

Abstract
Background and purpose:
Hydrogen sulphide (H2S) is a labile, endogenous metabolite of cysteine, with multiple biological roles. The development of sulphide-based therapies for human diseases will benefit from a reliable method of quantifying H2S in blood and tissues.
Experimental approach:
Concentrations of reactive sulphide in saline and freshly drawn whole blood were quantified by reaction with the thio-specific derivatization agent monobromobimane, followed by reversed-phase fluorescence HPLC and/or mass spectrometry. In pharmacokinetic studies, male rats were exposed either to intravenous infusions of sodium sulphide or to H2S gas inhalation, and levels of available blood sulphide were measured. Levels of dissolved H2S/HS- were concomitantly measured using an amperometric sensor.
Key results:
Monobromobimane was found to rapidly and quantitatively derivatize sulphide in saline or whole blood to yield the stable small molecule sulphide dibimane. Extraction and quantification of this bis-bimane derivative were validated via reversed-phase HPLC separation coupled to fluorescence detection, and also by mass spectrometry. Baseline levels of sulphide in blood were in the range of 0.4–0.9 µM. Intravenous administration of sodium sulphide solution (2–20 mg·kg−1·h−1) or inhalation of H2S gas (50–400 ppm) elevated reactive sulphide in blood in a dose-dependent manner. Each 1 mg·kg−1·h−1 of sodium sulphide infusion into rats was found to be pharmacokinetically equivalent to approximately 30 ppm of H2S gas inhalation.
Conclusions and implications:
The monobromobimane derivatization method is a sensitive and reliable means to measure reactive sulphide species in whole blood. Using this method, we have established a bioequivalence between infused sodium sulphide and inhaled H2S gas.
Keywords: sodium sulphide, hydrogen sulphide, pharmacokinetics, blood, bimane, monobromobimane, fluorescence detection, amperometric detection
Introduction
Hydrogen sulphide (H2S) is a colorless, flammable, water-soluble gas with the characteristic odour of rotten eggs. For many decades, H2S received attention as a toxic gas and as an environmental hazard (Reiffenstein et al., 1992). Recent work, however, has identified H2S as a gaseous biological mediator that is produced in mammalian organisms, including humans. H2S is synthesized endogenously in a variety of mammalian tissues via two pyridoxal-5′-phosphate-dependent enzymes responsible for metabolism of l-cysteine: cystathionine β-synthase and cystathionine γ-lyase (Stipanuk and Beck, 1982). The synthesis and action of H2S in biological systems have been frequently reviewed (Wang, 2003;Fiorucci et al., 2006; Szabo, 2007; Li and Moore, 2008).
A growing body of evidence demonstrates that sulphide affects fundamental biological pathways, including those involved with vascular function, cytoprotection against reactive species, CNS functions and cellular metabolism (see Wang, 2003; Fiorucci et al., 2006; Szabo, 2007; Li and Moore, 2008). Recently, novel signal transduction pathways have also been described that may be responsible for some of the cellular actions of sulphide (Mustafa et al., 2009).
In many pathophysiological conditions, administration of various formulations or precursors of sulphide is of therapeutic benefit (see Szabo, 2007; Wallace, 2007). Animal models of conditions where sulphide has been shown to be beneficial include myocardial infarction (Sivarajah et al., 2006; Elrod et al., 2007; Zhu et al., 2007; Rossoni et al., 2008; Sodha et al., 2008), acute respiratory distress syndrome (Esechie et al., 2008; 2009;), colitis (Fiorucci et al., 2006), pancreatitis (Bhatia et al., 2008) and gastric ulceration (Wallace et al., 2007).
A number of these animal studies, both in vitro and in vivo, have utilized IK-1001 (sodium sulphide for injection) to investigate the therapeutic potential of sulphide (Elrod et al., 2007; Szabo, 2007; Jha et al., 2008; Kiss et al., 2008; Simon et al., 2008; Sodha et al., 2008). Parenteral sulphide therapy necessitates the determination of the exposure levels that are associated with efficacy and potential toxicity. The potential use of IK-1001 in humans requires a pharmacokinetic assay by which the concentration and fate of intravenous sulphide may be reliably tracked and by which dosing regimens for sulphide may be designed.
Several different methods have been used previously to measure sulphide concentrations in biological systems. These include head space gas analysis (Ubuka, 2002); derivatization methods, for example, with pentafluorobenzyl bromide (Kage et al., 1988; Nagata et al., 1990) or N,N-dimethyl-p-phenylenediamine to form methylene blue (Ogasawara et al., 1993; Mok et al., 2004); spectrophotometry (Wei et al., 2008); and direct measurement in solution with a silver sulphide or polarographic sensor (Zhao et al., 2001; Doeller et al., 2005; Whitfield et al., 2008; Yang et al., 2008). Due in large part to the wide variety of experimental methods used, the various assays have yielded highly variable results with respect to determination of absolute concentrations of sulphide in blood and tissues, and there is no consensus in the field as to which measurement best represents ‘biologically available sulphide’.
Given sulphide's inherent sensitivity to oxidation and the reversible nature of chemically bound sulphide that is present in blood and tissues, we sought to develop a simple method that captures reactive sulphide quickly, without releasing chemically bound sulphur that may exist in a biological matrix. In this paper, we describe a method whereby available sulphide in whole blood or other biological fluids is rapidly derivatized under gentle conditions with excess monobromobimane. Chemical derivatization is followed by extraction of the resulting sulphide dibimane into ethyl acetate, after which the sulphide dibimane is concentrated into a known volume and quantified by separation using reversed-phase HPLC coupled to fluorescence detection. This derivatization method was subsequently applied to measure the levels of sulphide in the blood of animals, and was used to compare the pharmacokinetics of intravenous sulphide with the response elicited from inhaled H2S. To corroborate the monobromobimane derivatization assay, we also utilized a previously published method that can operate both in vitro and in vivo, and is based on H2S detection by a commercial amperometric sensor (Elrod et al., 2007; Koenitzer et al., 2007).
Methods
Sulphide dibimane synthesis
Sulphide dibimane was prepared by reaction of sulphide with 2.0 equivalents of monobromobimane. For example, 25 mL of 71 mM aqueous sodium sulphide solution (see below) was added dropwise with stirring at room temperature to a solution consisting of 1.0 g monobromobimane dissolved in 25 mL acetonitrile and 47 mL deoxygenated HEPES buffer (100 mM, pH 8.0). The solution was then extracted with 150 mL ethyl acetate, and the aqueous layer was re-extracted twice with 100 mL methylene chloride. The organic layers were combined and evaporated under nitrogen stream to give the crude product. The material was then purified by reversed-phase flash chromatography: RediSep Rf normal phase silica column, 40–60 µM, 40 g cartridge, flow rate 40 mL·min−1, gradient from CH2Cl2 (100%) to CH2Cl2:MeOH (75:25) over 25 min. The sulphide dibimane elutes at 12.3 min. Column-purified material was recrystallized from 50 mL boiling EtOH. Overall yield: 475 mg of a yellow powder (63%). NMR (CDCl3) δ 3.8 (s, 4H, CH2), 2.3 (s, 6H, CH3), 1.9 (s, 6H, CH3), 1.85 (s, 6H, CH3); m/z (M + H) 415.07.
Sodium sulphide solutions for in vitro and in vivo studies
Studies were conducted using a sodium sulphide solution adjusted to pH 7.5–8.0 and osmolarity 260–340 mOsm·kg−1 in order to make it physiologically acceptable for intravenous dosing. Solutions were prepared by bubbling H2S gas through an aqueous sodium hydroxide solution under anoxic conditions. Sodium chloride was used to balance the solution to iso-osmolarity. For example, in a nitrogen-filled glove box, 20% H2S gas was bubbled through 2.5 L of an argon-sparged solution of 64 mM NaOH/85 mM NaCl to a final pH of 7.6. This resulted in a solution of 70 mM sulphide with an osmolarity of 281 mOsm·kg−1. Solutions were stored at room temperature in glass vials of 10–50 mL with air-tight seals (stoppered and crimp-capped). The final sulphide concentration of each lot was determined by potentiometric titration with a lead perchlorate standard as below. Please note that all mention in the current report of investigations using ‘sodium sulphide’ (e.g. animal dosing with a given level of sodium sulphide) refers to an equimolar amount of the above physiologically balanced sulphide solution, and does not imply Na2S per se, which is highly alkaline (pH 14).
Potentiometric titration with lead perchlorate standard
Samples were diluted to the desired concentration in a final volume of 50 mL sulphide antioxidant buffer (SAOB, Orion# 941609, Thermo Fisher Scientific, Waltham, MA, USA). To ensure a precision of at least 1%, the dilution factor is selected such that the sample will be titrated with no less than 5 mL of 100 mM Pb(ClO4)2 (Orion# 948206) when using a 10 mL burette capable of dispensing 50 µL titrant drops. For example, if the sulphide concentration is expected to be 100 mM, at least 5 mL sample was diluted into SAOB; if 50 mM is expected, at least 10 mL sample should be diluted into SAOB. During dilution, components were added in the following order: 2× SAOB, distilled H2O, sample. While stirring, Pb(ClO4)2 was added using a 10 mL burette, and the mV reading (silver/sulphide electrode: Thermo Fisher Scientific #9616BN) recorded. In the beginning of the titration, the Pb(ClO4)2 solution was added rapidly (1 or 2 mL additions). As the inflection point was reached, volume addition was reduced to 50 µL per addition. The titration was continued until the mV signal was no longer significantly changing, and the results were plotted as (–)mV versus volume Pb(ClO4)2. Sulphide concentration is calculated from the volume of 100 mM Pb stock titrant required to reach the inflection point. Then, S2−= 100 mM [Pbv/Sv], where Pbv= volume Pb(ClO4)2 at inflection point and Sv= volume of sample in dilution.
34S-labelled sodium sulphide solutions for in vitro and in vivo studies
The following procedure is for making an aqueous solution of 34S-sodium sulphide (Na234S) using 34S powder and zinc dust. The reaction is carried out by first forming solid zinc sulphide (Zn34S) and then reacting it with concentrated hydrochloric acid (HCl) to evolve 34S-hydrogen sulphide (H234S). The H234S gas is captured in an alkaline solution, which is finally brought to pH 8.0 with HCl. Preparation of Zn34S was conducted as follows: to a 35 mL pressure vessel (Chemglass Inc., cat. #: CG-1880-02, Vineland, NJ, USA) was added 0.800 g zinc dust, carefully tapping the vessel and forming a small layer on the bottom. Using a mortar and pestle, 6.10 g zinc dust was pre-mixed with 2.0 g 34S powder and carefully added to form a second layer. Another layer of 0.800 g zinc dust was added on top of the Zn/34S layer, and the reaction vessel was tapped gently to settle the layers. The reaction vessel was purged with argon gas for 30 min, and sealed and transferred to an oil bath, where the vessel was heated for 1.5 h at 170°C. The reaction was cooled to room temperature, and the shiny grey solid was transferred out of the vessel to be ground with a mortar and pestle. H234S transfer set-up was then conducted as follows. The ground powder was added into a 250 mL two-necked round bottom flask with a stirring bar, and the side neck was sealed with a rubber septum. A large bent cannula was placed through the side septum and connected to a nitrogen gas source. The top neck of the flask was fitted with an addition funnel and capped with a glass adapter connected to PVC tubing. The tubing was then connected to a gas washing bottle with a stirring bar (trap #1). Trap #1 was further connected to another gas washing bottle (trap #2) using PVC tubing, and plastic clamps were placed between the tube connections. Both traps were filled with a solution of 250 mL 0.3N NaOH. H234S generation from Zn34S was performed as follows: 90 mL of degassed 6N HCl was added to the addition funnel, and the entire transfer system was purged for 45 min with positive nitrogen pressure such that gas bubbled through both traps. The 6N HCl solution was then added into the Zn34S reaction flask with stirring over a period of 30–40 min, approximately 2.5 mL·min−1. Just after the acid addition was begun, the nitrogen cannula was placed directly into the resulting grey suspension, and nitrogen was bubbled through the solution at a rate such that throughout the process, gas bubbled through both traps. After complete addition of acid, the greyish white suspension was stirred for 1 h at room temperature. Trap #1 was then clamped off, and the gas washing bottle was transferred to an anoxic glove box. Formulation of Na34S solution was made as follows. Using an auto-titrator, the sulphide concentration and the osmolarity of the solution in trap #1 were analysed (101.5 mM, 543 mOsm·kg−1). In an anoxic glove box, the solution was slowly brought to pH 8.0 with HCl, and the osmolarity was checked again (311 mOsm·kg−1). The resulting solution (51 mM) was checked for oxidation products via ion chromatography, and was then aliquotted into 30 mL crimp seal vials.
