

Abstract
Background and purpose:
A functional link between seizure-induced P-glycoprotein overexpression at the blood–brain barrier and therapeutic failure has been suggested by several studies using rodent epilepsy models and human epileptic tissue. Recently, we reported that interference with the mechanisms that up-regulate P-glycoprotein in response to seizure activity might provide a novel approach to control its expression in the epileptic brain. Based on these data, we hypothesized that blocking the appropriate signalling cascade by cyclooxygenase-2 inhibition should improve brain penetration of antiepileptic drugs and help to overcome drug resistance.
Experimental approach:
Effects of the selective cyclooxygenase-2 inhibitor celecoxib on the response to the P-glycoprotein substrate, phenobarbital, was evaluated in a chronic model of drug-resistant temporal lobe epilepsy in rats. Drug-resistant rats selected from this model exhibit a marked overexpression of P-glycoprotein in the hippocampus and other limbic brain regions.
Key results:
Responders and non-responders were selected from a group of rats with spontaneous recurrent seizures after prolonged treatment with phenobarbital at maximum tolerated doses. The efficacy of phenobarbital was re-evaluated following a 6 day treatment with celecoxib and the frequency of spontaneous recurrent seizures was significantly reduced in both groups of rats, phenobarbital responders or non-responders selected from the previous drug trial.
Conclusions and implications:
Pretreatment with the cyclooxygenase-2 inhibitor restored the anticonvulsant activity of phenobarbital in rats that failed to exhibit a relevant response before celecoxib treatment. Our data provide further support for a novel therapeutic approach to overcome transporter-mediated drug resistance in epilepsies.
Keywords: P-glycoprotein, pharmacoresistance, drug-refractoriness, epilepsy, seizure, blood–brain barrier, cyclooxygenase-2, phenobarbital, multidrug transporter
Introduction
Numerous studies using rodent epilepsy models and epileptic tissue from pharmacoresistant patients have revealed a strong correlation between seizure-induced overexpression of P-glycoprotein and resistance to antiepileptic drugs (Loscher and Potschka, 2005a). At the blood–brain barrier (BBB), P-glycoprotein mediates the efflux of different antiepileptic drugs into the capillary lumen thereby limiting their brain penetration and access to the target sites. Experimental proof-of-principle for the transporter hypothesis of pharmacoresistant epilepsy has been provided by studies in animal models demonstrating that it is possible to overcome drug resistance by modulating P-glycoprotein transport function (Clinckers et al., 2005; Brandt et al., 2006; van Vliet et al., 2006). A major drawback of this approach is that any strategy aiming to restore antiepileptic drug efficacy by inhibition of P-glycoprotein-mediated transport will also limit the protective function of this efflux transporter that is of specific physiological relevance in several blood–tissue barriers, haematopoetic cells and excretory organs.
Interference with the mechanism that up-regulates P-glycoprotein in response to seizure activity has recently been substantiated as a novel approach that would limit P-glycoprotein expression rates to basal, constitutive levels, thereby conserving its protective transport function (Zibell et al., 2009). Cyclooxygenase-2 (COX-2) proved to be one of the central factors that drives the endothelial transcriptional activation of the P-glycoprotein encoding gene in the epileptic brain in response to excess glutamate concentrations (Bauer et al., 2008). In vivo experiments demonstrated that, both, the non-selective COX inhibitor indomethacin as well as the COX-2 inhibitor celecoxib counteracted the status epilepticus-induced increase in capillary P-glycoprotein expression (Bauer et al., 2008; Zibell et al., 2009). Moreover, selective COX-2 inhibition controlled P-glycoprotein expression in rats with recurrent seizure activity and promoted delivery of the P-glycoprotein substrate phenytoin in these rats (van Vliet et al., 2010). Taken together, these data suggest that COX-2 inhibition would improve antiepileptic drug efficacy in individuals with P-glycoprotein-mediated pharmacoresistance. To test this hypothesis, we determined the effect of selective COX-2 inhibition on the response to the P-glycoprotein substrate phenobarbital in a chronic model of pharmacoresistant temporal lobe epilepsy in rats. In this model, pharmacosensitivity to phenobarbital is known to correlate with P-glycoprotein expression rates (Volk and Loscher, 2005). Rats with a poor or no response are known to exhibit enhanced BBB expression of P-glycoprotein, in comparison with drug-responsive animals.
Methods
Animals
All animal care and experimental protocols were approved by the Committees of the Government of Upper Bavaria and were in compliance with the European Communities Council Directive, 86/609/EEC, and the German Animal Welfare Act. Fifty-six female Sprague-Dawley rats (purchased with a weight range of 200–224 g, Harlan Netherlands, Horst, The Netherlands) were used for the experiments. Animals were kept under controlled environmental conditions (24–25°C, 50–60% humidity, 12 h dark/light cycle) with free access to tap water and standard feed. Before experiments, animals were allowed to adapt to the new environment for at least 1 week.
Induction of status epilepticus
A Teflon-isolated bipolar stainless steel electrode was stereotactically implanted into the right anterior basolateral nucleus of the amygdala (BLA) under anaesthesia as described previously (Pekcec et al., 2008) (stereotaxic coordinates in millimeter relative to Bregma: AP +2.2 L +4.8 DV +8.6). Six weeks after surgery, 48 rats were electrically stimulated for 25 min via the BLA electrode for induction of a self-sustained status epilepticus (Figure 1). The stimulation parameters used have been previously described (Pekcec et al., 2008) and were, in brief: stimuli consisting of 100 ms trains of 1 ms alternating positive and negative square-wave pulses (given at a frequency of 2 s−1, intra-train pulse frequency 50 s−1, peak pulse intensity 700 µA). In all rats, the electroencephalogram (EEG) was recorded via the BLA electrode before and following status epilepticus. The majority of rats (n = 30) exhibited a generalized status epilepticus. Eighteen rats developed a focal status epilepticus with repeated single generalized seizures. Self-sustained status epilepticus was limited to a duration of 4 h (including the 25 min of BLA stimulation) by diazepam (Diazepam-ratiopharm, from Ratiopharm, Ulm, Germany). Diazepam was injected i.p. at a dose of 10 mg·kg−1 body weight. If necessary, the application of this dose was repeated until seizure activity was completely suppressed and the EEG was normalized. Starting 8 weeks later, the 48 rats were monitored by EEG/video recordings in a pre-screening period for about 6 weeks until the first spontaneous seizures were detected as described recently (Brandt et al., 2006). Twelve rats that developed spontaneous recurrent seizures during the pre-screening period were randomly chosen used for selection of responders and non-responders by prolonged treatment with phenobarbital. The remaining 36 rats that did not display seizure activity during the pre-screening period were excluded from further analysis. Electrode-implanted control rats (n = 8) were not stimulated, but were subjected to all handling and experimental procedures associated with the stimulation.
Figure 1.
