA-1077 for in vivo study, because Y-27632 has been the most commonly used inhibitor for in vitro experimental studies for a long time, while HA-1077 has been clinically used for cerebrovascular disorders. The protective effect of HA-1077 was consistent with previous findings using both permanent ischaemia and transient focal ischaemia models in mice (Yamashita et al. 2007 and Rikitake et al., 2005 respectively). In addition to its protective effect on infarction, HA-1077 significantly attenuated the oedema formation and behavioural dysfunction observed after ischaemia. The protective effect of HA-1077 was more potent than that of EP3 receptor antagonist, possibly because HA-1077 protects the neurons from ischaemic injury not only through a reduction of inducible nitric oxide synthase (iNOS) expression (Li et al., 2009), but also through an increase in CBF by vasodilation through endothelial nitric oxide synthase expression (Rikitake et al., 2005). Therefore, EP3 may mediate nitric oxide production by induction of iNOS through activation of Rho kinase, and thereby induce apoptotic cell death (Nomura, 1998). As we were able to detect neuronal excitotoxicity by Rho kinase activation even in the cultured hippocampal slice system (Figure 6), we suggest that some of the stroke injury observed in our in vivo system was the result of direct damage through the activation of Rho kinase by the activation of EP3 receptors in the parenchymal lesion site. Actually, in the spinal cord, Rho kinase has been shown to mediate nitric oxide formation by EP3 receptor activation (Matsumura et al., 2005). However, the mechanisms by which EP3 receptors/Rho kinase affects inflammation and neuronal apoptosis remain to be identified.
In summary, we have shown that EP3 receptors, as effectors of mPGES-1 neurotoxicity, play a critical role in the infarction, oedema and behavioural dysfunctions observed after ischaemia through the activation of Rho kinase and/or Gi (Figure 9). Thus, our results suggest that the EP3 receptor, as well as mPGES-1, is one of the most promising novel targets for treatment of human stroke.
Acknowledgments
The authors are grateful to Mr Shintaro Miyazawa for his skilful technical assistance. This study was partially supported by a Grant-in-Aid for Young Scientists (B) (18790063, Y.I-M) from the Ministry of Education, Culture, Sports, Science and Technology of Japan and by The Mochida Memorial Foundation for Medical and Pharmaceutical Research (Y.I-M).
Glossary
Abbreviations:
- CBF
cerebral blood flow
- COX
cyclooxygenase
- iNOS
inducible nitric oxide synthase
- KO
knockout
- MAP
mean arterial pressure
- MBP
myosin-binding protein
- MCA
middle cerebral artery
- mPGES-1
microsomal prostaglandin E2 synthase
- OGD
oxygen–glucose deprivation
- PG
prostaglandin
- PGES
PGE2 synthase
- PI
propidium iodide
- PTX
Pertussis toxin
- RT-PCR
reverse transcriptase polymerase chain reaction
- TTC
triphenyltetrazolium chloride
- WT
wild-type
Conflict of interest
None.
References
- Ahmad AS, Ahmad M, de Brum-Fernandes AJ, Doré S. Prostaglandin EP4 receptor agonist protects against acute neurotoxicity. Brain Res. 2005;1066:71–77. doi: 10.1016/j.brainres.2005.10.068. [DOI] [PubMed] [Google Scholar]
- Ahmad AS, Saleem S, Ahmad M, Doré S. Prostaglandin EP1 receptor contributes to excitotoxicity and focal ischemic brain damage. Toxicol Sci. 2006;89:265–270. doi: 10.1093/toxsci/kfj022. [DOI] [PubMed] [Google Scholar]
- Ahmad M, Ahmad AS, Zhuang H, Maruyama T, Narumiya S, Doré S. Stimulation of prostaglandin E2-EP3 receptors exacerbates stroke and excitotoxic injury. J Neuroimmunol. 2007;184:172–179. doi: 10.1016/j.jneuroim.2006.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano H, Hayashi I, Endo H, Kitasato H, Yamashina S, Maruyama T, et al. Host prostaglandin E(2)-EP3 signaling regulates tumor-associated angiogenesis and tumor growth. J Exp Med. 2003;197:221–232. doi: 10.1084/jem.20021408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audoly LP, Tilley SL, Goulet J, Key M, Nguyen M, Stock JL, et al. Identification of specific EP receptors responsible for the hemodynamic effects of PGE2. Am J Physiol. 1999;277:H924–H930. doi: 10.1152/ajpheart.1999.277.3.H924. [DOI] [PubMed] [Google Scholar]
- Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19:819–834. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- Butcher SP, Bullock R, Graham DI, McCulloch J. Correlation between amino acid release and neuropathologic outcome in rat brain following middle cerebral artery occlusion. Stroke. 1990;21:1727–1733. doi: 10.1161/01.str.21.12.1727. [DOI] [PubMed] [Google Scholar]
- Dávalos A, Shuaib A, Wahlgren NG. Neurotransmitters and pathophysiology of stroke: evidence for the release of glutamate and other transmitters/mediators in animals and humans. J Stroke Cerebrovasc Dis. 2000;9:2–8. doi: 10.1053/jscd.2000.18908. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Hatae N, Sugimoto Y, Ichikawa A. Prostaglandin receptors: advances in the study of EP3 receptor signaling. J Biochem. 2002;131:781–784. doi: 10.1093/oxfordjournals.jbchem.a003165. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Ueyama T, Kajimoto T, Yagi K, Kohmura E, Saito N. Involvement of gamma protein kinase C in estrogen-induced neuroprotection against focal brain ischemia through G protein-coupled estrogen receptor. J Neurochem. 2005;93:883–891. doi: 10.1111/j.1471-4159.2005.03080.x. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Niwa K, Nogawa S, Zhao X, Nagayama M, Araki E, et al. Reduced susceptibility to ischemic brain injury and N-methyl-D-aspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice. Proc Natl Acad Sci USA. 2001;98:1294–1299. doi: 10.1073/pnas.98.3.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Ueno A, Naraba H, Matsuki N, Oh-ishi S. Intracellular Ca2+ increase in Neuro-2A cells and rat astrocytes following stimulation of bradykinin B2 receptor. Jpn J Pharmacol. 2000;84:140–145. doi: 10.1254/jjp.84.140. [DOI] [PubMed] [Google Scholar]
- Ikeda-Matsuo Y, Ikegaya Y, Matsuki N, Uematsu S, Akira S, Sasaki Y. Microglia-specific expression of microsomal prostaglandin E synthase-1 contributes to lipopolysaccharide-induced prostaglandin E2 production. J Neurochem. 2005;94:1546–1558. doi: 10.1111/j.1471-4159.2005.03302.x. [DOI] [PubMed] [Google Scholar]
- Ikeda-Matsuo Y, Ota A, Fukada T, Uematsu S, Akira S, Sasaki Y. Microsomal prostaglndin E synthase-1 is a critical factor of stroke-reperfusion injury. Proc Natl Acad Sci USA. 2006;103:11790–11795. doi: 10.1073/pnas.0604400103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda-Matsuo Y, Hirayama Y, Ota A, Uematsu S, Akira S, Sasaki Y. Microsomal prostaglandin E synthase-1 and cyclooxygenase-2 are both required for ischaemic excitotoxicity. Br J Pharmacol. 2010;159:1174–1186. doi: 10.1111/j.1476-5381.2009.00595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie A, Segi E, Sugimoto Y, Ichikawa A, Negishi M. Mouse prostaglandin E receptor EP3 subtype mediates calcium signals via Gi in cDNA-transfected Chinese hamster ovary cells. Biochem Biophys Res Commun. 1994;204:303–309. doi: 10.1006/bbrc.1994.2460. [DOI] [PubMed] [Google Scholar]
- Jadhav V, Jabre A, Lin SZ, Lee TJ. EP1- and EP3-receptors mediate prostaglandin E2-induced constriction of porcine large cerebral arteries. J Cereb Blood Flow Metab. 2004;24:1305–1316. doi: 10.1097/01.WCB.0000139446.61789.14. [DOI] [PubMed] [Google Scholar]