Derivatization of sulphide with monobromobimane
For each blood sample to be derivatized, no more than 2 h earlier, one 9 mL glass vial (Thermo Fisher Scientific, cat# 02-912-353, with Teflon-lined caps) was labelled and filled with 200 µL of 50 mM HEPES buffer (pH 8) (freshly sparged for at least 20 min with argon gas), followed by 200 µL of a 10 mM monobromobimane solution in acetonitrile. Charged vials were capped tightly and kept protected from light. For each sample, a graduated 1 mL syringe with needle was labelled and coated with heparin by rinsing the syringe with 1 mL of heparin sodium solution. Approximately 300 µL blood from jugular or sublingual venepuncture was drawn directly into the syringe, the syringe was held upright and any bubbles and rat blood in excess of 200 µL were pushed out of the syringe into a sterile wipe. With the needle below the solvent line, the content of the syringe (200 µL blood) was then dispensed into the 400 µL of monobromobimane/acetonitrile/HEPES buffer solution contained in the charged reaction vial. It took consistently between 30 and 60 s to collect the blood and dispense 200 µL into the derivatization solution. Immediately thereafter, reaction vials were capped tightly and placed on an orbital shaker plate for 10 min (Rototron RT10, Vulcon Technologies, Grandview, MO, USA) at 75% of max speed. Subsequently, 2.0 mL ethyl acetate was added, causing the blood to begin to congeal into a red spongy mass, and vials were immediately capped and tumbled for 10 min at 0.5 Hz on an end-over-end rotator (Rugged Rotator, cat. # 099ARD4512, BLD Science, Garner, NC, USA). After tumbling, the reaction vials were centrifuged gently at 350× g for 7 min to collect the spattered congealed blood, and to separate aqueous and organic layers. One millilitre of the organic layer was collected from each extraction, transferred to a 1.5 mL glass vial and the solvent was evaporated completely under a nitrogen stream. Acetonitrile (200 µL) was added to each vial, and the solvent was again evaporated to remove any traces of ethyl acetate. Failure to completely remove ethyl acetate causes severe peak broadening during HPLC analysis. The final dried residue (containing any sulphide dibimane from the the initial derivatization reaction) was stored at −20°C until analysed.
HPLC analysis
To quantify sulphide dibimane in a sample, the dried residue was re-suspended in 50 µL acetonitrile by vortexing of the capped vial for 10 s. The entire sample was transferred to an HPLC autosampler vial with a 200 µL glass sample insert, and the vial was closed with a penetrable cap. The vial was gently vortexed to remove any air bubbles in the glass insert. Then, 7 µL of the sample was injected onto a C18 reversed-phase column (Alltech Prevail C18 3 µM 4.6 × 150 mm column, Part # 99204, Alltech Associates Inc., Deerfield, IL, USA) and guard column (Alltech Prevail C18 5 µM 4.6 × 7.5 mm Part # 99286) on an Agilent 1200 HPLC system (Agilent, Santa Clara, CA, USA). Sulphide dibimane was eluted with the following elution profile: 80% H2O/20% CH3CN from 0 to 2 min, gradient to 64% H2O/36% CH3CN at 10 min, gradient to 4% H2O/96% CH3CN at 11 min, gradient to 4% H2O/91% CH3CN/5% methanol at 12 min, isocratic elution from 12 to 15 min. The eluent was analysed by UV absorption (λabs= 254 nm) and fluorescence (λex= 390 nm, λem= 475 nm). Typical retention times for sulphide dibimane and monobromobimane were ∼12.1 and ∼12.4 min respectively. The concentration of sulphide in a blood sample was calculated from the sulphide dibimane peak area, taking into account: (i) volume changes during sample processing; (ii) recovery of sulphide dibimane from blood via ethyl acetate extraction; and (iii) the specific fluorescence of sulphide dibimane. Extracting a 200 µL blood sample into 2.2 mL of organic layer (2 mL ethylacetate + 200 µL acetonitrile), evaporating 1 mL of the organic layer to dryness and then re-dissolving the residue in 50 µL acetonitrile result in an overall concentration factor of 1.82. Given that the average recovery of sulphide dibimane from blood under these conditions is 76% (see Results), that the specific fluorescence of sulphide dibimane is 2.77 FAU·pmol−1 on our system, and that 7 µL of sample is being injected, the resulting total conversion factor for measuring sulphide concentration in blood via this assay is 0.0373 µM·FAU−1.
Determination of the kinetic properties of the monobromobimane reaction
In a disposable 1 mL methacrylate cuvette, 0.333 mL HEPES (pH 8), 0.333 mL 1 mM monobromobimane in acetonitrile and 0.333 mL of a sodium sulphide solution (3–100 µM) in saline were mixed. Sulphide dibimane formation was monitored in a SpectraMax M2 spectrophotometer (Molecular Devices, Sunnyvale, CA, USA) as the increase in fluorescence intensity over 5 min (λex= 390 nm, λem= 475 nm). The results were used to derive the rate constant (k) for the formation of sulphide dibimane.
Interference studies
Sulphate (0–1.2 mM), thiosulphate (0–90 µM) or sulphite (0–10 µM) was added to 5 mL of fresh rat blood at the indicated concentrations prior to addition of 20 µM sodium sulphide. Then, 200 µL aliquots were removed for monobromobimane derivatization prior to and immediately after sodium sulphide addition.
In vivo pharmacokinetic studies in rats
The investigators adhered to, and all animal care and experimental procedures complied with, the guidelines in the ‘Guide for the Care and Use of Laboratory Animals’, prepared by the Institute of Laboratory Animal Resources Commission on Life Sciences National Research Council (1996).
For the intravenous sulphide-dosing experiments, male Sprague-Dawley rats (276–300 g, Taconic, Hudson, NY, USA) were implanted with jugular and/or femoral vein catheters. The rats were anaesthetized with 5% isoflurane gas until they were in a deep plane of anaesthesia, and then maintained on 2–3% isoflurane gas for the remainder of the surgery. The right external jugular vein was exposed by blunt dissection, and a beveled polyurethane catheter was inserted 3.5 mm towards the heart. For the femoral catheter placement, a blunt dissection of the left adductor muscles exposed the left femoral vein; a polyethylene catheter was inserted into the vessel and secured with three ligatures. Following catheter implantation, an i.v. dose of pentobarbital was given to maintain anaesthesia, and subsequent injections of pentobarbital were given i.p. when required.
Bolus dosing of intravenous sulphide
A 70 mM sodium sulphide solution (pH 7.6, 281 mOsm·kg−1) or equivalent volume of vehicle was injected as a bolus intravenous injection into the tail vein, using a syringe with a 27G needle. Blood was sampled from the jugular vein catheter before dosing and at various time-points after the injection.
Pharmacokinetic-based bioequivalence between parenterally infused sodium sulphide and inhaled H2S gas in rats
Sulphide in all blood samples was measured via derivatization with monobromobimane. For the studies involving continuous intravenous sulphide infusion, rats implanted with both jugular and femoral vein catheters were infused for 2 h from a syringe pump (Alaris Medical Systems, Carefusion, San Diego, CA, USA) delivering into the femoral vein a constant flow rate of sodium sulphide solution (70 mM, pH 7.6, 281 mOsm·kg−1, or diluted with anoxic saline, if needed). Blood was sampled from the jugular vein before dosing, as well as four times during the infusion. A final blood sample was collected 1 h after the infusion had ended. The rats were infused at various infusion rates (2, 5, 10 and 20 mg·kg−1·h−1 sodium sulphide or vehicle).
The studies involving H2S gas exposure were conducted inside a fume hood. Prior to H2S gas exposure, a blood sample was collected from the jugular vein catheter of each rat, and to establish baseline sulphide blood levels. Thereafter, each rat was placed into a 3000 cm3 glass chamber with two ports, one for gas delivery and the other to allow venting of the gas, and covered with a glass plate with a hole through which a tubing extension (0.99 mm o.d.) from the jugular vein catheter had been threaded. At least three animals were exposed to each H2S gas concentration (50, 100, 200, 300, 400 ppm, in room air) for a period of 2 h, and blood was collected from the jugular vein at 10, 30, 60 and 120 min. A final blood sample was collected 1 h after the gas exposure had ended. H2S gas concentrations were mixed using mass flow controllers (Sierra Instruments, Monterey, CA, USA). The gas atmospheres passed through the exposure chamber at a flow rate of one chamber volume per minute. After the 3 h experimentation period, the rats were killed by carbon dioxide asphyxiation.
Measurement of sulphide concentration in vitro using both an amperometric sensor and monobromobimane derivatization
The amperometric sensor used was adapted from Kraus and Doeller, 2004 and Doeller et al., 2005 (see also Benavides et al., 2007; Elrod et al., 2007; Koenitzer et al., 2007). Amperometric detection of sulphide in vitro was performed in an Oxygraph-2k respirometer (Oroboros Instruments, Innsbruck, Austria), which contained two 2.5 mL temperature-regulated chambers, each sealed with a PVDF stopper fashioned to contain an amperometric H2S gas macro sensor (WPI, Sarasota, FL, USA). The polarizing voltage of 155 mV was applied using a TBR4100 Free Radical Analyzer (WPI), and oxygen concentration in each chamber was monitored via a polarized oxygen sensor positioned diagonal to the bottom of the chamber. Sulphide concentration was monitored as a change in sensor current after the addition of sodium sulphide (10 or 20 µM) either to a 2.5 mL solution of 50 mM HEPES (pH 8), 155 mM NaCl, 1 mM DTPA and 3 mM glutathione, or to 2.5 mL of freshly drawn human blood or subsequent plasma (via immediate centrifugation), or to a 2.5 mL solution of 50 mM HEPES (pH 8), 155 mM NaCl, 5% human serum albumin (HSA). All solutions were kept stirred at 25°C. For each experiment, sensor calibration was performed by stepwise addition of sodium sulphide to a temperature- and pH-matched solution of deoxygenated 50 mM HEPES, 155 mM NaCl (pH 8 for HEPES experiments and pH 7.4 for blood, plasma and HSA experiments). For comparison of sulphide measurements by both electrochemical detection and monobromobimane derivatization methods, 400 µL of the 2.5 mL reaction volume from one chamber was drawn out at the indicated time-points with a 1 mL plastic syringe. Blood samples were immediately derivatized using the monobromobimane derivatization assay described above.
