

Abstract
Background and purpose:
Pancreatic cancer is a highly aggressive malignancy, and improvement in systemic therapy is necessary to treat this frequently encountered metastatic disease. The current targeted agents used in combination with gemcitabine improved objective response rates, but with little or no improvements in survival and also increased toxicities in pancreatic cancer patients. Recently, we showed that the triterpenoid cucurbitacin B inhibited tumour growth in pancreatic cancer cells by inhibition of the JAK/STAT pathway, and synergistically increased antiproliferative effects of gemcitabine in vitro.
Experimental approach:
The anti-tumour effects and toxicities of cucurbitacin B in combination with gemcitabine were tested against human pancreatic cancer cells in a murine xenograft model.
Key results:
Combined therapy with cucurbitacin B and gemcitabine at relatively low doses (0.5 mg·kg−1 and 25 mg·kg−1 respectively) resulted in highly significant tumour growth inhibition of pancreatic cancer xenografts (up to 79%). Remarkably, this therapy was well tolerated by the animals, as shown by histology of visceral organs, analysis of serum chemistry, full blood counts and bone marrow colony numbers. Western blot analysis of the tumour samples of mice who received both cucurbitacin B and gemcitabine, revealed stronger inhibition of Bcl-XL, Bcl-2 and c-myc, and higher activation of the caspase cascades, than mice treated with either agent alone.
Conclusions and implications:
Combination of cucurbitacin B and gemcitabine had profound anti-proliferative effects in vivo against xenografts of human pancreatic cancer cells, without any significant signs of toxicity. This promising combination should be examined in therapeutic trials of pancreatic cancer.
Keywords: cucurbitacin B, gemcitabine, pancreatic cancer, JAK/STAT pathway, murine xenograft model
Introduction
Pancreatic cancer is one of the most common invasive malignancies and the fourth leading cause of cancer-related mortality in the United States (Strimpakos et al., 2008). The most common tumour of the pancreas is adenocarcinoma, responsible for an estimated 37 680 newly diagnosed cases in 2008, and an associated 34 290 deaths from the disease in the same year in the USA. Gemcitabine (2′-deoxy-2′,2′-difluorocytidine) still remains the standard therapy for adjuvant and palliative pancreatic cancer, although the response rate is low, at less than 20% (Jemal et al., 2008; Strimpakos et al., 2008). Limited progress has been achieved despite intense research using combined treatments with chemotherapeutic agents, often along with biological agents targeting activated molecular pathways involved in pancreatic cancer (Langer, 1998; Jemal et al., 2008; Strimpakos et al., 2008). Due to a small improvement in median survival and therapy-related toxicities, the clinical benefit of these combinations remains uncertain. Therefore, developing novel therapeutic strategies are required to decrease the incidence and control the severity associated with this form of cancer.
The natural cucurbitacins constitute a group of triterpenoid substances which are well-known for their bitterness and toxicity. Structurally, they are characterized by the tetracyclic cucurbitane nucleus skeleton and can be divided into twelve groups (Chen et al., 2005). Cucurbitacins exhibit wide-ranging pharmacological effects in vivo, such as anti-inflammatory activities, and preventive and curative effects against CCl4-induced hepatotoxicity (Chen et al., 2005). In addition, some cucurbitacins are known for cytotoxicity and anti-cancer activity (Jayaprakasam et al., 2003; Chen et al., 2005). Recently, we demonstrated that cucurbitacin B (C32H46O8) markedly inhibited the proliferation of several human pancreatic cancer cell lines in vitro (Thoennissen et al., 2009). The growth-inhibitory effect of the compound was associated with a significant G2/M phase arrest and increased apoptosis by inhibition of the JAK/STAT pathway, which has been shown to be aberrant in pancreatic cancer (Toyonaga et al., 2003; Sahu and Srivastava, 2009). Therefore, cucurbitacin B provides a novel therapeutic strategy for this type of aggressive malignancy. Moreover, the triterpenoid significantly decreased pancreatic tumour growth in a xenograft model, and also synergistically potentiated the anti-proliferative effect of gemcitabine on pancreatic cancer cells in vitro (Thoennissen et al., 2009).
The aim of the present study was to evaluate the anti-proliferative activity of a combination of cucurbitacin B and gemcitabine in comparison with monotherapy regimens in the in vivo treatment of pancreatic tumour xenografts in athymic nude mice, and to find a rationale for further clinical studies in pancreatic cancer. To further explore this background, we conducted an analysis of possible treatment-related toxicities in peripheral blood, visceral organs and bone marrow.
Methods
Cell culture
The human pancreatic cancer cell line Panc-1 was obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA) and cultured as monolayer in DMEM medium (Life Technologies, Rockville, MD, USA) with 10% (v/v) heat-inactivated fetal bovine serum (Gemini Bio-Products, Calabasas, CA, USA). The adherent cells were detached from the flask surface using a solution of 2.5% trypsin and ethylenediaminetetra-acetate (EDTA).
Murine xenograft model and treatment groups
All animal care and experimental protocols were in accordance with the guidelines of Cedars-Sinai Research Institute and the National Institutes of Health. Human cancer xenografts in nude mice are a well-accepted animal model for pre-clinical evaluation of promising anti-cancer agents. Therefore, to extend our recent observations made in cultured pancreatic cancer cells, the effects of a combination of cucurbitacin B and gemcitabine on the growth of human pancreatic tumours were determined in a murine xenograft model. Five-week old female nu/nu athymic mice (weight: 20–22 g) from Harlan Sprague-Dawley, Inc., IN, were maintained in pathogen-free conditions and fed irradiated chow.
Xenografts were developed by injecting subcutaneously the human pancreatic cancer cell line Panc-1 (107 cells) in 0.2 mL of matrigel (Basement Membrane Matrix, High Concentration; BD Biosciences, NJ, USA) in both flanks of the animals. Mice (5 per cohort) were randomly assigned to six different treatment groups: (A) diluent control (DMSO); (B) gemcitabine (25 mg·kg−1); (C) low-dose cucurbitacin B (0.5 mg·kg−1); (D) high-dose cucurbitacin B (1 mg·kg−1); (E) low-dose combination (cucurbitacin B 0.5 mg·kg−1 plus gemcitabine 25 mg·kg−1); and (F) high-dose combination (cucurbitacin B 1 mg·kg−1 plus gemcitabine 25 mg·kg−1 body weight). Treatment was started the day after cell implantation with intraperitoneal (i.p.) applications of either diluent-control, cucurbitacin B (three times per week), and/or gemcitabine (two times per week) in a volume of 0.1 mL for each application. A summary of our treatment schedule is given in Table 1.