- Jakobsson PJ, Thoren S, Morgenstern R, Samuelsson B. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci USA. 1999;96:7220–7225. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashima K, Saji T, Murata T, Nagamachi M, Matsuoka T, Segi E, et al. The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J Clin Invest. 2002;109:883–893. doi: 10.1172/JCI14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh H, Negishi M, Ichikawa A. Prostaglandin E receptor EP3 subtype induces neurite retraction via small GTPase Rho. J Biol Chem. 1996;271:29780–29784. doi: 10.1074/jbc.271.47.29780. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Andreasson KI, Isakson PC, Worley PF. Cyclooxygenases and the central nervous system. Prostaglandins. 1997;54:601–624. doi: 10.1016/s0090-6980(97)00128-7. [DOI] [PubMed] [Google Scholar]
- Kawano T, Anrather J, Zhou P, Park L, Wang G, Frys KA, et al. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12:225–229. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- Kempski O, Shohami E, von Lubitz D, Hallenbeck JM, Feuerstein G. Postischemic production of eicosanoids in gerbil brain. Stroke. 1987;18:111–119. doi: 10.1161/01.str.18.1.111. [DOI] [PubMed] [Google Scholar]
- Lee JJ, Li L, Jung HH, Zuo Z. Postconditioning with isoflurane reduced ischemia-induced brain injury in rats. Anesthesiology. 2008;108:1055–1062. doi: 10.1097/ALN.0b013e3181730257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399:A7–A14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- Li J, Liang X, Wang Q, Breyer RM, McCullough L, Andreasson K. Misoprostol, an anti-ulcer agent and PGE2 receptor agonist, protects against cerebral ischemia. Neurosci Lett. 2008;438:210–215. doi: 10.1016/j.neulet.2008.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Huang XJ, He W, Ding J, Jia JT, Fu G, et al. Neuroprotective potential of fasudil mesylate in brain ischemia-reperfusion injury of rats. Cell Mol Neurobiol. 2009;29:169–180. doi: 10.1007/s10571-008-9308-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias-Perez IM, Zent R, Carmosino M, Breyer MD, Breyer RM, Pozzi A. Mouse EP3 a, b, and g receptor variants reduce tumor cell proliferation and tumorigenesis in vivo. J Biol Chem. 2008;283:12538–12545. doi: 10.1074/jbc.M800105200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough L, Wu L, Haughey N, Liang X, Hand T, Wang Q, et al. Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. J Neurosci. 2004;24:257–268. doi: 10.1523/JNEUROSCI.4485-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura S, Abe T, Mabuchi T, Katano T, Takagi K, Okuda-Ashitaka E, et al. Rho-kinase mediates spinal nitric oxide formation by prostaglandin E2 via EP3 subtype. Biochem Biophys Res Commun. 2005;338:550–557. doi: 10.1016/j.bbrc.2005.09.058. [DOI] [PubMed] [Google Scholar]
- Murakami M, Nakatani Y, Tanioka T, Kudo I. Prostaglandin E synthase. Prostaglandins Other Lipid Mediat. 2002;68–69:383–399. doi: 10.1016/s0090-6980(02)00043-6. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Kaneko T, Yamashita Y, Hasegawa H, Katoh H, Negishi M. Immunohistochemical localization of prostaglandin EP3 receptor in the rat nervous system. J Comp Neurol. 2000;421:543–569. doi: 10.1002/(sici)1096-9861(20000612)421:4<543::aid-cne6>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- Nogawa S, Zhang F, Ross ME, Iadecola C. Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J Neurosci. 1997;17:2746–2755. doi: 10.1523/JNEUROSCI.17-08-02746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura Y. A transient brain ischemia- and bacterial endotoxin-induced glial iNOS expression and NO-induced neuronal apoptosis. Toxicol Lett. 1998;102–103:65–69. doi: 10.1016/s0378-4274(98)00286-0. [DOI] [PubMed] [Google Scholar]