Measurement of blood sulphide levels in vivo using an amperometric sensor
For all in vivo work, amperometric H2S sensors (WPI Instruments) were first calibrated with a stepwise addition of sodium sulphide in a deoxygenated solution of 50 mM HEPES (pH 7.4), 155 mM NaCl and 1 mM DTPA at 37°C. Male Sprague-Dawley rats (350–450 g) were anaesthetized with an i.p. injection of pentabarbitol, and femoral vein catheters were implanted. The H2S sensor was then inserted into the right jugular vein, and advanced into the superior vena cava. Once a steady-state baseline current was obtained for the sensor, a 1 mg·kg−1 bolus of sodium sulphide was infused via the femoral vein catheter using a Harvard Apparatus PhD series infusion pump (Harvard Apparatus, Holliston, MA, USA) at various rates (infusion performed over 1 min, or 30, 20 or 10 s, which can be calculated to be equvalent with hourly infusion rates of 60, 120, 180 or 360 mg·kg−1·h−1 respectively).
Measurement of blood sulphide levels in vivo using both an amperometric sensor and monobromobimane derivatization
For in vivo experiments measuring blood sulphide levels via both electrochemical detection and monobromobimane derivatization, the experimental set-up described above was employed with the addition of a carotid artery catheter to draw blood directly from the rat into a monobromobimane reaction vial. A peristaltic pump was used to maintain the flow rate from the carotid artery at 200 µL·min−1, so that a monobromobimane derivatization reaction was generated every minute for 30 min. Once a steady-state baseline current was obtained for the sensor, the peristaltic pump was started, and baseline blood samples were taken for 5 min. After five baseline samples were generated, sodium sulphide was infused at 10 mg·kg−1·h−1 for 5 min, then increased to 20 mg·kg·h−1 for 5 min, and then to 60 mg·kg−1·h−1 for 5 min. At this point, sodium sulphide infusion was stopped, and blood was drawn for another 10 min after the end of the infusion. Blood samples were derivatized with monobromobimane as described above.
Measurement of H2S concentration in vivo using 34S labelled sulphide and the monobromobimane derivatization reaction
The rats with implanted jugular and femoral vein catheters were dosed with 34S-sodium sulphide by continuous i.v. infusion through the femoral vein catheter, using a syringe with a 23G Luer stub adapter (Intramedic, Cardinal Health, Dublin, OH, USA), an infusion line and an infusion pump (Medfusion 2001, Medex Inc., Smiths Medical, Norwell, MA, USA). The rats were dosed for 2 h at a constant infusion rate (2.31–3.00 mL·h−1, based on weight of animal). Blood was sampled from the jugular vein catheter twice before dosing (−30, 0 min), as well as four times during the infusion (10, 30, 60 and 120 min) and three times after dosing (130, 150 and 180 min) using a heparinized 1 mL polypropylene syringe with a 23G Luer Stub adapter. Then, 300 µL blood was collected at each time-point. Derivatization and extraction of the sulphide with monobromobimane were performed as described in the above section. Dried samples from the derivatization protocol were brought up in 50 µL 80/20 water/acetonitrile + 0.1% formic acid. For analysis, 5 µL was injected onto Thermo Finnigan Discovery MAX Triple Quad [liquid chromatography/mass spectrometry (LC/MS)], ESI positive ion mode (Thermo Fisher Scientific) with a Phenomenex guard column and column (Security Guard C18 4 × 2 mm, Gemini 5µ C18 110A 50 × 2.0 mm) (Torrance, CA, USA). Sulphide dibimane was eluted with the following elution profile: starting conditions of 100% 1 mM ammonium formate in water with 0.1% formic acid (Fisher Scientific, PartA666-500 and J.T. Baker Part#0129-01, Mallinckrodt Baker, Phillipsburg, NJ, USA), gradient to 100% acetonitrile with 0.1% formic acid (J.T. Baker, Part#9853-2 and Part#0129-01) from 0.1 to 5 min, and held for 0.5 min, then returned to starting conditions (flow rate 300 µL·min−1). The eluent was analysed with the LC/MS/MS for the following components (with typical retention times): sulphide dibimane ∼4.2 min (SRM 415.1 → 192.9), sulphide dibimane ∼4.2 min (with 34-Sulphide, SRM 417 → 192.9) and monobromobimane ∼4.3 min (SRM 271 → 192). Data analysis was performed with a component of the Xcaliber software version 2.0 SR2 (Thermo Finnigan); Quan Browser (version 2.0). A standard curve was used to quantify the unknown values of the samples.
In vivo microdialysis, derivatization and quantitation of sulphide dibimane by LC/MS
Male Sprague-Dawley rats were anaesthetized with ketamine, and a femoral vein catheter was implanted as described above. A cylindrical CMA-20 Elite microdialysis probe (0.5 mm o.d. × 10 mm length) from CMA Microdialisis AB (Stockholm, Sweden) with a polyarylethersulphone membrane (PAES, 20 kDa cut-off), was subsequently implanted in the jugular vein. Saline was passed through the dialysis probe at a rate of 10 µL·min−1, and 3.3 µL fractions were collected every 20 s from 1 min before the sodium sulphide injection to 5 min after the bolus into a mixture of 3.3 µL of 10 mM monobromobimane solution in acetonitrile plus 3.3 µL 50 mM HEPES buffer (pH 8). After collection, each sample was mixed by pipetting up and down three times. The samples were diluted and desalted before analysis on the LC/MS. The ∼10 µL of derivatized sample was diluted in 180 µL of water and loaded onto a preconditioned Waters Oasis HLB SPE column (Part#186001828BA, Waters Corporation, Milford, MA, USA). Preconditioning was done with first 200 µL methanol (EMD Chemicals, Part#MX0486-01, Gibbstown, NJ, USA) and 200 µL Millipore water (In-house Millipore WIFI system, Millipore, Billerica, MA, USA). The column was washed with 200 µL of a 5% methanol solution, and then the sample was eluted with 50 µL of 100% methanol. The eluent was dried down under nitrogen, and then brought up in 100 µL of 20/80 acetonitrile/water + 0.1% formic acid (samples were diluted 1/10). One microlitre of the diluted sample was separated on a Finnigan Surveyor Plus HPLC (Thermo Electron Corporation) with a Phenomenex Gemini C18 column (50 × 2.0 mm) and analysed on a Finnigan Quantum Discovery Max MS (Thermo Electron Corporation) with ESI source in positive mode. The sample was loaded in 1% formic acid/aqueous 1 mM ammonium formate, and separated over 5 min with a linear gradient to 1% formic acid in acetonitrile at a flow rate of 300 µL·min−1. Sulphide dibimane was quantitated using the SRM transition of 415.1 → 192.9, collision energy = 21, scan time = 0.2 s, ion source polarity: ESI (+), ESI needle voltage: 5000 V, sheath gas pressure: 60 arb units, auxillary gas pressure: 55 arb units, ion transfer capillary temperature 400°C, tube lens offset 96 V, Q2 pressure: 0.8 mtorr argon, scan width: 0.010 u, Q1–Q3 resolution: unit (0.7 u FWHM). Data analysis was conducted with a component of the Xcaliber software version 2.0 SR2 (Thermo Finnigan); Quan Browser (version 2.0). For quantitation, a standard curve is used to quantify the unknown values of the samples. The final concentration was adjusted by multiplying by the dilution factor.
Statistical analysis
All values are reported as mean ± SE mean with n representing the number of experimental observations per cohort. Statistical analysis was performed by anova followed by an unpaired t-test, using GraphPad Prism version 5.00 statistical software. Probability values of P < 0.05 were considered statistically significant.
Materials
Monobromobimane was purchased from Toronto Research Chemicals (cat. # M52500, North York, ON, Canada) or synthesized via the route of Kosower and Pazhenchevsky (1980). Sulphide dibimane was synthesized as described below. The pH-neutralized sodium sulphide solution was prepared anoxically as described below. H2S gas was purchased from Byrne Specialty Gases (Seattle, WA, USA) as 20% H2S, 80% nitrogen. Acetonitrile was purchased from Fisher Scientific (cat. #A21-4), as were ethyl acetate (cat. #E196-4) and HEPES (cat. # BP310, 99%, ‘enzyme grade’). Heparin sodium solution USP was purchased from Baxter Pharmaceuticals (cat. # NDC0641-2450-41, Deerfield, IL, USA). All other chemicals and reagents were obtained from Sigma-Aldrich (Milwaukee, WI, USA).
Results
Characterization of the monobromobimane derivatization method
Monobromobimane has been used extensively to quantify thiols by forming fluorescent derivatives containing one bimane molecule per thiol group (Fahey et al., 1981; Newton et al., 1981; Togawa et al., 1992). The nucleophilic substitution reaction occurs twice with sulphide, however, allowing two equivalents of monobromobimane to form the highly fluorescent sulphide dibimane (Figure 1). As bimane itself is relatively hydrophobic, sulphide dibimane is more hydrophobic than most monobimane derivatives of physiological thiols, and thus the dibimane derivative of sulphide can be separated by reversed-phase chromatography from other thiol bimane derivatives. The only more hydrophobic moiety we have encountered in the current study is the starting monobromobimane itself. Using a gradient from 80% H2O to 4% H2O/5% methanol with the balance as acetonitrile, we have developed an elution method that isolates sulphide dibimane (retention time: ∼12.1 min) from other biologically related bimane thiols, but elutes ahead of unreacted monobromobimane (retention time: ∼12.4 min) (Figure 2A). Because of the high fluorescence of sulphide dibimane compared to monobromobimane, (Figure 2A and B), the peak area of the eluted sulphide dibimane, when quantified by in-line fluorescence detection (λex= 390 nm, λem= 475 nm), can be quantitated despite a large molar excess of monobromobimane eluting nearby in the chromatogram.
Figure 1.
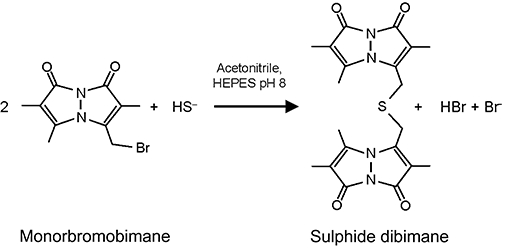
Reaction scheme for the derivatization of sulphide with monobromobimane to form sulphide dibimane.
Figure 2.