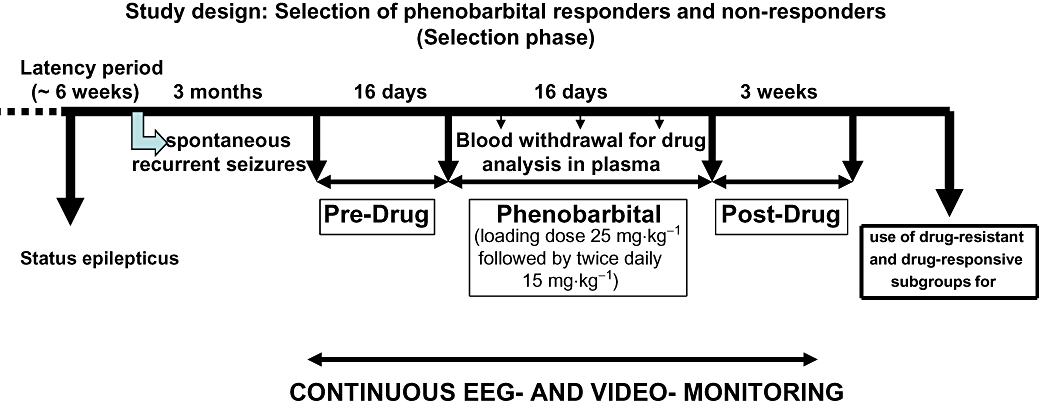
Schematic illustration of the selection of drug-refractory (non-responders) and drug-responsive rats (responders) from a post-status epilepticus model of temporal lobe epilepsy by prolonged administration of phenobarbital.
Detection of spontaneous seizures by continuous video/EEG recording
Rats were continuously monitored (24 h·day−1 for 7 days a week) using a combined full digital video- and EEG-detection system analogues to that described before (Pekcec et al., 2008). To allow optimal video observation, animals were housed in modified clear glass cages (40 × 40 × 40 cm) under controlled conditions (see above). For continuous day and night video-monitoring several infrared light sensitive cameras and a multiple-channel PCI analogue-digital converter (ABUS Security-Tech, Affing, Germany) were used. The sampling rate was 100 pictures·s−1. Data analysis was performed using the Digi-Protect Searcher 6.275 beta software (ABUS Security-Tech, Affing, Germany). EEGs were simultaneously recorded using 16 1-channel bio amplifiers (BioAmps) and two analogue-digital converters (PowerLab/800s) (all from ADInstruments, Hastings, East Sussex, UK). Data acquisition and analysis was performed using the Chart5 for Windows software (ADInstruments, Hastings, East Sussex, UK).
For detection of spontaneous seizures, the continuous EEG recordings were visually analysed for characteristic seizure-like activity. For evaluation of the seizure severity of an EEG-detected seizure the corresponding video recording was viewed. For rating of seizure severity of spontaneous seizures, Racine's scale was used: stage 1 was characterized by immobility and facial automatisms, stage 2 by head nodding, associated with more severe facial and mouth clonus (mastication), stage 3 by unilateral forelimb clonus, stage 4 by rearing and bilateral forelimb clonus, and stage 5 by rearing and falling accompanied by generalized tonic–clonic convulsions (Racine, 1972).
During the entire study, all spontaneous seizures that occurred during handling of the animals were additionally noted and added to the number of seizures evaluated by EEG/video monitoring. Only generalized seizures were evaluated and analysed in this study as the reliable video analysis of focal seizures would depend on the position of the animal in relation to the camera.
Experimental design: selection of phenobarbital non-responders
Based on preliminary experiments and a previous phenobarbital selection trial in epileptic rats (Volk and Loscher, 2005; Brandt et al., 2006) the following dosing protocol was used (Figure 1). For the present study 12 rats with spontaneous recurrent seizures received an i.p. bolus dose of 25 mg·kg−1 in the morning of the first treatment day, followed by an administration of 15 mg·kg−1 i.p. 10 h later, and then twice daily 15 mg·kg−1 i.p. for 16 subsequent days. For drug administration, the sodium salt of phenobarbital (Sigma, Steinheim, Germany) was dissolved in 0.9% saline and injected at a volume of 3 mL·kg−1. Before onset of phenobarbital treatment, baseline seizure frequency was determined over 16 days (pre-drug control period, which started about 3 months after induction of status epilepticus). Phenobarbital treatment was followed by a post-drug control period of 3 weeks (Figure 1). In this way, each animal served as its own control, accounting for differences between animals, for example regarding variability in baseline seizure frequency. During the pre-drug control period, 0.9% saline (3 mL·kg−1) was injected instead of phenobarbital. During drug treatment, rats were observed for obvious adverse effects. However, these adverse effects were not evaluated in detail based on scoring.
Experimental design: impact of celecoxib pretreatment on the efficacy of phenobarbital
The period between the end of selection of responders (n = 5) and non-responders (n = 7) and the commencement of pretreatment of celecoxib was about 3 months (Figure 2). In a previous study using the same model, multidrug resistance proved to be stable over a period of several months (Bethmann et al., 2007). The interim period was needed for analysis of EEG/video recordings from the selection phase and analysis of positron emission tomographic scannings (H. Bartmann et al., unpubl. data). During this period one rat (non-responder) developed a status epilepticus and died. This non-responder exhibited a moderate increase in seizure frequency during the phenobarbital treatment. Rats not responding to phenobarbital (n = 6) by a relevant reduction of seizure frequency (non-responders) as well as a group of rats responding to phenobarbital (n = 5) were used for pretreatment with celecoxib. The decision was taken to use a strict definition of pharmacoresistance, so that the group of non-responders comprised four rats that did not respond with a ≥50% reduction in seizure frequency. In addition, two rats were considered non-responders that repeatedly exhibited pronounced seizure clusters, which were not affected by phenobarbital treatment, and thus could not be considered to show a satisfactory treatment success. These two rats showed an overall reduction in seizure frequency of 57% and 61% respectively. Thus, the resulting cut-off was ≥65% reduction in seizure frequency to categorize an animal a responder.
Figure 2.
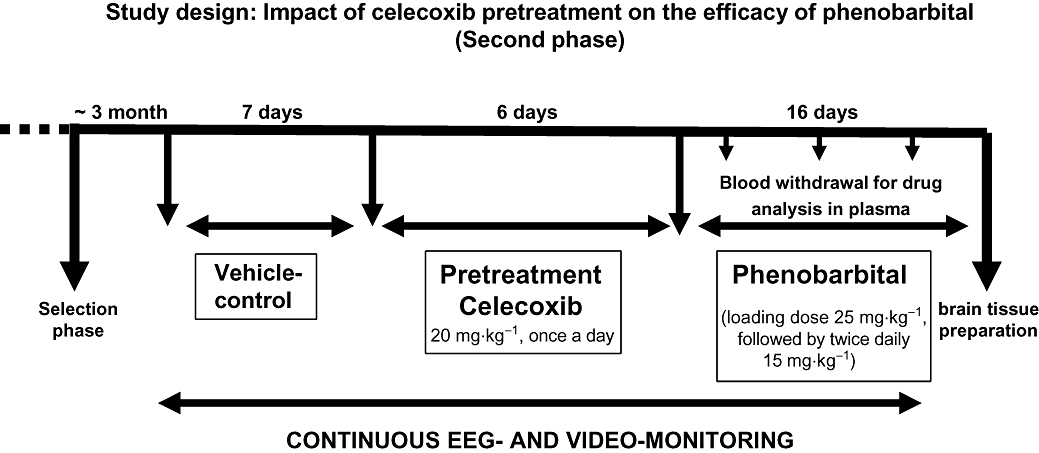
Schematic illustration of the second phenobarbital treatment phase following celecoxib pretreatment in rats previously designated drug-refractory (non-responders) and drug-responsive rats (responders).