Table 1.
Treatment groups and outcome
| No. | Treatment groups (5 mice/group) | Drug dose (mg·kg−1) body weight† | Final tumour volume (cm3) | Final tumour weight (g) | Percentage tumour inhibition (%) | Final body weight (g) |
|---|---|---|---|---|---|---|
| A | Control | 0 | 1.2 ± 0.2 | 1.8 ± 0.5 | 0 | 26.3 ± 1.1 |
| B | Gem | 25 | 0.8 ± 0.1 | 1.2 ± 0.4 | 32 | 25.7 ± 1 |
| C | CuB low | 0.5 | 0.9 ± 0.2 | 1.5 ± 0.4 | 18 | 25.8 ± 0.9 |
| D | CuB high | 1 | 0.4 ± 0.1 | 0.7 ± 0.4 | 63 | 25.1 ± 0.9 |
| E | Comb low (CuB + Gem) | 0.5 + 25 | 0.3 ± 0.03 | 0.4 ± 0.1 | 79 | 25.7 ± 1.2 |
| F | Comb high (CuB + Gem) | 1 + 25 | 0.2 ± 0.02 | 0.3 ± 0.1 | 83 | 23.7 ± 0.7* |
Control: diluent control group (A); Gem: gemcitabine treatment group (B); CuB low: low-dose cucurbitacin B treatment group (C); CuB high: high-dose cucurbitacin B treatment group (D); Comb low: low-dose combination treatment group (E); Comb high: high-dose combination treatment group (F). Data in the Table are means ± SD.
P < 0.05.
Gemcitabine was administered twice weekly; cucurbitacin B was administered three times weekly; both i.p.
Tumours (10 per group) were measured with vernier calipers every 3 days, and volume was calculated as described previously using the following formula: (length × width × depth) × 0.5236 (Luong et al., 2006). Each mouse was also weighed three times a week. After 43 days of treatment, the experiment was halted, all mice killed and dissected tumours were weighed. Tumour material was fixed in neutral-buffered formalin (10%), and embedded in paraffin wax. The inhibition rate of tumour growth was calculated using the following formula: tumour growth inhibition rate (%) = (1 −MT/MC) × 100, whereas MT and MC are the mean normalized tumour masses of treatment and control groups respectively (Sun et al., 2003). Metastatic disease was determined macroscopically at autopsy in all thoracic, abdominal, retroperitoneal and pelvic organs followed by histological examination.
Western blotting and immunoprecipitation assay
At the end of the experiment, equal amounts of each dissected xenograft tumour were also snap-frozen in liquid nitrogen and stored at −80°C. For Western blot and immunoprecipitation (IP) analysis, the frozen tumour tissue homogenates were prepared in SDS lysis buffer of 50 mM Tris-HCl (pH 7.4), 2% SDS (Solit et al., 2002) containing additional Halt protease and phosphatase inhibitor cocktail (Thermo Scientific, Rockford, IL, USA). We prepared pooled lysates out of the five mice (=10 tumours) per treatment group for Western blot and immunoprecipitation analysis. For Western blotting, protein lysates (50 µg) were boiled in Laemmli sample buffer (Bio-Rad Laboratories, Hercules, CA, USA), resolved by electrophoresis on 4–15% SDS-polyacrylamide gels, and transferred to PVDF membranes. Membranes were probed with antibodies from Cell Signalling Technology Inc. (MA, USA), as well as Santa Cruz Biotechnology (CA, USA) and developed using the enhanced chemiluminescence kit (Pierce, Rockford, IL, USA), whereas GAPDH was used as a control.
For the immunoprecipitation assay, 500 µg of protein lysate of each treatment group was incubated with specific antibodies and protein A/G agarose beads (Santa Cruz Biotechnology) at 4°C for 16 h. Precipitated proteins were washed three times with lysis buffer, eluted with sodium dodecyl sulphate sample buffer and subjected to Western blot analysis. All antibodies used in the present study can be found in Table S1.
RNA extraction and quantitative real-time RT-PCR
Total cellular RNA was extracted and pooled from equal amounts of 10 snap-frozen xenograft tumours from each treatment group using TRIzol® Reagent (Invitrogen, CA, USA) according to the manufacturer's protocol. Aliquots of total cellular RNA (2 µg) were subjected to first-strand cDNA synthesis using SuperScript II reverse transcriptase (Invitrogen, CA, USA). For real-time RT-PCR analysis, reactions were performed using HotMaster Taq DNA Polymerase (Eppendorf, Hamburg, Germany) and SYBRGreen I (Molecular Probes, Eugene, OR, USA). Reactions were performed in triplicate using an iCycler iQ system (Biorad, Hercules, CA, USA). For each sample, the amount of the target gene and reference gene was determined from standard curves. mRNA levels were normalized against endogenous GAPDH mRNA. Sequence of primers used for quantitative real-time RT-PCR for PEG10: forward, 5′-GCCTGGCTCAGGTGTGGGAC-3′ and reverse: 5′-CCTGGATCCTGGCCCCCTCT-3′; for GAPDH: forward, 5′-TTAGCACCCCTGGCCAAG-3′ and reverse: 5′-CTTACTCCTTGGAGGCCATG-3′.
Toxicity studies
Serum chemistry and blood count analysis
Heparinized blood samples (40 µL per mouse) were obtained on day +0 and at the end of treatment (day +43) by submandibular bleeding. For chemistry, samples were additionally centrifuged for 3 min, followed by separation of the serum. Blood results were obtained by the Hemagen Analyst® Benchtop Chemistry System (Hemagen Diagnostics, Inc., Columbia, MD, USA).
Colony formation assay
Bone marrow mononuclear cells were isolated after killing the mice, by flushing femurs with phosphate-buffered saline containing 2% heat-inactivated fetal bovine serum and 2 mM EDTA using a 22-gauge needle. Red blood cells were removed from the bone marrow cell isolates by lysis on ice for 5 to 10 min in 10 mM NH4Cl made in 10 mM Tris (pH 7.2). Bone marrow cells isolated from all six groups were plated in triplicate in a 6-well plate at 2 × 104 cells per well in semi-solid 1% methylcellulose medium (MethoCult GF M3434, StemCell Technologies, Vancouver, Canada) containing recombinant murine (rm) Stem Cell Factor, rm IL-3, recombinant human (rh) IL-6, and rh erythropoietin for growth of myeloid and erythroid colony-forming units. Number of colonies (BFU-E, Blast-forming unit-erythroid; CFU-GM, Colony-forming unit-granulocyte/macrophages; CFU-GEMM, Colony-forming unit granulocyte/ erythrocyte/ monocyte/megakaryocyte) were evaluated and counted under the microscope, 10 days after seeding.