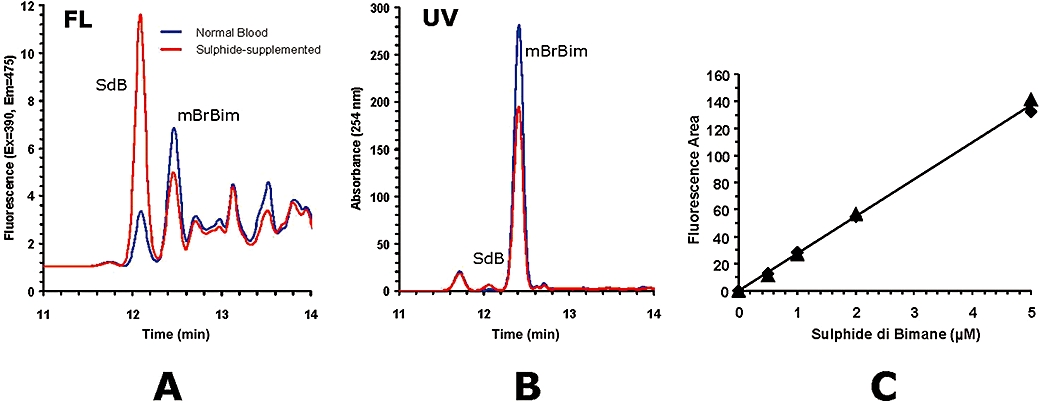
Typical chromatograms of monobromobimane derivatized and ethyl acetate-extracted blood samples. (A) Fluorescence trace (λex= 390 nm, λem= 475 nm) resulting from assay of naïve rat blood (solid line) and assay of rat blood supplemented with 5 µM sulphide (dotted line) (SdB, sulphide dibimane; mBrBim, monobromobimane). (B) Absorbance trace (λabs= 254 nm) of same samples. Sulphide concentration in the sample is calculated from the fluorescence area under the sulphide dibimane (SdB) peak at 12.1 min. (C) Linear response relationship between the fluorescence area of HPLC chromatograms as function of sulphide dibimane concentration, injecting 7 µL of standard sulphide dibimane solutions in acetonitrile. The specific fluorescence of the sulphide dibimane solution in acetonitrile is 2.77 FAU·pmol−1 sulphide dibimane.
Using these parameters, a linear and reproducible standard curve is obtained up to and beyond 5 µM of sulphide dibimane, with the limit of detection lower than 0.2 µM of sulphide dibimane (Figure 2C). In order to characterize the kinetic properties of the reaction, sulphide concentrations between 3 and 100 µM were reacted with 1 mM monobromobimane in acetonitrile/HEPES buffer, and the formation of sulphide dibimane was followed by fluorescence (ex = 390 nm, em = 475 nm). The reaction is first order relative to the initial substrate sulphide, with a rate constant k= 0.01 ± 0.0016 s−1 (Figure 3). On a mechanistic basis, both species H2S and HS- could react with monobromobimane depending on the exact pH and solvent conditions, but in this setting the precise chemical mechanism is irrelevant: the bimane reagent is in great excess, and thus the H2S ↔ HS- equilibrium will instantly be driven to the side of whichever species is being consumed more quickly.
Figure 3.
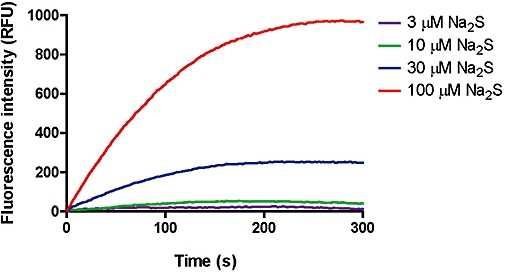
Kinetic analysis of the monobromobimane reaction. Sulphide concentrations between 3 and 100 µM were reacted with 1 mM monobromobimane in acetonitrile/HEPES buffer, and the subsequent formation of sulphide dibimane was followed by fluorescence (λex= 390 nm, λem= 475 nm). The reaction is first order relative to the substrate hydrosulphide, with a rate constant k= 0.01 ± 0.0016 s−1. Fluorescence was calibrated against standards of sulphide dibimane, and sulphide reacted completely with an excess of monobromobimane. Data show representative curves from three independent experiments.
The hydrophobicity of sulphide dibimane was exploited to reduce the complexity of the chromatogram by partitioning sulphide dibimane into ethyl acetate. The majority of fluorescent bimane derivatives formed on reaction with rat blood are insoluble, including most free thiols on proteins, peptides and small polar molecules, as well as other polar monobromobimane-reactive nucleophiles. Extraction into ethyl acetate, evaporation and re-constitution in HPLC solvent allow for an overall twofold concentration of the sulphide dibimane and concomitant increase in sensitivity. The extraction, concentration and re-solubilization procedure, as a whole, shows a reproducible recovery of sulphide dibimane from blood (76% on average) that is independent of the amount of sulphide dibimane over the relevant concentration range (Figure 4).
Figure 4.
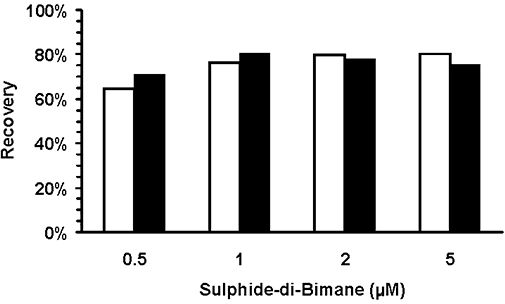
Recovery of sulphide dibimane from whole rat blood as a function of sulphide dibimane concentration. Duplicate blood samples were supplemented with sulphide dibimane, were processed via ethyl acetate extraction as prescribed above and were then analysed via HPLC. Average recovery of sulphide dibimane for the assay was 76%, and this recovery was shown to be independent of sulphide dibimane concentration. White and black bars show data from two independent experiments.
The monobromobimane reaction described herein is not influenced by any of the major metabolites of sulphide tested. When challenged with excess sulphite (Figure 5A), sulphate (Figure 5B) or thiosulphate (Figure 5C), neither false positives (sulphide dibimane from a different sulphur source) nor false negatives (chemical interference with the derivatization reaction) were observed.
Figure 5.
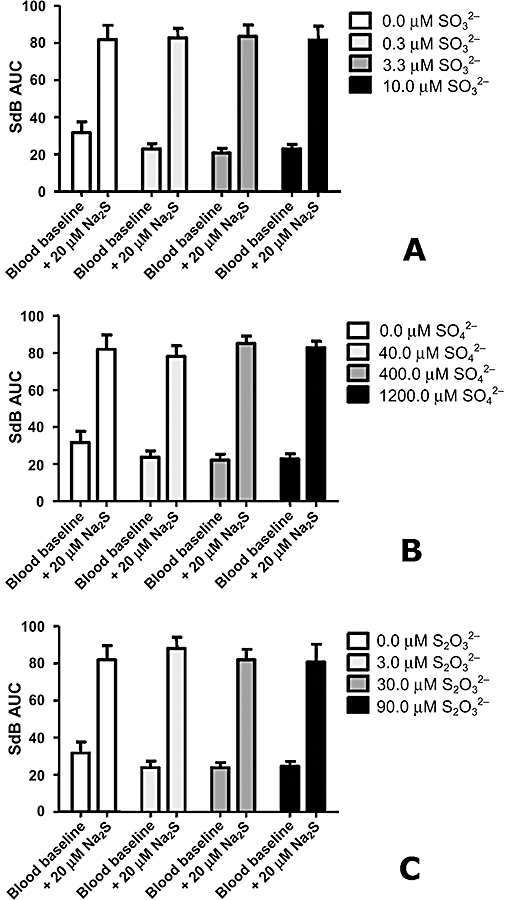
The major oxidation products of sulphide do not interfere with sulphide quantitation. Control rat blood or rat blood with added sodium sulphide was derivatized with monobromobimane in the presence of physiological concentrations of (A) sulphite (SO32–), (B) sulphate (SO42−) and (C) thiosulphate (S2O32−). No effect on measured sulphide levels was observed up to 10 µM sulphite, 1200 µM sulphate and 90 µM thiosulphate. Data represent mean ± SEM for three replicates.
Characterization of the pharmacokinetics of sulphide in rats subjected to an intravenous bolus of sodium sulphide
As measured via monobromobimane derivatization of freshly drawn blood samples, rats injected with a bolus of Na2S solution exhibited a dose-dependent increase in their blood levels of available sulphide (Figure 6). The basal concentration of sulphide in rats was approximately 0.7 µM (0.4–0.9 µM), and the half-life of available sulphide after intravenous bolusadministration at 4 mg·kg−1 was calculated to be approximately 6 min.
Figure 6.
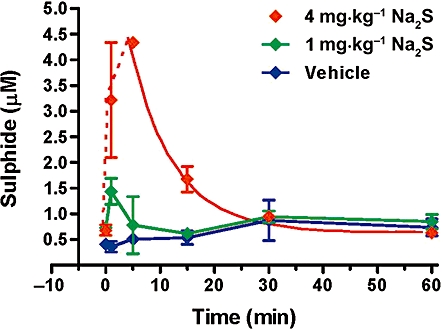
Measurement of blood levels of sulphide after a single intravenous bolus injection of sodium sulphide in rats. The dose of 1 mg·kg−1 sodium sulphide induced a transient elevation of blood sulphide levels at 1 min, while the dose of 4 mg·kg−1 sulphide produced an elevation for 30 min after the injection, with a calculated half-life of approximately 5.7 min. Data represent mean ± SEM values from n= 4 animals for each time-point.
Correlation of the pharmacokinetics of sulphide in rats subjected to either a continuous infusion of sodium sulphide or exposure to inhaled H2S
As shown in Figure 7A, blood sulphide levels in rats increase on continuous intravenous infusion of a sodium sulphide solution (2–20 mg·kg−1·h−1) for 2 h. Elevation of blood sulphide concentration is dose dependent, reaches a steady state within 2 h of sulphide infusion and, except for the highest dose, returns to baseline less than 1 h after discontinuation of dosing. Blood sulphide levels in rats also increase dose dependently after a 2 h exposure to 50–400 ppm of H2S gas in room air (Figure 7B). Blood sulphide levels appear to reach a steady state within 2 h of the start of H2S gas inhalation, and levels return to baseline within 1 h after sulphide dosing ceases and the animals are returned to room air. Based on these measurements, a monobromobimane-based bioequivalency curve for 2 h exposures can be constructed. As shown in Figure 8, concentration of available blood sulphide in rats increases linearly with 2 h exposures to increasing levels of either intravenous sodium sulphide infusion or continuous H2S gas inhalation. From inspection of the corresponding plots, sulphide infusion and H2S inhalation can thus be directly equated.
Figure 7.
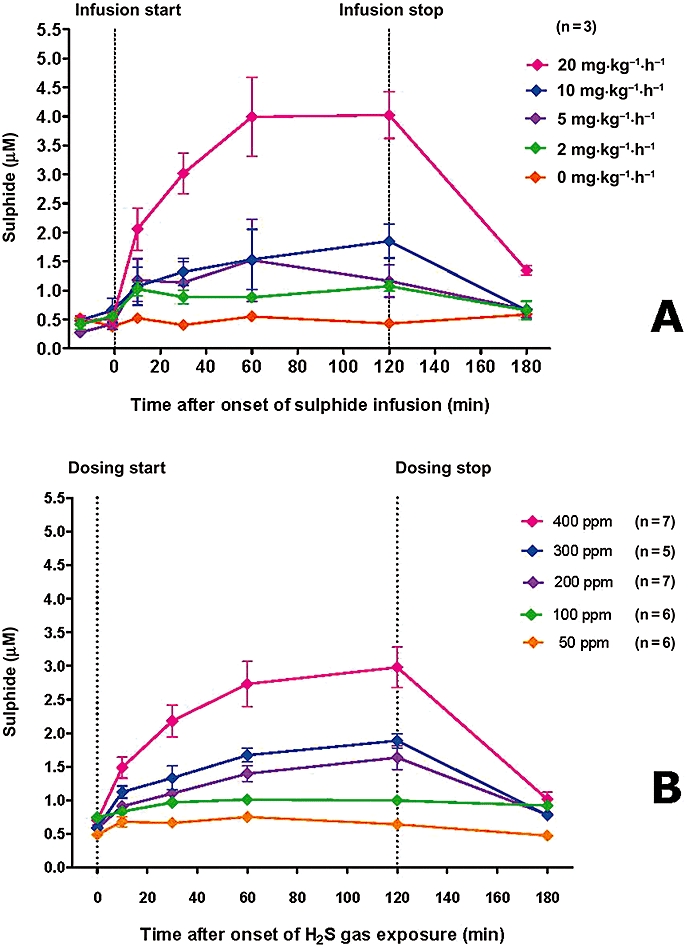
(A) Blood sulphide levels after exposure of rats to continuous intravenous infusions of sodium sulphide solution (2, 5, 10 or 20 mg·kg−1·h−1). (B) Blood sulphide levels after exposure of rats to air atmospheres containing varying concentrations of H2S gas (50, 100, 200, 300, 400 ppm). Data represent mean ± SD; n= 3 for each group. Exposures of 2 h for both infusion and inhalation experiments.