Before celecoxib treatment (Celebrex capsule, from Pfizer, Karlsruhe, Germany) a second vehicle period (7 days) was implemented to determine the current seizure frequency (Figure 2). In this period one non-responder died. The content of the celecoxib capsule was dissolved in phosphate buffered saline and administered by gavage (20 mg·kg−1 p.o. once daily) for 6 days. As intestinal side effects may occur due to subchronic COX-2 inhibition, all rats received pantoprazole (Pantozol, from Nycomed, Konstanz, Germany) 20 mg·kg−1 s.c. three times daily, as described elsewhere (Bonin et al., 2005). Despite pantoprazole treatment one responder and one non-responder died or had to be killed as a consequence of undesirable gastrointestinal side effects of celecoxib. These animals were not distinguished by a high seizure frequency.
Subsequent to pretreatment of celecoxib, phenobarbital was injected with the same dosing protocol that was used during the selection of responders or non-responder (Figure 2). In addition to the rats designated responders or non-responders, a control group (n = 8) of non-epileptic rats received phenobarbital administration over a period of 8 days. The sequence of experiments is shown in Figures 1 and 2.
Tissue preparation
Following the phenobarbital administration trial, the 16 rats (responder: n = 4, non-responder: n = 4, controls: n = 8) were decapitated. Brains were immediately removed, and separated into left and right hemispheres. The left hemisphere was dissected into various brain regions and frozen at −80°C for determination of phenobarbital brain concentrations. Homogenates of the parahippocampal cortex were used for analysis of phenobarbital concentrations. The right hemisphere was embedded in Tissue Freezing Medium® (Jung, Nussloch, Germany), frozen in liquid nitrogen and stored at –80°C. Brain tissue was cut in 14 µm slices using a cryostat (HM 560; Microm, Walldorf, Germany) and sections were mounted on HistoBond® adhesion slides (Marienfeld, Lauda-Koenigshofen, Germany) for immunohistochemistry.
Analysis of phenobarbital plasma concentration
Blood was sampled by retro-orbital puncture [after local anaesthesia with tetracaine hydrochloride (Opthocain N, Dr Winzer Pharma GmbH, Berlin, Germany)] 10 h after the first drug injection, 10 h after drug injection on days 10 and 16, for phenobarbital plasma analysis. The concentrations of phenobarbital were measured by high-performance liquid chromatography (HPLC) with ultraviolet detection. Briefly, 50 µL internal standard (50 µg·pentobarbitone per millilitre of H2O) and 300 µL ethanol 99% was added to a 50 µL plasma sample. Following centrifugation at 20 817×g for 15 min at 4°C, the supernatant was used for analysis. For brain tissue analysis 50 µL internal standard (20 µg·pentobarbitone per millilitre of H2O) was added to a 30 mg parahippocampal cortex sample. Following centrifugation (4°C, 20 817×g; 15 min) the mixture was homogenized with 300 µL 99% ethanol. The samples were injected in 20 µL at a time.
P-glycoprotein immunohistochemistry and image analysis
Brain sections of all rats (responder: n = 4, non-responder: n = 4, controls: n = 8) were processed simultaneously to obtain comparable staining intensities. Analysis of P-glycoprotein expression was performed using a monoclonal mouse antibody (C219; 1:100; Calbiochem, Darmstadt, Germany). The immunoreaction was visualized using nickel-intensified diaminobenzidine according to a previously described protocol (Volk et al., 2004b; Bauer et al., 2008).
P-glycoprotein staining of brain sections was analysed using a computer-assisted image analysis system as described previously (Zibell et al., 2009). The hardware consisted of an Olympus BH2 microscope with a Plan-Neofluar objective (Zeiss, Göttingen, Germany), a CCD color camera (Axiocam; Zeiss, Göttingen, Germany) and an AMD Athlon™ 64 processor-based computer with an image capture interface card (Axiocam MR Interface Rev.A; Zeiss, Göttingen, Germany). Brain sections were analysed at a 400× magnification. Captured images were 1300 × 1030 pixels in dimension and were processed using KS400 image analysis software (Windows Release 3.0; Carl Zeiss Vision, Halbergmoos Germany). Detailed image analysis methodology has been previously published (Volk et al., 2004a,b;). Briefly, prior to image analysis, a spatial calibration was performed and a signal threshold value was defined to exclude background signals. This signal threshold value was used for analysis of all sections within the same experiment. Thus, data reported reflect pixel density above the threshold. P-glycoprotein immunostaining was analysed in the hippocampal hilus and the parietal cortex. The area labelled for P-glycoprotein was evaluated using 3–10 fields of 43434 µm2 per subfield.
Statistical analysis
Data are expressed as mean ± SEM. Statistical differences between the controls, responders and non-responders were analysed by the Mann–Whitney U-test. Within group comparison in phenobarbital plasma levels were analysed by the Wilcoxon matched pairs test. All statistical tests were performed two-tailed. Differences between means were considered to be statistically significant when P < 0.05.
Results
Selection of responders and non-responders
In response to phenobarbital, complete control of seizures was achieved in three out of 12 animals. In two further animals, phenobarbital resulted in a reduction of seizure frequency by at least 65% (88% and 65% respectively). These five animals were categorized as responders (Table 1). Animals (n = 7) that did not show a corresponding anticonvulsant response were considered non-responders. As mentioned above two animals that would not match a less strict definition of a non-responder (i.e. less than 50% seizure reduction) were considered resistant as these still exhibited severe seizure clusters during phenobarbital treatment.
Table 1.