Examination of visceral organs
At the end of the experiment, visceral organs including liver, spleen and kidneys were also removed from each mouse. First, the organs were weighed, fixed in 10% buffered formalin, embedded in paraplast (Oxford Labware, St. Louis, MO, USA) and cut in 6 µm thick sections, followed by staining with haematoxylin and eosin (H&E) for histopathological examination.
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 4.02 for Windows, GraphPad Software, (San Diego, CA, USA, http://www.graphpad.com). Data of treatment groups were statistically analysed by one-way ANOVA including Bartlett's test for homogeneity of variances and Kolmogorov-Smirnov for normality. The methods of estimations included the standard deviation (±SD) of the sampling distribution; P-values <0.05 were considered statistically significant.
Materials
We used highly purified cucurbitacin B [C32H46O8; CKBP002; CK Life Sciences Int’l. (Holdings) Inc., Hong Kong, China] which was dissolved in dimethyl sulphoxide (DMSO) at a stock concentration of 10−2 M, and stored at −20°C. Gemcitabine (2′-deoxy-2′,2′-difluorocytidine, Gemzar; Eli Lilly, IN, USA) was stored at 4°C and dissolved in sterile PBS on the day of use. The chemical structure of the triterpenoid cucurbitacin B is shown in Thoennissen et al., 2009.
Results
Cucurbitacin B augmented the anti-proliferative activity of gemcitabine in Panc-1 tumour xenografts
We determined the potential of in vivo inhibition of pancreatic cancer growth using the combination of cucurbitacin B and gemcitabine compared with single-agent treatments. We implanted Panc-1 pancreatic tumour cells subcutaneously in both flanks of athymic nude mice, and divided the experimental mice into six treatment groups (Table 1). The experiment ended on day +43, determined by to the large tumour size in the diluent-treated control group. The percentage of tumour inhibition was lowest in the mice treated with either low-dose cucurbitacin B (group C, 18%, P= 0.12) or gemcitabine (group B, 32%, P= 0.02) compared with control mice (Table 1). In contrast, the tumour mass was inhibited by up to 62% in the group D mice that received high-dose cucurbitacin B (P= 0.008). However, the most impressive tumour inhibition was achieved in mice treated with the combinations of cucurbitacin B and gemcitabine. Combination group E with low-dose cucurbitacin B and gemcitabine resulted in 79% tumour inhibition (P < 0.001), and combination group F with high-dose cucurbitacin B and gemcitabine had 83% tumour inhibition (P < 0.001). Figure 1A displays the time-dependent inhibition of the pancreatic tumour volumes in all six groups over the treatment period of 43 days and the mean values of the final tumour weights are shown in Figure 1B. Notably, mice treated with the high-dose combination (group F) showed no significant differences in both the final tumour volumes (P= 0.32) as well as tumour weights (P= 0.36) in comparison with the mice treated with the low-dose combination (group E). Photographs of representative tumour pairs excised on day +43 are presented in Figure 2A.
Figure 1.
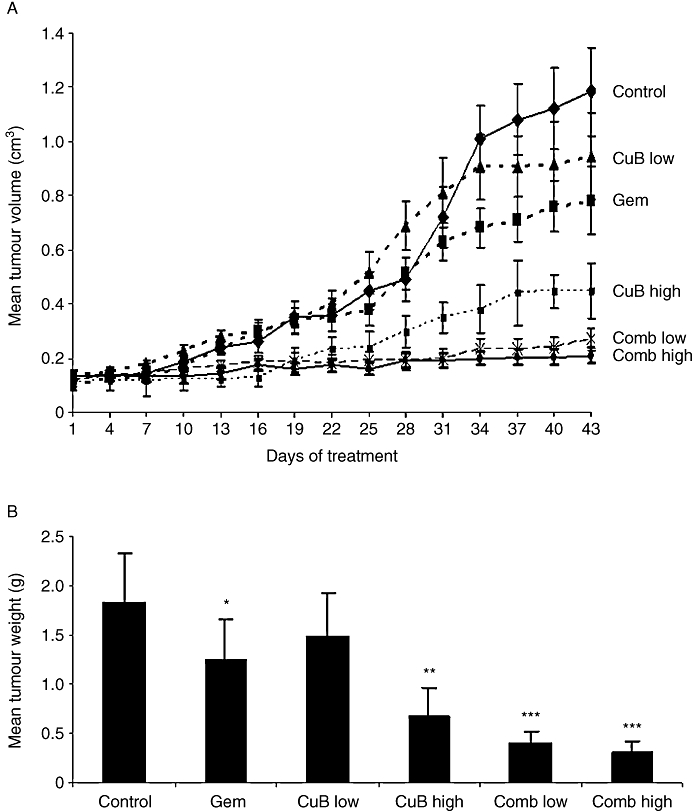
Cucurbitacin B augmented the anti-proliferative activity of gemcitabine in Panc-1 tumour xenografts. Panc-1 cells (1 × 107) were subcutaneously implanted into both flanks of 30 athymic nu/nu mice. Mice were then randomly divided into six groups and treated according to the treatment schedule described in Table 1. After 43 days of therapy, tumours from treated and untreated groups were dissected, weighed and their volumes were measured. (A) Time-dependent inhibition of growth of pancreatic tumours. Results represent the mean ± SD of tumour volumes of 10 tumours for each of six groups measured over 43 days. (B) Overall weight of the dissected tumours. Mean ± SD of overall tumour weights at autopsy (day +43). *P < 0.05; **P < 0.01; ***P < 0.001.
Figure 2.