Figure 8.
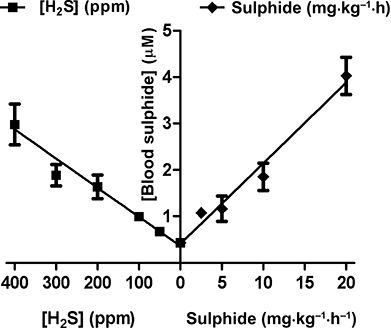
Correlation between levels of monobromobimane-reactive blood sulphide in rats after 2 h of either inhalation of H2S gas or constant infusion of sodium sulphide solution. Graph is a re-plotting of the 2 h data presented in Figure 7A and B, shown as mean ± SD; n= 3 for each group.
Comparison of sulphide levels in vitro as measured by amperometric detection and by the monobromobimane derivatization method
The reaction of 10 µM sodium sulphide in oxygen-depleted aqueous buffer with monobromobimane showed good correlation with the measurement of the same solution using an amperometric H2S sensor (Figure 9A). However, in fresh blood samples spiked with sodium sulphide, the monobromobimane reaction showed significantly higher levels of reactive sulphide than the levels of dissolved H2S/HS- that were measured by the H2S-sensitive probe, which registers only a fleeting spike of H2S (Figure 9B). Sulphide measurements between the two methods also showed a good correlation when sodium sulphide was added to fresh plasma (Figure 9C) or into a normoxic solution of 5% HSA in HEPES (Figure 9D).
Figure 9.
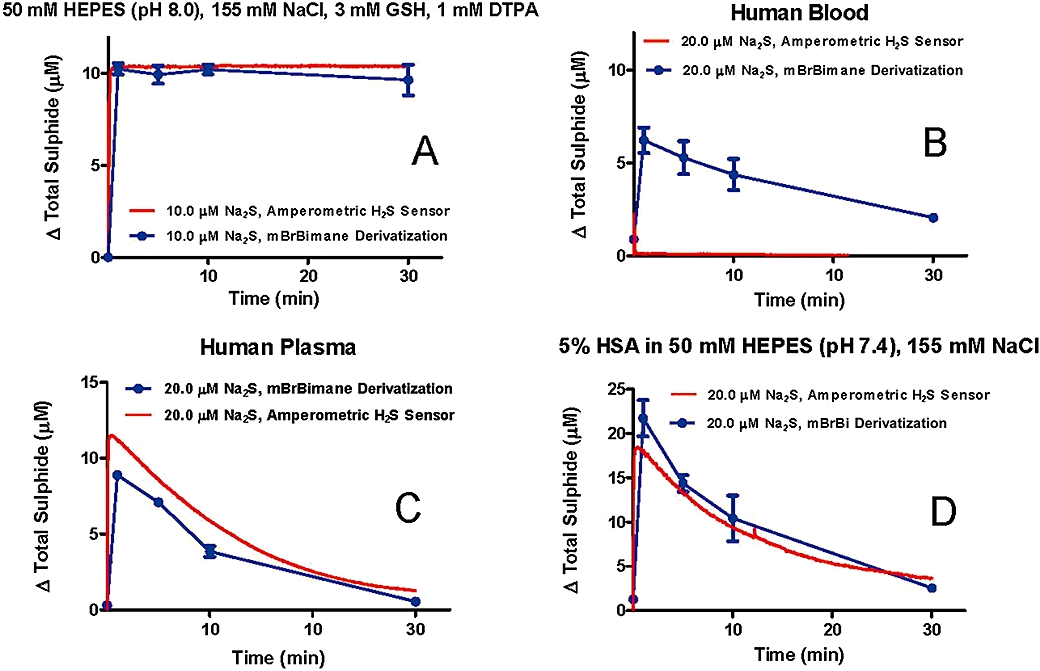
(A) Comparison of amperometric sensor and monobromobimane-based sulphide measurements after addition of 10 µM sodium sulphide to an oxygen-depleted buffered saline solution. Data represent mean ± SD of n= 3 replicates. (B) Comparison of amperometric sensor and monobromobimane-based sulphide measurements after addition of 20 µM sodium sulphide to human blood. (C) Comparison of amperometric sensor and monobromobimane-based sulphide measurements after addition of 20 µM sodium sulphide to human plasma. (D) Comparison of amperometric sensor and monobromobimane-based sulphide measurements after addition of 20 µM sodium sulphide to a solution containing 5% human serum albumin in 50 mM HEPES and 155 mM NaCl. Data represent mean ± SD of three replicates.
Comparison of sulphide levels in rats as measured by in vivo amperometric detection and by monobromobimane derivatization
During the initial experiments, anaesthetized rats with an in-dwelling jugular vein catheter housing the amperometric sulphide sensor were given sodium sulphide (1 mg·kg−1) as an intravenous bolus over 10, 20 or 30 s, or 1 min. The results show that the rate of administration of the sodium sulphide solution has a marked effect on the pharmacokinetics of dissolved H2S in vivo: the highest peak level of H2S/HS- (approximately 1 µM above baseline) was detected when the injection of sodium sulphide occurred over 10 s (Figure 10). When the same bolus was given over 1 min, almost no elevation of H2S above basal levels was seen.
Figure 10.
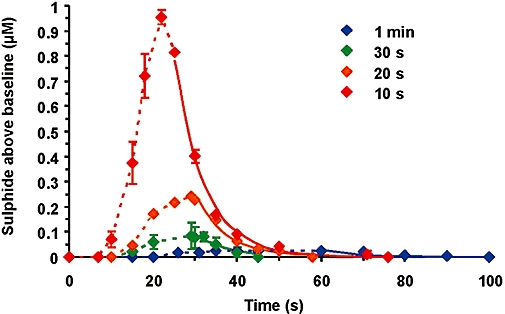
Measurement of sulphide in the vena cava of rats following intravenous injection of 1 mg·kg−1 sodium sulphide over 10, 20, 30 or 60 s, and detecting dissolved H2S using an amperometric sensor. Data represent mean ± SEM; n= 3.
In a subsequent study, sodium sulphide was administered in a ramping infusion regimen to rats with an additional catheter to remove blood samples for bimane derivatization. Sulphide levels measured by both the amperometric sensor and by the monobromobimane derivatization of fresh blood are compared in Figure 11. The results show that the available sulphide values measured by the monobromobimane reaction are substantially higher (10–30 µM) than the readings obtained with the amperometric sulphide sensor (<1 µM).
Figure 11.
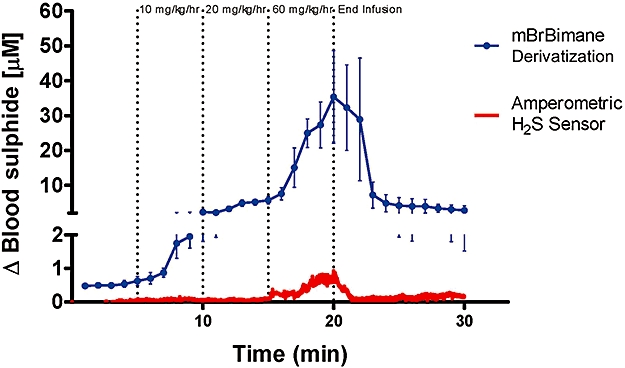
In vivo comparison of amperometric sensor and monobromobimane (mBrBimane)-based blood sulphide measurements in rats during stepwise elevations of sodium sulphide infusion at rates of 10, 20 and 60 mg·kg−1·h−1, each for 5 min. Data represent mean ± SEM; n= 3.
Measurement via derivatization with monobromobimane of levels of 34S-sulphide and 32S-sulphide in rat blood during 2 h infusion with 34S-sodium sulphide
In order to determine the relative contribution of exogenously administered and endogenously present sulphide to the levels of available blood sulphide measured via monobromobimane during sodium sulphide infusion, the rats received intravenous infusions of 34S-sulphide over 120 min at 10 mg·kg−1·h−1. Blood levels of reactive 34S-sulphide and reactive 32S-sulphide were measured by immediate monobromobimane derivatization of ex vivo blood samples, followed by quantitation via LC/MS of the resulting 34S-sulphide dibimane and 32S-sulphide dibimane concentrations. The results show a time-dependent increase in blood levels of available 34S-sulphide during dosing, while blood levels of available 32S-sulphide remain unchanged (Figure 12). On discontinuation of dosing, only available 34S-sulphide is eliminated, the latter with a half-life of approximately 20 min. Blood levels of available 32S-sulphide remained unchanged.
Figure 12.
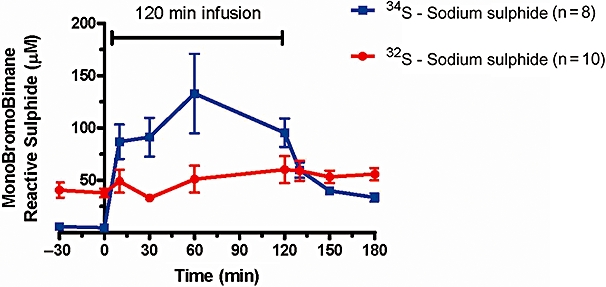
Measurement via derivatization with monobromobimane of 34S-sulphide and 32S-sulphide in rat blood during dosing with 34S-sodium sulphide. Rats received continuous intravenous infusions of 34S-sulphide over 120 min at 10 mg·kg−1·h−1. Blood concentrations of 34S-sulphide increased in a dose-dependent manner, while 32S-sulphide blood concentrations remained unchanged. Data represent mean ± SEM; n= 8–10.
Characterization of the pharmacokinetics of sulphide levels in rats by an in vivo microdialysis method coupled to monobromobimane derivatization
In this study, sodium sulphide solution was administered by intravenous bolus injection at 3 mg·kg−1. The microdialysis catheter has a cut-off of 20 kDa, allowing only small molecules and short peptides to pass into the monobromobimane reaction mixture. The microdialysis catheter thus prevents the reaction of monobromobimane with large protein or cell surface thiols. Within this controlled experimental setting, available sulphide concentrations in venous blood were estimated to peak at approximately 8 µM, and this available sulphide was estimated to have a biological half-life of 22 s (Figure 13).
Figure 13.
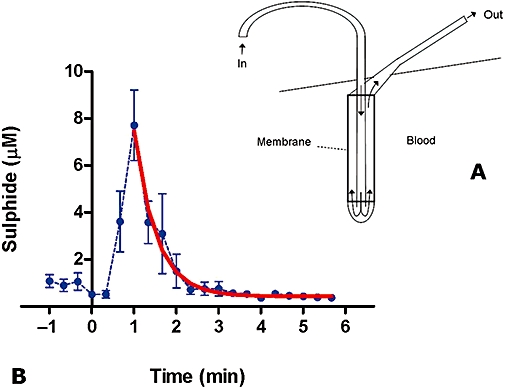
Concentrations of available sulphide in the venous blood of rats after an intravenous bolus injection of 3 mg·kg−1 sodium sulphide, as measured in the microdialysate via the monobromobimane assay. Values after the peak were analysed by non-linear regression and fitted to a curve (thick red line), yielding a half-life of 22 s. Data shown are means ± SEM; n= 4.