- Oka T, Oka K, Kobayashi T, Sugimoto Y, Ichikawa A, Ushikubi F, et al. Characteristics of thermoregulatory and febrile responses in mice deficient in prostaglandin EP1 and EP3 receptors. J Physiol. 2003;551:945–954. doi: 10.1113/jphysiol.2003.048140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikitake Y, Kim HH, Huang Z, Seto M, Yano K, Asano T, et al. Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke. 2005;36:2251–2257. doi: 10.1161/01.STR.0000181077.84981.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleem S, Kim YT, Maruyama T, Narumiya S, Doré S. Reduced acute brain injury in PGE2 EP3 receptor-deficient mice after cerebral ischemia. J Neuroimmunol. 2009;208:87–93. doi: 10.1016/j.jneuroim.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Kitagawa K, Yamagata K, Takemiya T, Tanaka S, Omura-Matsuoka E, et al. Amelioration of hippocampal neuronal damage after transient forebrain ischemia in cyclooxygenase-2-deficient mice. J Cereb Blood Flow Metab. 2004;24:107–113. doi: 10.1097/01.WCB.0000100065.36077.4A. [DOI] [PubMed] [Google Scholar]
- Shah ZA, Namiranian K, Klaus J, Kibler K, Dore S. Use of an optimized transient occlusion of the middle cerebral artery protocol for the mouse stroke model. J Stroke Cerebrovasc Dis. 2006;15:133–138. doi: 10.1016/j.jstrokecerebrovasdis.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Shum WW, Le GY, Jones RL, Gurney AM, Sasaki Y. Involvement of Rho-kinase in contraction of guinea-pig aorta induced by prostanoid EP3 receptor agonists. Br J Pharmacol. 2003;139:1449–1461. doi: 10.1038/sj.bjp.0705393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slawik H, Volk B, Fiebich B, Hüll M. Microglial expression of prostaglandin EP3 receptor in excitotoxic lesions in the rat striatum. Neurochem Int. 2004;45:653–660. doi: 10.1016/j.neuint.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Suzawa T, Miyaura C, Inada M, Maruyama T, Sugimoto Y, Ushikubi F, et al. The role of prostaglandin E receptor subtypes (EP1, EP2, EP3, and EP4) in bone resorption: an analysis using specific agonists for the respective EPs. Endocrinology. 2000;141:1554–1559. doi: 10.1210/endo.141.4.7405. [DOI] [PubMed] [Google Scholar]
- Takadera T, Shiraishi Y, Ohyashiki T. Prostaglandin E2 induced caspase-dependent apoptosis possibly through activation of EP2 receptors in cultured hippocampal neurons. Neurochem Int. 2004;45:713–719. doi: 10.1016/j.neuint.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Thornhill J, Asselin J. The effect of head cooling on the physiological responses and resultant neural damage to global hemispheric hypoxic ischemia in prostaglandin E2 treated rats. Brain Res. 1999;825:36–45. doi: 10.1016/s0006-8993(99)01210-x. [DOI] [PubMed] [Google Scholar]
- Uematsu S, Matsumoto M, Takeda K, Akira S. Lipopolysaccharide-dependent prostaglandin E2 production is regulated by the glutathione-dependent prostaglandin E2 synthase gene induced by the Toll-like receptor 4/MyD88/NF-IL6 pathway. J Immunol. 2002;168:5811–5816. doi: 10.4049/jimmunol.168.11.5811. [DOI] [PubMed] [Google Scholar]
- Ushikubi F, Segi E, Sugimoto Y, Murata T, Matsuoka T, Kobayashi T, et al. Impaired febrile response in mice lacking the prostaglandin E receptor subtype EP3. Nature. 1998;395:281–284. doi: 10.1038/26233. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Kawamori T, Nakatsugi S, Ohta T, Ohuchida S, Yamamoto H, et al. Inhibitory effect of a prostaglandin E receptor subtype EP(1) selective antagonist, ONO-8713, on development of azoxymethane-induced aberrant crypt foci in mice. Cancer Lett. 2000;156:57–61. doi: 10.1016/s0304-3835(00)00440-7. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Shiraishi M, Fukami K, Tanabe A, Ikeda-Matsuo Y, Naito Y, et al. MARCKS regulates lamellipodia formation induced by IGF-1 via association with PIP2 and β-actin at membrane microdomain. J Cell Physiol. 2009;220:748–755. doi: 10.1002/jcp.21822. [DOI] [PubMed] [Google Scholar]
- Yamashita K, Kotani Y, Nakajima Y, Shimazawa M, Yoshimura S, Nakashima S, et al. Fasudil, a Rho kinase (ROCK) inhibitor, protects against ischemic neuronal damage in vitro and in vivo by acting directly on neurons. Brain Res. 2007;1154:215–224. doi: 10.1016/j.brainres.2007.04.013. [DOI] [PubMed] [Google Scholar]