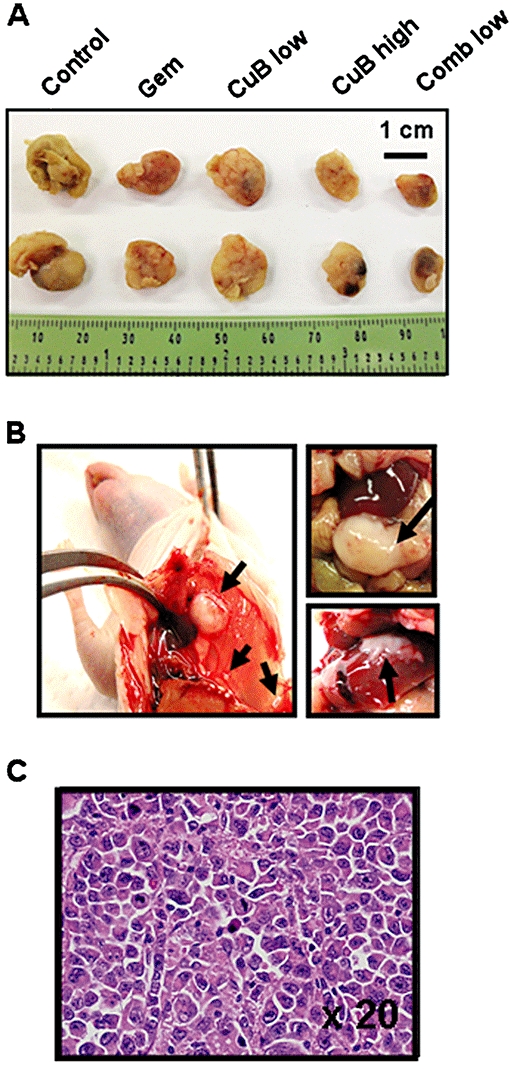
Panc-1-tumours and metastatic spread of Panc-1-tumours in control group and gemcitabine-treated cohort. Necropsy photographs of mice bearing tumour metastases. (A) Photographs of representative tumour pairs. (B) Left panel: macroscopic example of subphrenic and hepatic metastases of a control mouse; upper right panel: macroscopic example of a metastatic bulk tumour located in the abdomen of a control mouse; lower right panel: hepatic metastasis from a mouse treated with gemcitabine alone. Arrows indicate the metastatic spread. (C) Histology of the abdominal metastatic bulk tumour of the control mouse shown in Figure 2B, demonstrating pleiomorphic pancreatic cancer cells with atypical nuclei within the ductal structure (HE stain, ×20).
At autopsy, no metastases were found in the groups treated either with single high-dose cucurbitacin B (group D), low-dose combination (group E) or high-dose combination (group F) respectively. By comparison, four out of five mice of the control cohort had metastatic spread in the axillary lymph nodes, peritoneum, liver and spleen which was confirmed by histopathological examination. Representative examples of macroscopic subphrenic and hepatic metastases, as well as the histology of a metastatic bulk tumour located in the abdomen of a control mouse are presented in Figures 2B and C. Additionally, two of five mice that were treated with monotherapy with either gemcitabine (group B), as well as two of five mice treated with low-dose cucurbitacin B (group C) developed metastases localized in the peritoneum and liver. An example of one hepatic metastasis from a mouse treated with gemcitabine alone is indicated in Figure 2B (lower right panel).
Increased inhibition of c-myc and Bcl-family members by the combination of cucurbitacin B and gemcitabine
To evaluate the in vivo protein expression of pancreatic cancer cells undergoing different treatments, we analysed cell lysates by Western blotting. As both combination therapies gave similar results, only tumours of mice treated with the low-dose combination are represented in Figure 3. Activated STAT3 and JAK2 were clearly inhibited in tumours of mice treated either with high-dose cucurbitacin B or the combination regimes as compared with total STAT3 and JAK2 protein respectively. c-myc is one of the downstream targets of the JAK/STAT pathway. Interestingly, the drug combination decreased the expression of this proto-oncogene more potently than cucurbitacin B alone, whereas gemcitabine alone had no effect on its expression (Figure 3A). Moreover, the drug combination decreased the protein levels of the anti-apoptotic Bcl-XL and Bcl-2 (Figure 3A), and increased the activity of the pro-apoptotic caspase-3 and -9 (Figure 3B) to a greater extent than the monotherapies. Accordingly, pro-caspase nine enzyme levels were down-regulated and poly(ADP-ribose) polymerase (PARP) cleavage products subsequently increased. We also evaluated the protein expression of the phosphorylated forms of Raf kinase, extracellular signal regulated kinase (ERK) and Akt, as well as the nuclear forms of NFkB; no significant difference in the amount of protein among the various groups was noted (data not shown). Therefore, we suggest that the combination of cucurbitacin B and gemcitabine achieved its in vivo anti-tumour activity at least in part by inhibition of the JAK/STAT pathway, and not through blocking activated Ras/Raf or the phosphatidylinositol three kinase (PI3K)/Akt pathways.
Figure 3.
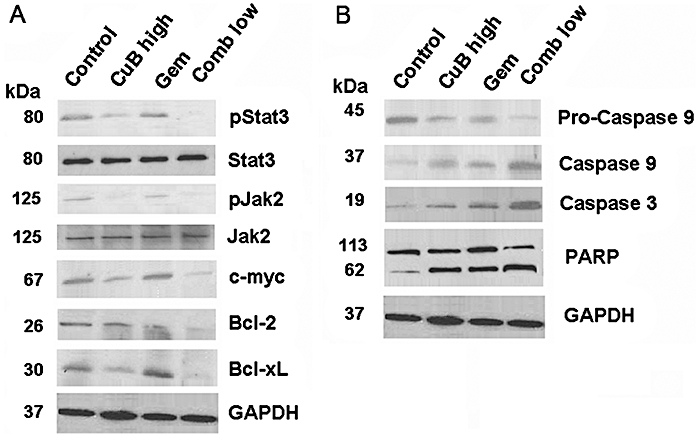
Increased inhibition of c-myc and Bcl-family members by the combination of cucurbitacin B and gemcitabine in vivo. (A and B) Tumour lysates were prepared on day +43 from xenograft mice with Panc-1 tumours. Western blotting was performed to analyse the in vivo effect of cucurbitacin B and gemcitabine on the JAK/STAT pathway and its downstream targets. Each blot indicates combined tumour lysates of five mice (=10 tumours) per treatment group. All Western blots have been performed three times to validate the results.
The ability of c-myc to promote proliferation, transformation and apoptosis requires dimerization with Max (Amati et al., 1993). Hence, we also investigated the level of interaction between c-myc and its activator Max, and between its inhibitor Mad1 and Max. We carried out immunoprecipitation assays with lysates isolated from the tumour xenografts by using anti-c-myc for IP and detection with anti-Max. We showed that c-myc interacted clearly less with Max in the tumour lysates of mice treated either with high-dose cucurbitacin B alone or with the low-dose combination compared with control or gemcitabine alone (Figure 4A). In contrast, by using anti-Mad1 for IP and detection with Max, the interaction of Mad1 with Max was slightly increased in the IP-samples treated with the low-dose combination compared with control, high-dose cucurbitacin B or gemcitabine alone (Figure 4B).
Figure 4.