Discussion
Sulphide is highly oxygen sensitive, rapidly forming thiosulphate, sulphite and sulphate in the presence of any free oxygen. In addition, under various conditions, H2S may be freely dissolved in solution (as H2S/HS-), it may be loosely bound to other thiols present (e.g. within a persulphide, RSnH, or a polysulphide, RSnR), it may be strongly bound to other thiols present (e.g. within a disulphide, RS2R), it may be covalently bound to carbon (e.g. as RSH or RSR) or it may be coordinated to metals (e.g. iron centres) (Ubuka, 2002; Mueller, 2006; Benavides et al., 2007; Szabo, 2007). It is thus, from the outset, a challenging proposition to attempt to characterize sulphide in all but the simplest matrices, and to measure sulphide in a biological setting one must attempt to carefully define the likely scope of the assay.
There is currently no consensus in the literature with respect to the type of assay that would most accurately provide a measure of ‘biologically available sulphide’, and thus a great variety of assays have been utilized for this purpose. Difference in the methodology of various assays is the most likely explanation for the fact that there is no agreement even as to the order of magnitude of sulphide concentration in blood and tissues (see Table 1: Kage et al., 1988; Nagata et al., 1990; Togawa et al., 1992; Ogasawara et al., 1993; Zhao et al., 2001; Mok et al., 2004; Wei et al., 2008; Whitfield et al., 2008; Yang et al., 2008). These methods often incorporate reducing agents or acid, which can liberate more strongly bound sulphur in addition to easily available sulphide (see Furne et al., 2008). Other difficulties in comparison stem from the length of time that methods expose matrices to incubation in the presence of air oxygen, and in some cases the use of a diffusion-limited transfer of sulphide across liquid/gas or liquid/solid phase boundaries.
Table 1.
Detection of sulphide levels in blood: comparison of various published values and corresponding methods
| Animal species |
Blood treatment |
Sulphide detection |
Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Organic solvent | Reducing agent | Strong acid | Metal | Detection method | Mean sulphide detected (µM) | Sulphide type measured | |||
| Blood in vivo | Rat | Polarographic sensor | Not detected | Free H2S/HS- dissolved in solution | Whitfield et al., 2008 | ||||
| Whole blood | Rat | EtOAC + toluene + TDMBA | PFBenzylBr derivative of sulphide | Not detected | Free H2S/HS- dissolved in solution | Kage et al., 1991 | |||
| Whole blood | Rat | CH3CN + EtOAc | MonoBrBimane derivative of sulphide | 0.7 | Free and reversibly bound H2S/HS- | Current report | |||
| Serum | Human | Phosphoric acid, 20% | MonoBrBimane derivative of sulphide | Not detected | Free H2S/HS- dissolved in solution | Togawa et al., 1992 | |||
| Serum | Human | DTT, 20 mM | Phosphoric acid, 20% | MonoBrBimane derivative of sulphide | 1.3 | Protein-bound sulphide | Togawa et al., 1992 | ||
| Serum | Human | Phosphoric acid, 20% | Thionine derivative of sulphide | Not detected | Free H2S/HS- dissolved in solution | Ogasawara et al., 1993 | |||
| Serum | Human | DTT, 20 mM | Phosphoric acid, 20% | Thionine derivative of sulphide | 1.1 | Protein-bound sulphide | Ogasawara et al., 1993 | ||
| Serum | Rat | Silver probe surface | Electrochemical | 46 | Unknown | Zhao et al., 2001 | |||
| Plasma | Mouse | HCl, 7 M | ZnOAc + FeCl3 | Methylene blue derivative of sulphide | 30 | Unknown | Mok et al., 2004 | ||
CBS, cystathionine β-synthase; CSE, cystathionine γ-lyase; PVC, polyvinylchloride; FAU, fluorescence arbitrary units; DTPA, diethylene triamine pentaacetic acid; GSH, glutathione; LC/MS, liquid chromatography/mass spectrometry; PAES, polyacrylethersulphone; HSA, human serum albumin; TDMBA, tetradecyldimethylbenzyl ammonium chloride; DTT, dithiothreitol; PFBenzylBr, pentafluorobenzylbromide.
None of the existing methods of sulphide determination met our defined need for an assay that could be adapted to the scale of samples required in pharmacokinetic studies, yet would maintain a high specificity and sensitivity for available sulphide in complex biological solutions such as whole blood. To fill this need, we made the initial assumption that ‘biologically available sulphide’ would be essentially equivalent to ‘reactive sulphide’, where the latter is defined as reactive to a moderate nucleophilic acceptor. The basis for the derivatization described in this work is therefore the reaction of sulphide with two equivalents of monobromobimane (Figure 1).
Through optimization of reaction conditions first with pure samples and then with whole blood, we found that reaction conditions of 1/3 acetonitrile, 1/3 50 mM HEPES buffer (pH 8) and 1/3 sample gave rapid, clean conversion of sulphide to sulphide dibimane in the presence of excess monobromobimane. It was anticipated that in whole blood, an excess of the derivatization reagent would be necessary to ensure full conversion of all reactive sulphide present, because it is well known that monobromobimane will react with a wide range of sulphydryls, including the abundant background concentrations of reactive cysteines on HSA and glutathione. We thus titrated baseline rat blood with monobromobimane for a 10 min reaction period, and found that 200 µL of 5 mM monobromobimane was taken up almost completely by 200 µL of whole rat blood (data not shown). However, beyond 5 mM, excess monobromobimane was present at the end of the reaction period. To overcome this baseline sink of reactive species in the assay, 200 µL of 10 mM monobromobimane is used to derivatize each 200 µL of blood. This assures that in our range of interest (0.2–5 µM sulphide), we are starting with an approximately 1000-fold excess of derivatization reagent for conversion of sulphide to sulphide dibimane in rat blood.
As already mentioned, due to the high reactivity of sulphide with oxygen, it is of utmost importance to capture any available sulphide in blood as rapidly as possible. To establish the validity of the bimane assay for that aspect, initial experiments were conducted varying the time of reaction from 10 min to 1, 5 or 20 min, and quenching any excess monobromobimane derivatization reagent with mercaptoethanol at the appropriate time. None of these changes in timing had an effect on sulphide dibimane production from standard test samples of 1 or 5 µM sulphide (data not shown). Kinetic traces of sulphide dibimane (Figure 3) confirmed that under the conditions of our assay, 10 µM sulphide was fully reacted within the first minute, and that even 100 µM sulphide was fully reacted within 5 min. Furthermore, under the conditions of our assay, the reaction is first order relative to the substrate hydrosulphide, meaning that the chemically limiting step is reaction of sulphide with the first equivalent of monobromobimane. The subsequent reaction to form the bis-bimane derivative (sulphide dibimane) is rapid in comparison and does not play a kinetic role. Taken together, the above data allow us to state that our assay conditions of 10 mM monobromobimane reacting at room temperature for 10 min (in the designated solvent system of 33% acetonitrile and 33% 50 mM HEPES buffer, pH 8) are sufficient for complete and reproducible conversion of 0.2–5 µM reactive sulphide to sulphide dibimane.
During in vivo dosing with sulphide, it is inevitable that the oxidation products thiosulphate, sulphite and sulphate will be produced, and thus assay specificity in the presence of these metabolites had to be established. During initial dosing of rodents with sodium sulphide, blood plasma levels of thiosulphate, sulphite and sulphate were monitored via ion chromatography, and as expected, levels of each increased with increased sulphide dosing (data not shown). In our hands, typical baseline concentrations observed in rat plasma via ion chromatography are 0.5–2.0 µM thiosulphate, 0–1.0 µM sulphite and 600–900 µM sulphate. During sulphide dosing at 10 mg·kg−1·h−1, corresponding concentrations were measured as 50 µM thiosulphate, 5 µM sulphite and 1100 µM sulphate. To confirm that these salt levels do not cause interference (false positive or negative peaks) during monobromobimane derivatization, we assayed standard sulphide samples in the presence of each compound. At concentrations of oxidized sulphide in line with or above those seen in the blood of sulphide-dosed animals, the detected area of the resultant sulphide dibimane peaks was not changed (Figure 5A–C).
Having designed a chemical derivatization protocol that reliably and quantitatively reacts with available sulphide in blood, the challenge remained to maintain sensitivity of the assay such that large numbers of samples could nevertheless be measured with precision. In order to maximize the sulphide dibimane signal in a setting where sulphide is derivatized directly from the complex matrix of whole blood, an ethyl acetate extraction and concentration procedure was used. The procedure detailed above separated sulphide dibimane from the majority of other fluorescent species formed – protein/peptide cysteines that are also derivatized by bimane, but are not soluble in an organic medium – and then removed the ethyl acetate. When reconstituted for HPLC analysis, there is an overall twofold concentration in volume, thereby doubling the sulphide dibimane signal. The extraction, concentration and re-solubilization procedure, as a whole, shows an average 76% recovery of sulphide dibimane from blood (Figure 4) that is reproducible and concentration independent over our range of interest of 0.2–5 µM sulphide dibimane.
The final measurement of sulphide dibimane (and thus the original reactive sulphide) was made via the well-known fluorescent properties of the sulpho-bimane moiety. To distinguish post-reaction the derivatized sulphide dibimane from the excess of starting monobromobimane, a gradient HPLC method was developed using water and methanol in acetonitrile to separate the analytes in question. In this setting, the standard curve for sulphide dibimane shows excellent linearity over the range of interest of 0.2–5 µM (Figure 2C). Thus, having explored the various aspects of the monobromobimane derivatization method detailed above, we have concluded that we could use this assay to measure the reactive sulphide of biological samples in a range of 0.2–5 µM.
Initial pharmacokinetic findings in rodents were straightforward: using the monobromobimane assay to measure aliquots of fresh blood sampled from jugular vein catheters in rats, baseline levels of available blood sulphide were determined to be approximately 0.7 µM, and intravenous bolus dosing of sodium sulphide resulted in a dose-dependent increase in sulphide blood levels (Figure 6). Levels of available blood sulphide reached at least 1.5 µM in the rats that received 1 mg·kg−1 bolus dose of sodium sulphide, and reached at least 4.5 µM sulphide in rats given the 4 mg·kg−1 bolus dose. The half-life of this available sulphide was calculated to be approximately 6 min following intravenous bolus administration of sodium sulphide at 4 mg·kg−1. Levels of available blood sulphide were also measured during continuous intravenous infusions of sodium sulphide solution (2–20 mg·kg−1·h−1, administered for 2 h). Again, using the monobromobimane assay to measure aliquots of fresh blood sampled from jugular vein catheters in rats (Figure 7A), elevation of blood sulphide concentration during infusion was found to be dose dependent, and available blood sulphide appeared to reach a steady state within 2 h of the start of sulphide infusion. After infusion was stopped, sulphide levels returned to baseline within 1 h post-discontinuation.