Mean seizure frequency per day of responders and non-responders
|
Selection phase |
Second treatment phase |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
Pre-drug |
Phenobarbital |
Vehicle |
Celecoxib |
Phenobarbital |
||||||
| Mean±SEM | n | Mean±SEM | n | Mean±SEM | n | Mean±SEM | n | Mean±SEM | n | |
| Data of all animals included in the respective experimental phase | ||||||||||
| Responder | 0.86 ± 0.30 | 5 | 0.14 ± 0.10 | 5 | 1.40 ± 0.91 | 5 | 0.63 ± 0.40 | 5 | 0.50 ± 0.34 | 4 |
| Non-responder | 3.14 ± 1.68 | 7 | 2.63 ± 1.75 | 7 | 6.31 ± 4.65 | 5 | 6.17 ± 2.79 | 5 | 2.20 ± 2.20 | 4 |
| Data of those animals that completed all experimental phases | ||||||||||
| Responder | 1.03 ± 0.32 | 4 | 0.17 ± 0.13 | 4 | 1.75 ± 1.09 | 4 | 0.79 ± 0.47 | 4 | 0.50 ± 034 | 4 |
| Non-responder | 4.80 ± 2.77 | 4 | 4.01 ± 3.04 | 4 | 7.68 ± 5.74 | 4 | 6.46 ± 3.47 | 4 | 2.20 ± 2.20 | 4 |
The Table gives the average seizure frequency per day during the different treatment phases of the selection phase (pre-drug: 16 days, phenobarbital: 16 days) and second treatment phase (vehicle: 7 days; celecoxib: 6 days; phenobarbital: 16 days) for both groups of rats, designated responders and non-responders according to the first phenobarbital treatment phase. Please note that four animals died during different experimental phases, so that the number of animals included changed. To allow a better comparison, data of the animals that completed all experimental phases are given separately.
One rat of the non-responder group showed no change in seizure frequency as compared with the vehicle control phase. Two rats exhibited a moderate increase in seizure frequency by 25% and 8%. Four further rats exhibited a reduction in seizure frequency by 17%, 47%, 57% and 61% respectively. Figure 3A and Table 1 give the seizure data of all animals and those animals that completed all experimental phases (n = 4 responders; n = 4 non-responders). Analysis of plasma drug concentrations of the rats that survived till the end of experiment demonstrated that concentrations within the therapeutic range (15–40 µg·mL−1) were maintained in all rats throughout the period of treatment (Figure 4A).
Figure 3.
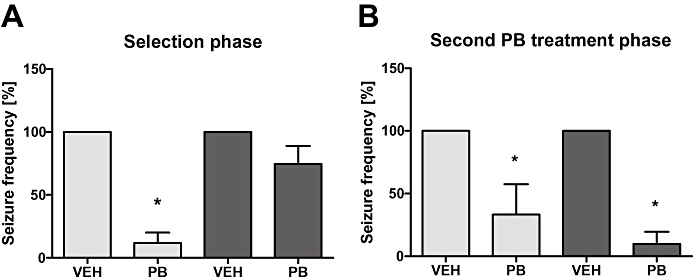
Effects of phenobarbital (PB) on spontaneous recurrent seizures in responders (n = 4) and non-responders (n = 4) epileptic rats (A and B). In this figure only those animals were included, that survived till the end of experiment. (A) Spontaneous recurrent seizures were recorded over a period of 16 days before onset of phenobarbital treatment (pre-drug control) followed by drug treatment for 16 days. Spontaneous recurrent seizures were continuously (24 h·day−1, 7 days·week−1) recorded in all eight rats over the whole time of this experiment. (A) illustrates the average of seizure reduction (mean ± SEM) in responders and non-responders during phenobarbital treatment in comparison with the vehicle (VEH) control period during the selection phase. Note the striking reduction of seizure frequency by phenobarbital in responders compared with non-responders. *Significantly lower than vehicle period. (B) Following a second vehicle control period of 7 days and constitutive application of celecoxib over 6 days, the effect of a 16 day phenobarbital treatment on spontaneous recurrent seizures was evaluated during the second treatment phase. (B) illustrates the average of seizure reduction (mean ± SEM) in responders (n = 4) and non-responders (n = 4) during this second phenobarbital treatment following after celecoxib pretreatment in comparison with the vehicle control period. Note that both groups of rats, responders and non-responders exhibit a significant reduction of seizure frequency by phenobarbital. *Significantly lower than respective vehicle period.
Figure 4.
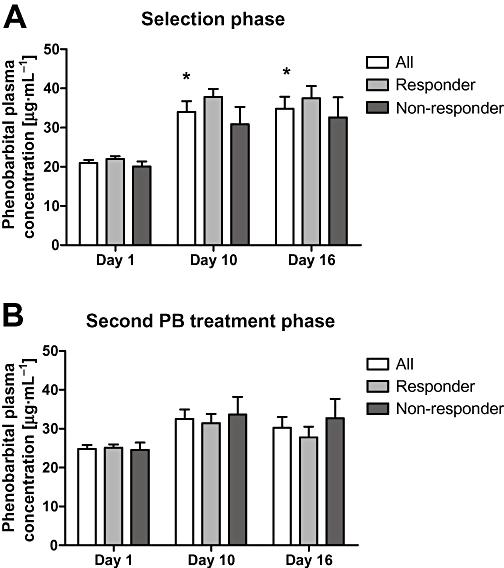
Schematic illustration of the phenobarbital (PB) plasma concentrations from the blood samples taken at days 1, 10 and 16 of the phenobarbital treatment during (A) the selection phase and (B) the second treatment phase. Statistical analysis between responders (n = 4) and non-responders (n = 4) within the selection phase and second treatment phase by the Wilcoxon matched pairs test, and inter-group analysis between the two trials by the Wilcoxon matched pairs test did not indicate a significant difference in phenobarbital plasma levels between groups or between treatment phases. When considering all animals included in the selection phase phenobarbital plasma concentrations significantly increased on days 10 and 16 compared with the first day of treatment. Data are given as mean ± SEM. *Significantly higher than day 1.
In the whole group of rats included during the selection phase (n = 12; responder: n = 5; non-responder n = 7), average phenobarbital concentrations 10 h after drug application in the morning of the days 1, 10 and 16 were 20.7 (range 14.4–22.81), 30.1 (range 19.8–50.3) and 34.3 (range 16.4–52.8) µg·mL−1 respectively. Phenobarbital concentrations in plasma at days 10 and 16 were significantly (P < 0.001) higher than drug levels on day 1, indicating drug accumulation during prolonged treatment. Plasma concentrations did not differ significantly between responders and non-responders. Average plasma concentrations determined 10 h after the bolus administration of phenobarbital amounted to 21.98 ± 0.69 µg·mL−1 in responders (n = 5) and 19.97 ± 1.12 µg·mL−1 in non-responders (n = 7). Side effects of phenobarbital comprising ataxia and sedation were comparable in both groups of rats as far as judged by observation without further evaluation by scoring analysis.
The mean number of seizures per day during the 16 day pre-drug phase was lower in the responder group than in the non-responder group (Table 1). Two of the non-responders exhibited a very high seizure frequency during the vehicle control phase (mean seizure frequency per day ≥6.3), while the other non-responders did not differ from responders in seizure frequency during the vehicle control phase. The mean number of seizures per day during the 16 day phenobarbital treatment phase was markedly less in the responder group than in the non-responder group (Table 1). Table 1 also gives the mean number of seizures per day for all animals and the animals that completed the analysis till the end of all experimental phases (n = 4 responders; n = 4 non-responders). Moreover, the analysis of video/EEG recordings did not reveal any differences in seizure severity. As described previously in the same model (Volk and Loscher, 2005), the type of spontaneous recurrent seizures resembled stage 4 or 5 seizures of the Racine scale (Racine, 1972).