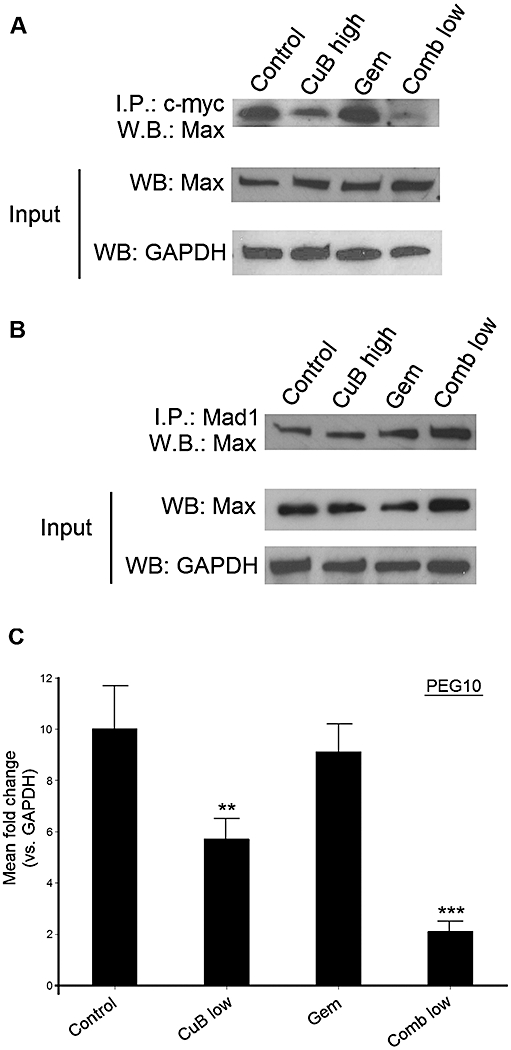
Influence of cucurbitacin B in combination with gemcitabine on c-myc activator Max, inhibitor Mad1 and downstream target PEG10. (A) Immunoprecipitation assay of tumour lysates for c-myc and its activator Max. Top: Immunoprecipitation by anti-c-myc and detection by anti-Max. Middle: Western blotting with anti-Max. Bottom: Western blotting with anti-GAPDH. (B) Immunoprecipitation assay of tumour lysates for Mad1 and Max. Top: Immunoprecipitation by anti-Mad1 and detection by anti-Max. Middle: Western blotting with anti-Max1. Bottom: Western blotting with anti-GAPDH. A/B Each lysate indicates combined tumour lysates from five mice (=10 tumours) per treatment group. (C) Quantitative real-time RT-PCR of pooled cDNA isolated from 10 tumours per treatment group. Experiments were performed in triplicate. Data shown are means ± SD. **P < 0.01; ***P < 0.001. All experiments have been performed three times to validate the results.
Moreover, we isolated RNA out of the snap-frozen xenograft tumours of each treatment group, and analysed the transcriptional level of PEG10, one known downstream target gene of c-myc. Treatment with either high-dose cucurbitacin B or the low-dose combination therapy resulted in a significantly reduced expression of PEG10 (up to fourfold in high-dose cucurbitacin B, and 7.5-fold in low-dose combination therapy) compared with control or treatment with gemcitabine alone (P < 0.01 and P < 0.001 respectively; Figure 4C).
Toxicity studies
The aim of this study was not only to estimate the in vivo anti-pancreatic tumour activity of both cucurbitacin B and gemcitabine, but also to investigate their potential for toxic side effects when combined. All mice were examined and weighed three times a week during treatment. We noted no significant weight loss in mice receiving monotherapy (groups B, C and D) and the low-dose combination (group E) compared with the control mice (Table 1). In contrast, mice receiving the high-dose combination (group F) showed a significant 10% reduction of body weight compared with the control mice at study closure (P= 0.02).
The weights of the liver, spleen and kidneys were also evaluated after the animals were killed on day +43. The values were not significantly different in any of the drug-treated groups compared with the control cohort (Table S2). Histopathological examinations of these organs showed no signs of treatment-related toxicities in any of the mice (liver fibrosis, expansion of red pulp, renal tubular damage or glomerular congestion; data not shown). Additionally, treatment either with single or combination therapies did not cause any significant change in blood parameters. Total blood counts [white blood cells (WBC), haemoglobin (HB) and platelets (PLT)] were in the normal range for all the mice on day +43 compared with day +0 (Table S3). The following serum chemistry parameters were also taken on day +0 and +43 of treatment, and were in the normal range for all mice groups, including glutamate oxaloacetate transaminase (GOT), glutamate pyruvate transaminase (GPT), γ-glutamyl transpeptidase (γGT), total bilirubin, creatinine, glucose levels (unstarved) and cholesterol (Table S3), as well as alkaline phosphatase, phosphate, chloride, calcium, potassium, total protein, albumin, uric acid, creatine kinase and triglycerides (data not shown).
The colony-forming unit assay (CFU-C Assay) is a very sensitive method to analyse impaired function of hematopoietic stem and progenitor cells (Young et al., 1987). In order to examine underlying bone marrow toxicities, we plated bone marrow of three mice per treatment group for 10 days in triplicate in semi-solid methylcellulose. Colony numbers of all treated animals, with either mono- or combination therapies, showed significantly decreased colony counts compared with control mice (Figure 5). But interestingly, colony numbers for BFU-E, CFU-GM and CFU-GEMM from mice treated either with gemcitabine alone (group B) or cucurbitacin B alone (group C and D) were similar to those from mice treated with the low-dose combination (group E, all P≥ 0.2). On the other hand, counts for BFU-E and CFU-GM were significantly decreased in mice treated with the high-dose combination (group F) compared with mice treated with the single drug and low-dose combination (BFU-E: P= 0.025; CFU-GM: P < 0.001; Figure 5). Counts for CFU-GEMM colonies were comparable in all treatment groups. The colony counts for all groups are provided in Table S4.
Figure 5.