Thus, intravenous administration of sodium sulphide to rats, both by bolus dosing and by constant infusion, exhibited pharmacokinetics that were dose dependent and that were consistent with a short half-life of the compound in blood. Expanding the purview of the monobromobimane assay, similar pharmacokinetics have subsequently been measured following exogenous sulphide infusion in mice, beagle dogs, domestic pigs and cynomolgus monkeys (data not shown; all studies done under documented animal welfare approval according to local and US and/or EU regulations). Across all species studied, we observed a short half-life of available sulphide in blood, and we observed a dose-dependent relationship between sodium sulphide administration and subsequent elevation of available blood sulphide. Furthermore, when baseline levels of sulphide are measured with monobromobimane, all animal species studied yielded basal blood concentrations of sulphide between 0.3 and 1.2 µM.
Given that the bulk of animal and human sulphide exposure data to date describes the effects of H2S gas, we sought to use the monobromobimane derivatization assay detailed above to construct a relative bioequivalency curve between exposure to atmospheric H2S gas and dosing with intravenous sodium sulphide. By plotting available blood sulphide concentrations reached by both sodium sulphide infusion and H2S inhalation over 2 h (Figure 7A and B), it is apparent that both methods of exposure give rise to linear dose dependency over the dose range investigated. Furthermore, because blood sulphide increases linearly with increasing levels of either exposure method, intravenous sodium sulphide infusion and continuous H2S gas inhalation can themselves be linearly correlated. Comparison of the two opposing plots in Figure 8 yields the following metric, a rough bioequivalency formula for a 2 h exposure of rats to sulphide: each 1 mg·kg−1·h−1 of intravenous sodium sulphide dosing for 2 h is approximately equivalent to 30 ppm of gaseous H2S inhalation for 2 h. It must be stressed, of course, that this calculation assumes that the reactive blood sulphide measured via monobromobimane is a valid indicator of the ‘biologically available sulphide’ that is responsible for the in vivo effects of sulphide administration.
To tie the monobromobimane assay firmly to existing methods of sulphide analysis, and to probe the exact nature of the ‘available sulphide’ being measured in this chemical derivatization reaction, we made use of a second assay that operates through completely different means: measurement of dissolved H2S via an amperometric sensor. The amperometric sensor (WPI Instruments) detects current change from an electrochemical reaction at its head, and is the same model that Kraus and colleagues used in their studies (e.g. Doeller et al., 2005). It consists of a platinum anode and cathode bathed in a solution of ferricyanide that is retained inside an H2S-permeable membrane. The membrane and characteristic electrochemistry give the sensor a high selectivity and sensitivity for H2S relative to other physiological gases (CO2, O2, CO, etc.). However, as the membrane is permeable to H2S gas and not to HS- ions, the instrument must always be calibrated at a specific pH such that the total concentration of free sulphide (H2S + HS-) is properly accounted for. The sensor has been used in previous studies to measure the production and biological ‘consumption’ of H2S in biological matrices (Doeller et al., 2005; Benavides et al., 2007) While the instrumentation required for this electrochemical method of detection is not easily amenable to handling large numbers of samples for pharmacokinetics, and while absolute determination of sulphide levels via the amperometric sensor requires constant calibration, this method is nevertheless very sensitive in its ability to detect relative changes in dissolved H2S over a short period of time. In addition, the H2S sensor can be placed directly into the bloodstream of the rat, allowing ‘in-line’ measurements in an in vivo setting.
To begin, we used the amperometric sensor to confirm the ability of the monobromobimane assay to accurately measure concentrations of sulphide dissolved in aqueous buffer. When sodium sulphide was added to an oxygen-depleted buffer, the bimane assay measured an almost identical curve to that of the immersed sulphide sensor (Figure 9A). When we repeated the latter experiment in a matrix of fresh blood, however, the monobromobimane reaction measured significantly higher levels of reactive sulphide than the levels of dissolved H2S that were registered by the H2S-sensitive probe (Figure 9B). The amperometric sensor recorded a brief initial spike (<10 s) on addition of 20 µM sodium sulphide to the stirring solution, but thereafter essentially no signal for H2S/HS- was measured. During this same period, a significant concentration of reactive sulphide was available to monobromobimane, and this observation, consistent with previous findings (Doeller et al., 2005), suggests that when administered to whole blood, H2S is rapidly taken into a ‘sink’. From the absence of signal from the amperometric sensor, this sink is marked as something other than dissolved, unassociated H2S, but the sink is ‘reversible’ in that its sulphide can be captured, still in the −2 oxidation state, by a relatively mild substitution reaction with monobromobimane. To further characterize this free sulphide-scavenging property of blood, we added sodium sulphide to solutions of either human plasma or a buffered 5% HSA solution. In both of these solutions, sulphide measurement by either amperometric detection or chemical derivatization was similar (Figure 9C and D). The fact that the centrifugation step of producing plasma removes the substance that differentiates the H2S sensor from the monobromobimane assay suggests the formation of a sulphydrated protein product (e.g. a protein persulphide) from which monobromobimane can still remove sulphide prior to its oxidation.
The above experiment retains the caveat of measuring sulphide in stirred blood in an in vitro setting. Thus, we placed the amperometric sensor directly into the vena cava in vivo by means of an in-dwelling catheter. Bolus dosing experiments with sodium sulphide confirmed that an H2S/HS- signal disappears rapidly in vivo, as well with a 1 mg·kg−1 bolus dose visible to the probe only when administered in less than 60 s (Figure 10). By extending this experiment to a design where continuous infusion of sodium sulphide was performed and the rate of infusion increased in a stepwise fashion, we conclude that the in-line H2S sensor could be used simultaneously with blood sampling for the bimane assay. As shown in Figure 11, the amperometric probe registers essentially no signal until a rate of 60 mg·kg−1·h−1 is reached. Derivatization of the freshly drawn blood with monobromobimane, however, measures reactive sulphide already at the lowest infusion rate, and at the highest infusion rate measures over 30 µM of available sulphide. Thus, the ‘reversible sulphide sink’ noted from the blood experiments in vitro appears to be present in vivo as well.
Similar to our in vitro conclusions, the ‘sink’ observed via the bimane assay during a bolus (Figure 6) or infusion (Figure 11) of sodium sulphide solution is shown not to be H2S/HS-per se. The presence of dissolved, unassociated H2S in the rat bloodstream after 60 s in the case of bolus dosing, and up to 20 mg·kg−1·h−1 in the case of continuous infusion is ruled out by the amperometric sensor data of Figures 10 and 11 respectively. While oxidation by whole blood to thiosulphate certainly contributes to the rapid disappearance of sulphide in vivo (Kage et al., 1991), the postulated in vivo sulphide ‘sink’ that we propose to be the substrate of the monobromobimane assay is unlikely to be composed of oxidized sulphide, because we have shown that neither sulphite, sulphate nor thiosulphate, the three main oxidation products of sulphide, produce sulphide dibimane in the presence of monobromobimane.
Given that we propose a reversible sulphide ‘sink’ to explain levels of monobromobimane-reactive sulphide measured during intravenous sodium sulphide dosing, we sought to refine the hypothesis based on the lability with which sulphur atoms in the sink are exchanged with other pools of sulphide in vivo. When rats are dosed with 34S-sodium sulphide, levels of 34S-sulphide in blood increase directly, and levels of 32S-sulphide remain unchanged (Figure 12). These data are consistent with the hypothesis that the sulphide of the available sulphide sink is loosely bound, but that it is not in rapid exchange with any endogenous pool of sulphide during the time frame of the experiments. Interestingly, after the end of dosing, only 34S-sulphide disappears and the blood levels of 34S-sulphide fall below those of 32S-sulphide once more. This observation suggests that the pool of derivatizable sulphide which provides the endogenous baseline signal is not of the exactly the same molecular structure as that of the ‘reversible sulphide sink’ which is utilized on dosing with sodium sulphide. While the structures may be related (in particular, both might be persulphides RSnH on different peptide cysteines), if both types were identical, upon cessation of 34S-sodium sulphide infusion, levels of both 34S-sulphide and 32S-sulphide would fall until their sum equaled the original basal level of available sulphide. However, as noted, only 34S-sulphide dissipates, so this sulphide must be present in a slightly more reactive molecular environment than the endogenous 32S-sulphide which is measured in baseline blood.
In another in vivo study, sodium sulphide was administered by intravenous bolus injection of 3 mg·kg−1 to a rat implanted with an in-line microdialysis catheter (Figure 13A). Measuring levels of blood sulphide via derivatization of aliquots from the catheter showed a strong and rapid response to the bolus (8 µM sulphide within the first minute), with a subsequent half-life of 20–30 s (Figure 13B). By comparison of this graph to the response of the in-line amperometric sensor in a similar bolus dosing (Figure 10), the level of sulphide detected via monobromobimane cannot be due to dissolved H2S gas alone. Furthermore, we have noticed a substantial difference between the half-life of sulphide in vivo, as determined from the monobromobimane-based method (approximately 6 min), and the half-life of sulphide, as determined by the microdialysis method (approximately 20–30 s). The existence of the proposed blood-based ‘sulphide sink’ may explain this difference: while the whole blood monobromobimane assay detects all forms of sulphur available for the reaction, the microdialysis catheter incorporates a filter with a cut-off of 20 kDa. If the nature of the ‘sulphide sink’ in the blood is, at least in part, protein(s) of 20+ kDa on which sulphide is reversibly bound (e.g. via a reactive cysteine), this bound sulphide would not be available for the monobromobimane reaction on the other side of the membrane. Hence, the reaction would in this case capture only diffusible molecules, such as small sulphydrated cysteine peptides and H2S gas, both of which would be expected to have a short half-life compared to large sulphydrated proteins that could stabilize their persulphides through local electronic environments.
The results of this paper are consistent with several recent publications on sulphide in vivo. In a separate study from this laboratory examining parenteral sodium sulphide dosing (Insko et al., 2009), the half-life of free H2S in blood as measured by the concentration of H2S in exhaled air appears to be short (approximately 10 s), in agreement with the hypothesis that the monobromobimane reaction detects additional reactive sulphur species in blood beyond free H2S/HS-. A recent study by Snyder and coleagues also corroborates the theory of a ‘sulphide sink’ and begins to specifically identify the putative sulphide-sequestering molecules which our bimane investigations indicate: H2S was found to physiologically modify cysteines in a large number of proteins by S-sulphhydration (Mustafa et al., 2009), a finding that is consistent with the conclusions of the present paper, and suggests specific components that may be responsible for the in vitro sulphide consumption effect differentiating Figure 9A (sulphide + saline) from Figure 9B (sulphide + blood).
In conclusion, we describe here a novel method for measuring sulphide in whole blood based on chemical derivatization of samples with excess monobromobimane. While we have shown that the bimane assay can derivatize H2S/HS- dissolved in solution at sub-micromolar levels, the presence of persistent unassociated H2S in the blood of sodium sulphide-treated animals is ruled out by the negative signal of an amperometric sensor. Therefore, another source must be proposed for the significant concentration of blood sulphide that is available to react with monobromobimane following administration of Na2S solution. Our findings are consistent with the existence in blood of a ‘reversible sulphide sink’, and we posit that the bimane assay of this paper measures not only free H2S/HS- dissolved in blood, but also one or more forms of available sulphide that are ‘loosely’ or ‘reversibly’ bound in vivo, and can thus be liberated, still in the −2 oxidation state, by the relatively mild (HEPES buffer, pH 8) substitution reaction in which sulphide dibimane is generated from monobromobimane. We further propose that, given the observed effect of a 20 kDa filter on the sulphide sink, its chemical structure may take the form of a persulphide RSnH on the cysteine residues of one or more proteins. Finally, because the mild chemical nature of the monobromobimane derivatization reaction suggests that the bimane assay is measuring sulphide that is likely to be reversibly held under physiological conditions, we propose that the monobromobimane assay presented here may be useful to estimate the concentration of biologically available sulphide in vivo.