The severity of the initial, electrically induced status epilepticus was not different between both groups of rats. Thus, the same severity and duration of status epilepticus resulted in epileptic rats differing by their pharmacosensitivity to phenobarbital.
Spontaneous seizure activity during the celecoxib pretreatment phase
In the 7 day vehicle control phase, the mean number of seizures per day recorded is shown in Table 1; note that one non-responder died during this celecoxib pretreatment phase. Again the higher mean value in the non-responder group was related to two animals that stood out by their high seizure frequency. The number of seizures in both groups was comparable to the vehicle control phase preceding the first phenobarbital treatment phase. Table 1 summarizes the data of all animals included (n = 5 responder, n = 5 non-responder) and the animals that completed all experimental phases (n = 4 responders; n = 4 non-responders).
In order to determine whether seizure frequency was affected by celecoxib or the additional treatment with pantoprazole, behaviour and EEG were continuously recorded during this pretreatment phase. It needs to be noted that this phase was limited to 6 days as considered appropriate for the main aim of our study. Therefore conclusions regarding putative pro- or anticonvulsant effects of the pretreatment are hampered by the short investigation period. Mean seizure frequency per day did not differ between the celecoxib phase reaching a mean of 3.22 ± 0.16 [mean ± SEM, responders 0.63 ± 0.40 (n = 5), non-responders 6.17 ± 2.79 (n = 5)] seizures during the 6 day monitoring and the preceding vehicle control phase. Considering individual data the changes observed during celecoxib pretreatment were rather divergent. Four animals including two responders and two non-responders showed a reduction in seizure frequency. Five animals including two responders and three non-responders exhibited an increase in seizure frequency as compared with the preceding control phase. One responder showed no change in seizure frequency. Regarding severity and duration of seizures, no alterations were observed during the celecoxib pretreatment phase, compared with the preceding vehicle control phase.
Effect of celecoxib pretreatment on the anticonvulsant response to phenobarbital
Following the celecoxib pretreatment phase, phenobarbital treatment significantly decreased the seizure frequency in both responders (n = 4) as well as non-responders (n = 4) (Figure 3B). In three animals categorized non-responders based on the first phenobarbital treatment phase, phenobarbital now resulted in a complete seizure control. In one non-responder that stood out by its high seizure frequency (mean seizure frequency per day during the vehicle control phases: 24.86 ± 1.98, range between 18 and 33 seizures per day) seizure frequency was reduced by >65% as compared with the preceding vehicle control phase. In contrast, this animal has shown an increase in seizure frequency by 8% during the first phenobarbital treatment phase.
Side effects of phenobarbital did not differ from those observed during the first phenobarbital treatment phase. Analysis of phenobarbital plasma concentrations during the second phenobarbital treatment phase and the comparison with respective data from the first phenobarbital treatment phase did not reveal any impact of the preceding celecoxib treatment (Figure 4).
Concentrations of phenobarbital in plasma of rats following celecoxib premedication and a group of phenobarbital-treated non-epileptic control rats were additionally analysed 60 min after the last i.p. administration of 15 mg·kg−1 phenobarbital. The concentrations of phenobarbital in the plasma were comparable in celecoxib-pretreated responder and non-responder rats and the control group (mean ± SEM 50.1 ± 8.9 µg·mL−1 in responder rats vs. 55.3 ± 17.8 µg·mL−1 in non-responder rats vs. 57.4 ± 11.5 µg·mL−1 in control group). In homogenates of the parahippocampal cortex, the analysis of phenobarbital concentrations did not reveal any significant differences (mean ± SEM 19.98 ± 2.45 µg·g−1 in responder rats, 25.9 ± 2.01 µg·g−1 in non-responder rats, 21.6 ± 1.17 µg·g−1 in control group) in line with our hypothesis regarding the expected impact of celecoxib pretreatment.
P-glycoprotein expression rates following celecoxib pretreatment
Based on our hypothesis that COX-2 inhibition prevents P-glycoprotein induction by recurrent seizures during the celecoxib treatment phase, it was expected that P-glycoprotein expression would not differ between those rats designated phenobarbital responders and non-responders based on the first phenobarbital treatment phase: Moreover, we expected that expression rates in these groups would approximate to control levels, based on the effect of celecoxib. The analysis was focused on two brain regions, in which seizure activity is well known to up-regulate P-glycoprotein expression.
Immunolocalization of P-glycoprotein was observed in microvessel endothelial cells of all rats, regardless of their treatment (Figure 5A–C). The DAB staining method was chosen for the present experiments as it exclusively labels endothelial P-glycoprotein. Therefore, the P-glycoprotein-labelled areas determined by computer-assisted analysis can be completely attributed to endothelial P-glycoprotein expression (Volk et al., 2005).
Figure 5.
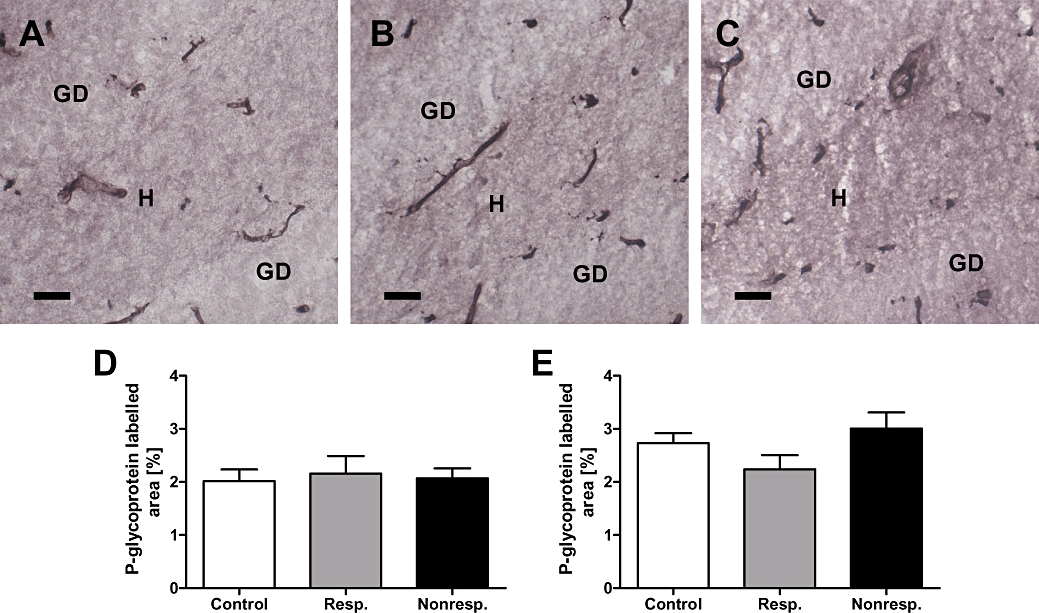
Celecoxib treatment of responders (n = 4) and non-responders (n = 4) decreases P-glycoprotein expression to control level. (A) Representative immunostaining of P-glycoprotein in the hippocampal dentate hilus [H] of a non-epileptic control rat (DG, dentate gyrus). (B) P-glycoprotein immunostaining in the hippocampal dentate hilus of an epileptic rat responding to phenobarbital (responder) during the first and second treatment phase. Note that brain tissue samples have been taken following celecoxib pretreatment and the second phenobarbital treatment phase. (C) P-glycoprotein immunostaining in the hippocampal hilus of an epileptic rat that was classified as a non-responder during the first phenobarbital treatment phase. Note that this rat responded to phenobarbital following celecoxib pretreatment, that is, during the second phenobarbital treatment phase and that brain tissue samples have been taken following celecoxib pretreatment and the second phenobarbital treatment phase. P-glycoprotein immunoreactivity in capillaries proved to be comparable between the three groups of rats. Quantitative analysis of P-glycoprotein immunostaining in the hippocampal dentate hilus (D) and the parietal cortex (E). Note that celecoxib treatment reduced P-glycoprotein levels to control levels in responders as well as non-responders. Data are given as mean ± SEM. Scale bar = 100 µm.