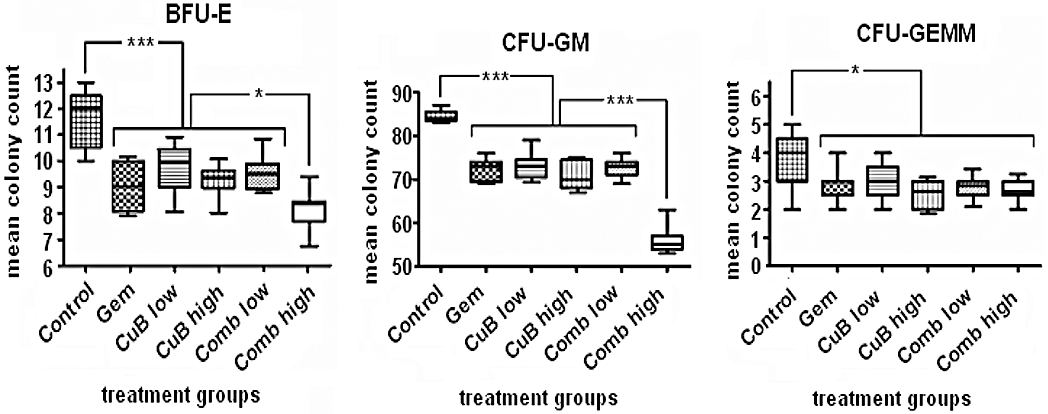
Analysis of bone marrow toxicity after drug treatment measured by clonogenic assays. Bone marrow cells isolated from all treatment groups (day +43) were plated in triplicates in a 6-well plate at density 2 × 104 per well in semi-solid 1% methylcellulose medium. Box and whisker plots display colony counts and their median 10 days after seeding. Bars, range of the data: *P < 0.05; **P < 0.01; ***P < 0.001. BFU-E, Blast-forming unit-erythroid; CFU-GEMM, colony-forming unit granulocyte/erythrocyte/monocyte/megakaryocyte; CFU-GM, colony-forming unit-granulocyte/macrophages.
Discussion and conclusions
Pancreatic cancer is one of the most challenging of solid organ malignancies due to aggressive and chemoresistant tumour biology, and a propensity for late presentation with inoperable metastatic disease (Strimpakos et al., 2008). The nucleoside analogue gemcitabine, a fluorinated pyrimidine related to cytosine arabinoside, is commonly used in pancreatic cancer therapy. Nevertheless, treatment with gemcitabine, the most potent chemotherapeutic agent against this cancer to date, is not curative, and resistance almost always occurs (Burris et al., 1997; Strimpakos et al., 2008; Vulfovich and Rocha-Lima, 2008). Although salvage chemotherapy with 5-fluorouracil (5-FU)/oxaliplatin-based regimens has brought benefit to patients with refractory advanced stage of the disease improvements in first-line therapy for pancreatic cancer have proven difficult (Oettle et al., 2005; Mitry et al., 2006). The vast majority of studies which explored the use of combined treatments consisting of chemotherapeutic agents together with biological substances such as the humanized antibody bevacizumab or the multi-targeted kinase inhibitor sorafenib, failed to demonstrate advantages over gemcitabine, mainly due to toxicities (Kindler et al., 2007; Wallace et al., 2007; Strimpakos et al., 2008). A trial in advanced pancreatic cancer conducted by the National Cancer Institute of Canada showed a statistical survival advantage with the addition of the oral epidermal growth factor receptor-inhibitor erlotinib to gemcitabine (Moore et al., 2007). Sadly, however, the median survival difference between this combination and gemcitabine alone was only 2 weeks. Nevertheless, the US Food and Drug Administration approved this combination for first-line treatment of advanced pancreatic cancer. Even if these results represent only a small step forward, the approval provides an important proof of concept regarding the use of newer ‘targeted’ therapies in managing this deadly disease.
Cucurbitacins possess a broad range of potent biological activity and are known for their anti-cancer properties (Musza et al., 1994). The antiproliferative activity of cucurbitacin B against human breast cancer, glioblastoma multiforme, and myeloid leukaemia cells has already been shown by our group (Haritunians et al., 2008; Wakimoto et al., 2008; Yin et al., 2008). Recently, we demonstrated that this compound had anti-tumour activity against human pancreatic cancer (Thoennissen et al., 2009). Cucurbitacin B inhibited the activation of JAK2, STAT3, as well as STAT5 in pancreatic cancer cells, and decreased in vivo pancreatic tumour growth by 69% at a dose of 1 mg·kg−1 body weight. Besides, some preliminary in vitro experiments revealed a synergistic anti-proliferative effect of cucurbitacin B and gemcitabine on the pancreatic cancer cell line MiaPaca-2 and Panc-1 in vitro (Thoennissen et al., 2009).
The main goal of our present study was to confirm this finding in an in vivo model for human pancreatic cancer treated with the combination of both cucurbitacin B and gemcitabine. To investigate the anti-tumour potency of cucurbitacin B in combination, we used two different combination regimens consisting of cucurbitacin B in either low or high concentration. The higher dose of 1 mg·kg−1 cucurbitacin B is known to be well tolerated by mice as monotherapy as shown in our previous studies (Wakimoto et al., 2008; Thoennissen et al., 2009). The dose of gemcitabine (25 mg·kg−1 body weight) is also known to be safe for mice (Wakimoto et al., 2008), and is equivalent to 1000 mg·m−2 used in humans diagnosed with pancreatic cancer. Both combination therapies potently inhibited by up to 83% the tumour-growth in our xenograft model, whereas the monotherapies with either gemcitabine or low-dose cucurbitacin B had less anti-tumour activity. In addition, metastatic spread was inhibited by the drug combinations. Alone, cucurbitacin B (1 mg·kg−1) still had remarkable activity resulting in a 62% primary tumour reduction and no detectable metastasis, which is comparable to our recent findings (Thoennissen et al., 2009). Evaluation of the effects of this compound on the apoptotic-related pathways associated with pancreatic cancer showed a potent inhibition of activated JAK2 and STAT3 by the combination treatment, as well as single agent cucurbitacin B. Additionally, c-myc, the downstream target of STAT3 was more prominently inhibited by the combination therapies compared with cucurbitacin B alone. Amplification and over-expression of c-myc is a common event in various neoplasias including pancreatic cancer. Dysregulation of this gene is suggested to be involved in both development and progression of pancreatic adenocarcinoma (Schleger et al., 2002; Levens, 2003; Patel et al., 2004).
The activation of c-myc is also dependent on its activator Max (Amati et al., 1993) as well as its inhibitors Mad1 and Mxi1. Moreover, binding of c-myc to Max has been shown to be dependent on the expression of Mad1, because both c-myc and Mad1 compete for Max (Ayer et al., 1993). For this reason, Mad1/Max complexes function as repressors of c-myc activity as shown by the fact that Mad1/Max inhibits c-myc transactivation of reporter plasmid constructs containing the c-myc consensus element (Blackwood et al., 1992; Zervos et al., 1993). Additionally, Mad1 suppressed c-myc transforming activity in rat embryo fibrobroblasts (Lahoz et al., 1994). In our study, we demonstrated that the treatment of Panc1 xenograft tumours with either high-dose cucurbitacin B or a low-dose combination with gemcitabine effectively reduced the interaction of c-myc with its activator Max compared with control or treatment with gemcitabine alone. Furthermore, the combination regime slightly increased the interaction of Mad1 with Max, which form the c-myc inhibitor complex, as compared with control, high-dose cucurbitacin B or gemcitabine alone. In conclusion, treatment of the pancreatic xenograft tumours with cucurbitacin B in combination with gemcitabine not only inhibited c-myc but also impaired its activity. We could emphasize this effect by showing the highly reduced expression levels of PEG10, a direct target of c-myc (Li et al., 2006), after combined treatment. Interestingly, the down-regulation of PEG10 has already been associated with decreased proliferation in hepatoma and pancreatic cancer cells (Li et al., 2006). Gemcitabine alone had no influence on either activated JAK2/STAT3 or c-myc and its activator Max or repressor Mad1.