Glossary
Abbreviations:
- GSH
glutathione
- HEPES
(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HPLC
high performance liquid chromatography
- H2S
hydrogen sulphide
- HSA
human serum albumin
- LC/MS
liquid chromatography/mass spectrometry
- ppm
parts per million
Conflicts of interest
All authors of this paper are employees and stockholders of Ikaria Inc., a for-profit organization involved in the commercialization of a H2S formulation as a human therapeutic agent.
References
- Benavides GA, Squadrito GL, Mills RW, Patel HD, Isbell TS, Patel RP, et al. Hydrogen sulfide mediates the vasoactivity of garlic. Proc Natl Acad Sci U S A. 2007;104:17977–17982. doi: 10.1073/pnas.0705710104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia M, Sidhapuriwala JN, Sparatore A, Moore PK. Treatment with H2S-releasing diclofenac protects mice against acute pancreatitis-associated lung injury. Shock. 2008;29:84–88. doi: 10.1097/shk.0b013e31806ec26. [DOI] [PubMed] [Google Scholar]
- Doeller JE, Isbell TS, Benavides G, Koenitzer J, Patel H, Patel RP, et al. Polarographic measurement of hydrogen sulfide production and consumption by mammalian tissues. Anal Biochem. 2005;341:40–51. doi: 10.1016/j.ab.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, et al. Hydrogen sulfide attenuates myocardial ischemia–reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci U S A. 2007;104:15560–15565. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esechie A, Kiss L, Olah G, Horvath EM, Hawkins HK, Szabo C, et al. Protective effect of hydrogen sulfide in a murine model of combined burn and smoke inhalation-induced acute lung injury. Clin Sci. 2008;115:91–97. doi: 10.1042/CS20080021. [DOI] [PubMed] [Google Scholar]
- Esechie A, Enkhbaatar P, Traber DL, Jonkam C, Lange M, Hamahata A, et al. Beneficial effect of a hydrogen sulphide donor (sodium sulphide) in an ovine model of burn- and smoke-induced acute lung injury. Br J Pharmacol. 2009;158:1442–1453. doi: 10.1111/j.1476-5381.2009.00411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahey R, Newton G, Dorian R, Kosower E. Analysis of biological thiols: quantitative determination of thiols at the picomole level based upon derivatization with monobromobimane and separation by cation-exchange chromatography. Anal Biochem. 1981;111:357–365. doi: 10.1016/0003-2697(81)90573-x. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Distrutti E, Cirino G, Wallace JL. The emerging roles of hydrogen sulfide in the gastrointestinal tract and liver. Gastroenterology. 2006;131:259–271. doi: 10.1053/j.gastro.2006.02.033. [DOI] [PubMed] [Google Scholar]
- Furne J, Saeed A, Levitt MD. Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1479–R1485. doi: 10.1152/ajpregu.90566.2008. [DOI] [PubMed] [Google Scholar]
- Insko MA, Deckwerth TL, Hill P, Toombs CF, Szabo C. Detection of exhaled hydrogen sulphide gas in rats exposed to intravenous sodium sulphide. Br J Pharmacol. 2009;157:944–951. doi: 10.1111/j.1476-5381.2009.00248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Institute of Laboratory Animal Resources Commission on Life Sciences National Research Council. National Research Council: Guide for the Care and Use of Laboratory Animals; Institute of Laboratory Animal Resources; Commission on Life Sciences. Washington, DC: National Academy Press; 1996. Available at http://www.nap.edu/openbook.php?record_id=5140 (accessed 1 February 2010) [Google Scholar]
- Jha S, Calvert JW, Duranski MR, Ramachandran A, Lefer DJ. Hydrogen sulfide attenuates hepatic ischemia–reperfusion injury: role of antioxidant and antiapoptotic signaling. Am J Physiol Heart Circ Physiol. 2008;295:H801–H806. doi: 10.1152/ajpheart.00377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kage S, Nagata T, Kimura K, Kudo K. Extractive alkylation and gas chromatographic analysis of sulfide. J Forensic Sci. 1988;33:217–222. [PubMed] [Google Scholar]
- Kage S, Nagata T, Kudo K. Determination of thiosulfate in body fluids by GC and GC/MS. J Anal Toxicol. 1991;15:148–150. doi: 10.1093/jat/15.3.148. [DOI] [PubMed] [Google Scholar]
- Kiss L, Deitch EA, Szabó C. Hydrogen sulfide decreases adenosine triphosphate levels in aortic rings and leads to vasorelaxation via metabolic inhibition. Life Sci. 2008;83:589–594. doi: 10.1016/j.lfs.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenitzer JR, Isbell TS, Patel HD, Benavides GA, Dickinson DA, Patel RP, et al. Hydrogen sulfide mediates vasoactivity in an O2-dependent manner. Am J Physiol Heart Circ Physiol. 2007;292:H1953–H1960. doi: 10.1152/ajpheart.01193.2006. [DOI] [PubMed] [Google Scholar]
- Kosower EM, Pazhenchevsky B. Bimanes. 5. Synthesis and properties of syn- and anti-1,5-diazabicyclo[3.3.0]octadienediones (9,10-dioxabimanes) J Am Chem Soc. 1980;102:4983–4993. [Google Scholar]
- Kraus DW, Doeller JE. Sulfide consumption by mussel gill mitochondria is not strictly tied to oxygen reduction: measurements using a novel polarographic sulfide sensor. J Exp Biol. 2004;207:3667–3679. doi: 10.1242/jeb.01212. [DOI] [PubMed] [Google Scholar]
- Li L, Moore PK. Putative biological roles of hydrogen sulfide in health and disease: a breath of not so fresh air? Trends Pharmacol Sci. 2008;29:84–90. doi: 10.1016/j.tips.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Mok YY, Atan MS, Yoke Ping C, Zhong Jing W, Bhatia M, Moochhala S, et al. Role of hydrogen sulphide in haemorrhagic shock in the rat: protective effect of inhibitors of hydrogen sulphide biosynthesis. Br J Pharmacol. 2004;143:881–889. doi: 10.1038/sj.bjp.0706014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller EG. Trafficking in persulfides: delivering sulfide in biosynthetic pathways. Nat Chem Biol. 2006;2:185–194. doi: 10.1038/nchembio779. [DOI] [PubMed] [Google Scholar]
- Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, et al. H2S signals through protein S-sulfhydration: a novel posttranslational modification. Sci Signal. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata T, Kage S, Kimura K, Kudo K, Noda M. Sulfide concentrations in postmortem mammalian tissues. J Forensic Sci. 1990;35:706–712. [PubMed] [Google Scholar]
- Newton G, Dorian R, Fahey R. Analysis of biological thiols: derivatization with monobromobimanes and separation by reverse phase high performance liquid chromatography. Anal Biochem. 1981;114:383–387. doi: 10.1016/0003-2697(81)90498-x. [DOI] [PubMed] [Google Scholar]
- Ogasawara Y, Ishii K, Togawa T, Tanabe S. Determination of bound sulfur in serum by gas dialysis/high-performance liquid chromatography. Anal Biochem. 1993;215:73–81. doi: 10.1006/abio.1993.1556. [DOI] [PubMed] [Google Scholar]
- Reiffenstein RJ, Hulbert WC, Roth SH. Toxicology of hydrogen sulfide. Annu Rev Pharmacol Toxicol. 1992;32:109–134. doi: 10.1146/annurev.pa.32.040192.000545. [DOI] [PubMed] [Google Scholar]
- Rossoni G, Sparatore A, Tazzari V, Manfredi B, Del Soldato P, Berti F. The hydrogen sulfide-releasing derivative of diclofenac protects against ischaemia–reperfusion injury in the isolated rabbit heart. Br J Pharmacol. 2008;153:100–109. doi: 10.1038/sj.bjp.0707540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon F, Giudici R, Duy CN, Schelzig H, Oter S, Groger M, et al. Hemodynamic and metabolic effects of hydrogen sulfide during porcine ischemia/reperfusion injury. Shock. 2008;30:359–364. doi: 10.1097/SHK.0b013e3181674185. [DOI] [PubMed] [Google Scholar]
- Sivarajah A, McDonald MC, Thiemermann C. The production of hydrogen sulfide limits myocardial ischemia and reperfusion injury and contributes to the cardioprotective effects of preconditioning with endotoxin, but not ischemia in the rat. Shock. 2006;26:154–161. doi: 10.1097/01.shk.0000225722.56681.64. [DOI] [PubMed] [Google Scholar]
- Sodha NR, Clements RT, Feng J, Liu Y, Bianchi C, Horvath EM, et al. The effects of therapeutic sulfide on myocardial apoptosis in response to ischemia–reperfusion injury. Eur J Cardiothorac Surg. 2008;33:906–913. doi: 10.1016/j.ejcts.2008.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stipanuk MH, Beck PW. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem J. 1982;206:267–277. doi: 10.1042/bj2060267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C. Hydrogen sulfide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- Togawa T, Ogawa M, Nawata M, Ogasawara Y, Kawanabe K, Tanabe S. High performance liquid chromatographic determination of bound sulfide and sulfite and thiosulfate at their low levels in human serum by pre-column fluorescence derivatization with monobromobimane. Chem Pharm Bull. 1992;40:3000–3004. doi: 10.1248/cpb.40.3000. [DOI] [PubMed] [Google Scholar]
- Ubuka T. Assay methods and biological roles of labile sulfur in animal tissues. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;781:227–249. doi: 10.1016/s1570-0232(02)00623-2. [DOI] [PubMed] [Google Scholar]
- Wallace JL. Hydrogen sulfide-releasing anti-inflammatory drugs. Trends Pharmacol Sci. 2007;28:501–505. doi: 10.1016/j.tips.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Dicay M, McKnight W, Martin GR. Hydrogen sulfide enhances ulcer healing in rats. FASEB J. 2007;21:4070–4076. doi: 10.1096/fj.07-8669com. [DOI] [PubMed] [Google Scholar]
- Wang R. The gasotransmitter role of hydrogen sulfide. Antioxid Redox Signal. 2003;5:493–501. doi: 10.1089/152308603768295249. [DOI] [PubMed] [Google Scholar]
- Wei HL, Zhang CY, Jin HF, Tang CS, Du JB. Hydrogen sulfide regulates lung tissue-oxidized glutathione and total antioxidant capacity in hypoxic pulmonary hypertensive rats. Acta Pharmacol Sin. 2008;29:670–676. doi: 10.1111/j.1745-7254.2008.00796.x. [DOI] [PubMed] [Google Scholar]
- Whitfield NL, Kreimier EL, Verdial FC, Skovgaard N, Olson KR. Reappraisal of H2S/sulfide concentration in vertebrate blood. Am J Physiol Regul Integr Comp Physiol. 2008;294:1930–1970. doi: 10.1152/ajpregu.00025.2008. [DOI] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YZ, Wang ZJ, Ho P, Loke YY, Zhu YC, Huang SH, et al. Hydrogen sulfide and its possible roles in myocardial ischemia in experimental rats. J Appl Physiol. 2007;102:261–268. doi: 10.1152/japplphysiol.00096.2006. [DOI] [PubMed] [Google Scholar]