In line with our hypothesis, the area labelled for P-glycoprotein in the hippocampal hilus and parietal cortex did not differ between the three groups, that is, phenobarbital-treated non-epileptic control rats as well as rats designated phenobarbital responders or non-responders based on the first phenobarbital treatment phase (Figure 5). In the hilus, the mean area labelled for P-glycoprotein amounted to 2.01 ± 0.22 (mean ± SEM) in control rats, 2.16 ± 0.33 (mean ± SEM) in responders and 2.07 ± 0.19 in non-responders (Figure 5D).
Discussion and conclusions
Here, we provide experimental support for a novel therapeutic concept to overcome pharmacoresistance in epilepsies. In a chronic rat model of pharmacoresistant temporal lobe epilepsy, COX-2 inhibition restored the sensitivity to the antiepileptic drug phenobarbital in rats that showed a poor or no response to phenobarbital in a preceding test of efficacy.
Based on previous studies, the model used was considered highly suitable to test our hypothesis, that is, whether blocking the signalling cascade that mediates seizure-associated up-regulation of P-glycoprotein results in improved efficacy of antiepileptic drugs. Using the same experimental approach and selection procedure, it has been demonstrated that phenobarbital non-responders selected from the post-status epilepticus model exhibit a marked overexpression of the drug efflux transporter P-glycoprotein in brain capillary endothelial cells in limbic brain regions including the hippocampus (Volk and Loscher, 2005). As phenobarbital is a substrate of P-glycoprotein (Schuetz et al., 1996; Potschka et al., 2002), the increased expression is likely to result in sub-therapeutic concentrations of this antiepileptic drug at its brain target sites (Volk and Loscher, 2005). Final proof for the role of BBB P-glycoprotein overexpression in pharmacoresistance in this model came from a study, in which co-administration of the highly selective, third generation, P-glycoprotein inhibitor, tariquidar restored the anticonvulsant efficacy of phenobarbital without altering its plasma concentrations (Brandt et al., 2006).
In line with these previous studies, we were able to identify subgroups of rats that significantly differed in their pharmacoresponse to phenobarbital in the chronic phase of this post-status epilepticus model. Data obtained from these rats suggested that prevention of P-glycoprotein up-regulation as a promising alternative strategy to overcome P-glycoprotein-mediated pharmacoresistance. Our efforts to develop alternate strategies to overcome transporter-mediated pharmacoresistance are driven by general concerns regarding the tolerability of chronic P-glycoprotein inhibition in epileptic patients. First, limitations of the combination of a P-glycoprotein inhibitor with an antiepileptic drug may be associated with the intended aim. An influence on pharmacokinetics of the antiepileptic drug might not only affect the target brain region. Enhanced drug concentrations in other brain regions may worsen adverse effects. Second, multidrug transporters such as P-glycoprotein are involved in a series of physiological functions including protection from xenobiotics (Huls et al., 2009). Several xenobiotics taken up by the body may be more harmful in the presence of efflux transporter inhibitors due to enhanced distribution and retarded excretion. Based on our recent success to identify a glutamate/NMDA receptor/COX-2 signalling pathway that up-regulates endothelial P-glycoprotein in the epileptic brain, we therefore suggested targeting of these signalling events as an alternative approach (Bauer et al., 2008). The role of COX-2, which stood out as one of the most obvious targets in the signalling cascade, was initially confirmed by in vitro experiments in isolated brain capillaries (Bauer et al., 2008). In these experiments selective COX-2 inhibition counteracted the induction of P-glycoprotein in response to excessive glutamate concentrations, whereas selective COX-1 inhibition did not elicit an effect. Moreover, the effect of glutamate exposure on P-glycoprotein expression and transport function was abolished when capillaries from COX-2 deficient mice were used. In line with these in vitro data, prevention of the transcriptional activation of the P-glycoprotein encoding gene was achieved by pretreatment with the non-selective COX inhibitor indomethacin and the COX-2 inhibitor celecoxib in the acute phase of the pilocarpine status epilepticus model (Bauer et al., 2008; Zibell et al., 2009). Recent experimental data showed that it is also possible to reduce and control P-glycoprotein expression levels in rats in which an overexpression of P-glycoprotein has already been induced by previous seizures, that is, in the chronic phase of a post-status epilepticus model (van Vliet et al., 2010). Now, we provide further support that COX-2 must be considered as a target to overcome drug-refractoriness in epilepsies. From earlier studies it is known that P-glycoprotein expression is transiently induced, returning to basal levels if no further induction occurs (Seegers et al., 2002). Therefore, P-glycoprotein expression should be reduced to basal levels, if we prevent activation of the signalling pathway by further recurrent seizure activity using a treatment paradigm with sub-chronic COX-2 inhibition. In support of this hypothesis, P-glycoprotein expression reached comparable levels in phenobarbital-treated non-epileptic rats as well as rats designated phenobarbital responders or non-responders based on the first phenobarbital treatment phase. In this context it needs to be considered that we are comparing present data with historical data that revealed a difference between responders and non-responders and, because of the extremely laborious and time-consuming experiments, we were unable to include an additional phenobarbital-selected group without celecoxib treatment. Although the lack of corresponding control groups without celecoxib treatment was unavoidable for practical reasons, the comparison with historical data must be considered as a deficiency in the present study, which needs to be kept in mind regarding final conclusions.