Additionally, Western blot analysis of the tumour samples from mice treated with the drug combination revealed stronger inhibition of Bcl-XL and Bcl-2 and a greater expression of the key ‘executioner’ caspase 3, caspase 9 and PARP cleavage activity compared with mice treated with single agents alone. The RAS/RAF, PI3K/AKT and NFkB signalling pathways appeared unchanged after exposure to either single or combination therapies suggesting that modulation of these pathways was not required for the inhibitory function of the drug combination in pancreatic cancer cells.
Anaemia and neutropenia are one of the most common effects observed in vivo during treatment with cytotoxic regimes. Erythropoiesis as well as granulopoiesis are responsible for red and white blood cell production by cell proliferation and differentiation of specific progenitors like BFU-Es, CFU-GMs and CFU-GEMMs. Clonogenic assays are an essential tool for predicting the degree of a putative in vivo haematopoietic toxicity in drug treated subjects (Young et al., 1987; Pessina et al., 2005). The mice cohort receiving the high-dose cucurbitacin B (1 mg·kg−1) in combination with gemcitabine (25 mg·kg−1) had not only significantly lower body weights, but also significantly reduced colony numbers of murine bone marrow (BFU-E and CFU-GM) compared with the other treatment groups, which we consider as an early sign of synergistic haematopoietic toxicity. However, peripheral blood indices were still in normal range at the end of therapy (day +43). In contrast, the drug combination including the lower-dose of cucurbitacin B and gemcitabine (25 mg·kg−1) was associated with similar levels of haematopoietic clonogenic cell counts and body weights as the mice receiving monotherapies.
In summary, we showed for the first time a remarkable in vivo anti-tumour effect of the JAK/STAT inhibitor cucurbitacin B in combination with gemcitabine against human pancreatic cancer. Low-dose of cucurbitacin B in combination with gemcitabine produced only mild toxicity. Therefore, this combination therapy with strong anti-pancreatic cancer activity and low toxicity provides a potent rationale for its use in clinical trials against human pancreatic cancer.
Acknowledgments
H.P.K. is the holder of the Mark Goodson endowed Chair in Oncology Research and is a member of the Jonsson Cancer and Molecular Biology Institute, UCLA, Los Angeles, USA. We also thank the Jung-Stiftung für Wissenschaft und Forschung (Hamburg, Germany) (G.B.I.), Deutsche Forschungsgemeinschaft (Bonn, Germany) (TH 1438/1-1 to N.H.T.), and CK Life Sciences, Inc. (Hong Kong, China) (M.T.) for financial support.
Glossary
Abbreviations:
- BFU-E
blast-forming unit-erythroid
- CFU-C Assay
colony-forming unit assay
- CFU-GEMM
colony-forming unit granulocyte/erythrocyte/monocyte/megakaryocyte
- CFU-GM
colony-forming unit-granulocyte/macrophages
Conflicts of interest
M.T. is an employee of CK Life Sciences, Inc.
Supporting information
Additional Supporting Information may be found in the online version of this article.
Table S1 Antibody list.
Table S2 Overview inner organ weights.
Table S3 Overview blood counts and serum chemistry.
Table S4 Overview CFU-counts.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Amati B, Brooks MW, Levy N, Littlewood TD, Evan GI, Land H. Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell. 1993;72:233–245. doi: 10.1016/0092-8674(93)90663-b. [DOI] [PubMed] [Google Scholar]
- Ayer DE, Kretzner L, Eisenman RN. Mad: a heterodimeric partner for Max that antagonizes Myc transcriptional activity. Cell. 1993;72:211–222. doi: 10.1016/0092-8674(93)90661-9. [DOI] [PubMed] [Google Scholar]
- Blackwood EM, Lüscher B, Eisenman RN. Myc and Max associate in vivo. Genes Dev. 1992;1:71–80. doi: 10.1101/gad.6.1.71. [DOI] [PubMed] [Google Scholar]
- Burris HA, 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- Chen JC, Chiu MH, Nie RL, Cordell GA, Qiu SX. Cucurbitacins and cucurbitane glycosides: structures and biological activities. Nat Prod Rep. 2005;22:386–399. doi: 10.1039/b418841c. [DOI] [PubMed] [Google Scholar]
- Haritunians T, Gueller S, Zhang L, Badr R, Yin D, Xing H, et al. Cucurbitacin B induces differentiation, cell cycle arrest, and actin cytoskeletal alterations in myeloid leukemia cells. Leuk Res. 2008;32:1366–1373. doi: 10.1016/j.leukres.2008.01.019. [DOI] [PubMed] [Google Scholar]
- Jayaprakasam B, Seeram NP, Nair MG. Anticancer anti-inflammatory activities of cucurbitacins from Cucurbita andreana. Cancer Lett. 2003;189:11–16. doi: 10.1016/s0304-3835(02)00497-4. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- Kindler HL, Niedzwiecki D, Hollis D, Oraefo E, Schrag D, Hurwitz H, et al. A double-blind, placebo-controlled, randomized phase III trial of gemcitabine (G) plus bevacizumab (B) versus gemcitabine plus placebo (P) in patients (pts) with advanced pancreatic cancer (PC): a preliminary analysis of Cancer and Leukemia Group B (CALGB) J Clin Oncol ASCO 2007 Annual Meeting. 2007;25:4508. [Google Scholar]
- Lahoz EG, Xu L, Schreiber-Agus N, DePinho RA. Suppression of Myc, but not E1a, transformation activity by Max-associated proteins, Mad and Mxi1. Proc Natl Acad Sci USA. 1994;91:5503–5507. doi: 10.1073/pnas.91.12.5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer R. Drug delivery and targeting. Nature. 1998;392:5–10. [PubMed] [Google Scholar]