Following pretreatment with the COX-2 inhibitor celecoxib the efficacy of phenobarbital was significantly improved in all four rats that were previously categorized non-responders. Phenobarbital now resulted in a mean reduction in seizure frequency of 80 ± 11% in this subgroup of rats with three animals even exhibiting a complete seizure control. As expected based on the lower P-glycoprotein expression rates characterizing phenobarbital responders (Volk and Loscher, 2005) the significant response to phenobarbital remained unaffected by celecoxib pretreatment in the group of responders.
However quite different mechanisms might have contributed to the enhancement in the anticonvulsant efficacy in the non-responder group. For instance, celecoxib or the addition of the gastroprotectant, pantoprazole, might have affected drug metabolizing enzymes thereby interfering with phenobarbital metabolism and excretion. However, phenobarbital concentrations determined at the end of the treatment did not differ significantly between the two phenobarbital treatment phases, arguing against any effect on enzyme inhibition. Another possible disease-modifying effect must be considered. Neuroinflammatory events are generally considered to contribute to epileptogenesis or disease progression (Vezzani and Granata, 2005). Therefore the anti-inflammatory drug celecoxib might exert disease-modifying effects. In our present study, a generalized attenuation of disease severity by celeccoxib can be ruled out as the mean seizure frequency tended to increase, rather than decrease, during the celecoxib treatment phase.
When considering these data as a departure point to plan translational studies, one needs to bear in mind that the clinical use of COX-2 inhibitors can be associated with an enhanced risk for renal, cardiovascular and cerebrovascular events. In addition, there are other data on the effect of COX-2 inhibition on seizure susceptibility. These data range from the description of beneficial neuroprotective effects and anticonvulsant effects to aggravation of seizures (Kulkarni and Dhir, 2009), raising further concerns regarding tolerability issues and demanding a thorough validation of safety and tolerability. During the celecoxib pretreatment phase, seizure frequency tended to increase in both subgroups of rats, that is, responders and non-responders. However, the difference to the preceding vehicle control phase did not reach significance. Moreover, pronounced inter-individual differences were obvious with three animals showing a reduction in seizure frequency and seven animals showing an increase. Conclusions regarding putative proconvulsant effects of celecoxib are definitely limited by the short celecoxib pretreatment phase we used here, but which was considered most suitable for the main aim of the study. Thus, further testing is necessary to carefully evaluate both the benefits and risks with the use of COX-2 inhibitors in epileptic patients and also test effects of the addition of COX-2 inhibition to different antiepileptic drug regimens. Regarding a putative impact on seizure susceptibility in temporal lobe epilepsy, we are currently analysing the effect of COX-2 inhibitors on seizure thresholds and seizure parameters in the amygdala kindling model.
Bearing in mind putative drawbacks of the approach, it is of specific interest to elucidate the complete cascade that drives transcriptional activation of P-glycoprotein in response to epileptic seizures aiming to identify other targets. Among the downstream signalling events, we recently identified prostaglandin E2 EP1 receptors as another target that controls or down-regulates P-glycoprotein expression in the epileptic brain (Pekcec et al., 2009).
These present data further support COX-2 inhibition as a strategy to improve efficacy of antiepileptic drugs and to overcome pharmacoresistance in a subpopulation of patients. Provided that the effect is related to an impact on P-glycoprotein expression, it needs to be kept in mind that the clinical relevance of P-glycoprotein overexpression still remains to be determined. A large series of studies in rodent epilepsy models and in epileptic tissue of pharmacoresistant patients suggested an association between seizure-induced P-glycoprotein overexpression and resistance to antiepileptic drugs (Loscher and Potschka, 2005b). In line with these data a recent PET study in patients with pharmacoresistant epilepsy gave some evidence for an excessive P-glycoprotein transport function in brain regions that are part of the epileptic network (Langer et al., 2007). Moreover, studies in animal models of pharmacoresistant epilepsy demonstrated that it is possible to overcome drug-refractoriness by modulation of P-glycoprotein transport function (Clinckers et al., 2005; Brandt et al., 2006; van Vliet et al., 2006). Up to now, evidence exists that phenytoin, phenobarbital, lamotrigine, levetiracetam as well as an active metabolite of oxcarbazepine are subject to BBB efflux by the human isoform of P-glycoprotein (Luna-Tortos et al., 2008). Thus, controlling transporter expression in the epileptic brain by COX-2 inhibition should have a beneficial effect on brain penetration and efficacy of these antiepileptic drugs. However, final conclusions regarding the promises of the approach have to await further progress with positron emission tomography methods aiming to image P-glycoprotein transport function in individual patients. Moreover, other mechanisms of pharmacoresistance such as alterations in target sites need to be considered as well (Remy and Beck, 2006). In this context, it will be especially important to evaluate whether patients with predominance of one resistance mechanism exist in whom blocking this mechanism would be sufficient to restore antiepileptic drug efficacy.
In conclusion, our findings provide further support for prevention of seizure-associated P-glycoprotein induction by COX-2 inhibition as a suitable procedure to restore pharmacosensitivity. Considering a series of previous studies it is likely that this effect is based on an interference with P-glycoprotein up-regulation in response to ongoing repeated seizure activity. Based on these data, transient COX-2 inhibition is suggested as a novel therapeutic approach to overcome pharmacoresistance in epilepsies. Regarding tolerability of COX-2 inhibitor treatment phases in epileptic patients, further experimental studies are necessary. Moreover, it needs to be noted that the present study did not provide direct proof that the inhibition of COX-2 contributed to the improvement of phenobarbital efficacy. Future studies might comprise similar experiments in conditional COX-2 knockout mice, in which the effect of COX-2 deficiency on pharmacosensitivity could be analysed.
Acknowledgments
We thank Inge Sautter, Marion Fisch, Katharina Gabriel, Heidrun Zankl and Regina Rentsch for their excellent technical assistance. This research was supported by the grant DFG PO 681/4-1 (to HP) from the German Research Foundation.
Glossary
Abbreviations:
- BBB
blood–brain barrier
- BLA
basolateral nucleus of the amygdala
- COX-2
cyclooxygenase-2
Conflicts of interest
The authors have no conflicts of interest.