- Levens DL. Reconstructing MYC. Genes Dev. 2003;17:1071–1077. doi: 10.1101/gad.1095203. [DOI] [PubMed] [Google Scholar]
- Li CM, Margolin AA, Salas M, Memeo L, Mansukhani M, Hishoosh H, et al. PEG10 is a c-MYC target gene in cancer cells. Cancer Res. 2006;2:665–672. doi: 10.1158/0008-5472.CAN-05-1553. [DOI] [PubMed] [Google Scholar]
- Luong QT, O'Kelly J, Braunstein GD, Hershman JM, Koeffler HP. Antitumor activity of suberoylanilide hydroxamic acid against thyroid cancer cell lines in vitro and in vivo. Clin Cancer Res. 2006;12:5570–5577. doi: 10.1158/1078-0432.CCR-06-0367. [DOI] [PubMed] [Google Scholar]
- Mitry E, Ducreux M, Ould-Kaci M, Boige V, Seitz JF, Bugat R, et al. Oxaliplatin combined with 5-FU in second line treatment of advanced pancreatic adenocarcinoma. Results of a phase II trial. Gastroenterol Clin Biol. 2006;30:357–363. doi: 10.1016/s0399-8320(06)73188-8. [DOI] [PubMed] [Google Scholar]
- Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- Musza LL, Speight P, McElhiney S, Barrow CJ, Gillum AM, Cooper R, et al. Cucurbitacins, cell adhesion inhibitors from Conobea scoparioides. Nat Prod. 1994;57:1498–1502. doi: 10.1021/np50113a004. [DOI] [PubMed] [Google Scholar]
- Oettle H, Pelzer U, Stieler J, Hilbig A, Roll L, Schwaner I, et al. Oxaliplatin/folinic acid/5-fluorouracil [24 h] (OFF) plus best supportive careversus best supportive care alone (BSC) in second-line therapy ofgemcitabine-refractory advanced pancreatic cancer (CONKO 003) J Clin Oncol. 2005;23:4031. ASCO Annual Meeting 2005 Proc (Post-Meeting Edition) [Google Scholar]
- Patel JH, Loboda AP, Showe MK, Showe LC, McMahon SB. Analysis of genomic targets reveals complex functions of MYC. Nat Rev Cancer. 2004;4:562–568. doi: 10.1038/nrc1393. [DOI] [PubMed] [Google Scholar]
- Pessina A, Malerba I, Gribaldo L. Hematotoxicity testing by cell clonogenic assay in drug development and preclinical trials. Curr Pharm Des. 2005;11:1055–1065. doi: 10.2174/1381612053381648. [DOI] [PubMed] [Google Scholar]
- Sahu RP, Srivastava SK. The Role of STAT-3 in the Induction of Apoptosis in Pancreatic Cancer Cells by Benzyl Isothiocyanate. J Natl Cancer Inst. 2009;101:176–193. doi: 10.1093/jnci/djn470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleger C, Verbeke C, Hildenbrand R, Zentgraf H Bleyl U. c-MYC activation in primary and metastatic ductal adenocarcinoma of the pancreas: incidence, mechanisms, and clinical significance. Mod Pathol. 2002;15:462–469. doi: 10.1038/modpathol.3880547. [DOI] [PubMed] [Google Scholar]
- Solit DB, Zheng FF, Drobnjak M, Münster PN, Higgins B, Verbel D, et al. 17-Allylamino-17-demethoxygeldanamycin induces the degradation of androgen receptor and HER-2/neu and inhibits the growth of prostate cancer xenografts. Clin Cancer Res. 2002;8:986–993. [PubMed] [Google Scholar]
- Strimpakos A, Saif MW, Syrigos KN. Pancreatic cancer: from molecular pathogenesis to targeted therapy. Cancer Metastasis Rev. 2008;27:495–522. doi: 10.1007/s10555-008-9134-y. [DOI] [PubMed] [Google Scholar]
- Sun FX, Tohgo A, Bouvet M, Yagi S, Nassirpour R, Moossa AR, et al. Efficacy of camptothecin analog DX-8951f (exatecan mesylate) on human pancreatic cancer in an orthotopic metastatic model. Cancer Res. 2003;63:80–85. [PubMed] [Google Scholar]
- Thoennissen NH, Iwanski GB, En-Gal S, Okamoto R, Lin P, Abbassi S, et al. Cucurbitacin B induces apoptosis by inhibition of the JAK/STAT pathway and potentiates antiproliferative effects of gemcitabine on pancreatic cancer cells. Cancer Res. 2009;69:5876–5884. doi: 10.1158/0008-5472.CAN-09-0536. [DOI] [PubMed] [Google Scholar]
- Toyonaga T, Nakano K, Nagano M, Zhao G, Yamaguchi K, Kuroki S, et al. Blockade of constitutively activated Janus kinase/signal transducer and activator of transcription-3 pathway inhibits growth of human pancreatic cancer. Cancer Lett. 2003;201:107–116. doi: 10.1016/s0304-3835(03)00482-8. [DOI] [PubMed] [Google Scholar]
- Vulfovich M, Rocha-Lima C. Novel advances in pancreatic cancer treatment. Expert Rev Anticancer Ther. 2008;8:993–1002. doi: 10.1586/14737140.8.6.993. [DOI] [PubMed] [Google Scholar]
- Wakimoto N, Yin D, O'Kelly J, Haritunians T, Karlan B, Said J, et al. Cucurbitacin B has a potent antiproliferative effect on breast cancer cells in vitro and in vivo. Cancer Sci. 2008;99:1793–1797. doi: 10.1111/j.1349-7006.2008.00899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JA, Locker G, Nattam S, Kasza K, Wade-Oliver K, Vokes EE, et al. Sorafenib (S) plus gemcitabine (G) for advanced pancreatic cancer (PC): a phase II trial of the University of Chicago Phase II Consortium. J Clin Oncol. 2007;25:4608. 2007 ASCO Annual Meeting Proc Part I. [Google Scholar]
- Yin D, Wakimoto N, Xing H, Lu D, Huynh T, Wang X, et al. Cucurbitacin B markedly inhibits growth and rapidly affects the cytoskeleton in glioblastoma multiforme. Int J Cancer. 2008;123:1364–1375. doi: 10.1002/ijc.23648. [DOI] [PubMed] [Google Scholar]
- Young GA, Croaker G, Vincent PC, Forrest P, Morris TC. The CFU-C assay in patients with neutropenia and, in particular, drug associated neutropenia. Clin Lab Haematol. 1987;9:245–253. doi: 10.1111/j.1365-2257.1987.tb00088.x. [DOI] [PubMed] [Google Scholar]
- Zervos AS, Gyuris J, Brent R. Mxi1, a protein that specifically interacts with Max to bind Myc–Max recognition sites. Cell. 1993;72:223–232. doi: 10.1016/0092-8674(93)90662-a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.