References
- Bauer B, Hartz AM, Pekcec A, Toellner K, Miller DS, Potschka H. Seizure-induced up-regulation of P-glycoprotein at the blood-brain barrier through glutamate and cyclooxygenase-2 signaling. Mol Pharmacol. 2008;73:1444–1453. doi: 10.1124/mol.107.041210. [DOI] [PubMed] [Google Scholar]
- Bethmann K, Brandt C, Loscher W. Resistance to phenobarbital extends to phenytoin in a rat model of temporal lobe epilepsy. Epilepsia. 2007;48:816–826. doi: 10.1111/j.1528-1167.2007.00980.x. [DOI] [PubMed] [Google Scholar]
- Bonin EA, Campos AC, Coelho JC, Matias JE, Malafaia O, Jonasson TH. Effect of pantoprazole administered subcutaneously on the healing of sutured gastric incisions in rats. Eur Surg Res. 2005;37:250–256. doi: 10.1159/000087872. [DOI] [PubMed] [Google Scholar]
- Brandt C, Bethmann K, Gastens AM, Loscher W. The multidrug transporter hypothesis of drug resistance in epilepsy: proof-of-principle in a rat model of temporal lobe epilepsy. Neurobiol Dis. 2006;24:202–211. doi: 10.1016/j.nbd.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Clinckers R, Smolders I, Meurs A, Ebinger G, Michotte Y. Quantitative in vivo microdialysis study on the influence of multidrug transporters on the blood-brain barrier passage of oxcarbazepine: concomitant use of hippocampal monoamines as pharmacodynamic markers for the anticonvulsant activity. J Pharmacol Exp Ther. 2005;314:725–731. doi: 10.1124/jpet.105.085514. [DOI] [PubMed] [Google Scholar]
- Huls M, Russel FG, Masereeuw R. The role of ATP binding cassette transporters in tissue defense and organ regeneration. J Pharmacol Exp Ther. 2009;328:3–9. doi: 10.1124/jpet.107.132225. [DOI] [PubMed] [Google Scholar]
- Kulkarni SK, Dhir A. Cyclooxygenase in epilepsy: from perception to application. Drugs Today (Barc) 2009;45:135–154. doi: 10.1358/dot.2009.45.2.1322481. [DOI] [PubMed] [Google Scholar]
- Langer O, Bauer M, Hammers A, Karch R, Pataraia E, Koepp MJ, et al. Pharmacoresistance in epilepsy: a pilot PET study with the P-glycoprotein substrate R-[(11)C]verapamil. Epilepsia. 2007;48:1774–1784. doi: 10.1111/j.1528-1167.2007.01116.x. [DOI] [PubMed] [Google Scholar]
- Loscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci. 2005a;6:591–602. doi: 10.1038/nrn1728. [DOI] [PubMed] [Google Scholar]
- Loscher W, Potschka H. Role of drug efflux transporters in the brain for drug disposition and treatment of brain diseases. Prog Neurobiol. 2005b;76:22–76. doi: 10.1016/j.pneurobio.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Luna-Tortos C, Fedrowitz M, Loscher W. Several major antiepileptic drugs are substrates for human P-glycoprotein. Neuropharmacology. 2008;55:1364–1375. doi: 10.1016/j.neuropharm.2008.08.032. [DOI] [PubMed] [Google Scholar]
- Pekcec A, Fuest C, Muhlenhoff M, Gerardy-Schahn R, Potschka H. Targeting epileptogenesis-associated induction of neurogenesis by enzymatic depolysialylation of NCAM counteracts spatial learning dysfunction but fails to impact epilepsy development. J Neurochem. 2008;105:389–400. doi: 10.1111/j.1471-4159.2007.05172.x. [DOI] [PubMed] [Google Scholar]
- Pekcec A, Unkruer B, Schlichtiger J, Soerensen J, Hartz AM, Bauer B, et al. Targeting prostaglandin E2 Ep1 receptors prevents seizure-associated P-glycoprotein up-regulation. J Pharmacol Exp Ther. 2009;330:939–947. doi: 10.1124/jpet.109.152520. [DOI] [PubMed] [Google Scholar]
- Potschka H, Fedrowitz M, Loscher W. P-Glycoprotein-mediated efflux of phenobarbital, lamotrigine, and felbamate at the blood-brain barrier: evidence from microdialysis experiments in rats. Neurosci Lett. 2002;327:173–176. doi: 10.1016/s0304-3940(02)00423-8. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Remy S, Beck H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain. 2006;129:18–35. doi: 10.1093/brain/awh682. [DOI] [PubMed] [Google Scholar]
- Schuetz EG, Beck WT, Schuetz JD. Modulators and substrates of P-glycoprotein and cytochrome P4503A coordinately up-regulate these proteins in human colon carcinoma cells. Mol Pharmacol. 1996;49:311–318. [PubMed] [Google Scholar]
- Seegers U, Potschka H, Loscher W. Transient increase of P-glycoprotein expression in endothelium and parenchyma of limbic brain regions in the kainate model of temporal lobe epilepsy. Epilepsy Res. 2002;51:257–268. doi: 10.1016/s0920-1211(02)00156-0. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Granata T. Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia. 2005;46:1724–1743. doi: 10.1111/j.1528-1167.2005.00298.x. [DOI] [PubMed] [Google Scholar]
- van Vliet EA, van Schaik R, Edelbroek PM, Redeker S, Aronica E, Wadman WJ, et al. Inhibition of the multidrug transporter P-glycoprotein improves seizure control in phenytoin-treated chronic epileptic rats. Epilepsia. 2006;47:672–680. doi: 10.1111/j.1528-1167.2006.00496.x. [DOI] [PubMed] [Google Scholar]
- van Vliet EA, Zibell G, Pekcec A, Schlichtiger J, Edelbroek PM, Holtman L, et al. COX-2 inhibition controls P-glycoprotein expression and promotes brain delivery of phenytoin in chronic epileptic rats. Neuropharmacology. 2010;58:404–412. doi: 10.1016/j.neuropharm.2009.09.012. [DOI] [PubMed] [Google Scholar]
- Volk HA, Loscher W. Multidrug resistance in epilepsy: rats with drug-resistant seizures exhibit enhanced brain expression of P-glycoprotein compared with rats with drug-responsive seizures. Brain. 2005;128:1358–1368. doi: 10.1093/brain/awh437. [DOI] [PubMed] [Google Scholar]
- Volk HA, Burkhardt K, Potschka H, Chen J, Becker A, Loscher W. Neuronal expression of the drug efflux transporter P-glycoprotein in the rat hippocampus after limbic seizures. Neuroscience. 2004a;123:751–759. doi: 10.1016/j.neuroscience.2003.10.012. [DOI] [PubMed] [Google Scholar]
- Volk HA, Potschka H, Loscher W. Increased expression of the multidrug transporter P-glycoprotein in limbic brain regions after amygdala-kindled seizures in rats. Epilepsy Res. 2004b;58:67–79. doi: 10.1016/j.eplepsyres.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Volk H, Potschka H, Loscher W. Immunohistochemical localization of P-glycoprotein in rat brain and detection of its increased expression by seizures are sensitive to fixation and staining variables. J Histochem Cytochem. 2005;53:517–531. doi: 10.1369/jhc.4A6451.2005. [DOI] [PubMed] [Google Scholar]
- Zibell G, Unkruer B, Pekcec A, Hartz AM, Bauer B, Miller DS, et al. Prevention of seizure-induced up-regulation of endothelial P-glycoprotein by COX-2 inhibition. Neuropharmacology. 2009;56:849–855. doi: 10.1016/j.neuropharm.2009.01.009. [DOI] [PubMed] [Google Scholar]