

Abstract
Background and purpose:
Current strategies to ameliorate cardiac ischaemic and reperfusion damage, including block of the sodium-hydrogen exchanger, are therapeutically ineffective. Here we propose a different approach, block of the persistent sodium current (INaP).
Experimental approach:
Left ventricular pressure was measured as an index of functional deficit in isolated, Langendorff perfused, hearts from adult rats, subjected to 30 min global ischaemia and reperfusion with vehicle only (control) or riluzole (1–10 µM) in the perfusate. Cell shortening and intracellular Ca2+ concentrations [Ca2+]i were measured in adult rat isolated myocytes subjected to hypoxia and re-oxygenation. The block of transient and persistent sodium currents by concentrations of riluzole between 0.01 and 100 µM were assessed in rat isolated myocytes using patch clamp techniques.
Key results:
In perfused hearts, riluzole produced a concentration-dependent cardioprotective action, with minor protection from 1 µM and produced rapid and almost complete recovery upon reperfusion from 3 and 10 µM. In isolated myocytes, riluzole at 3 and 10 µM greatly attenuated or prevented the hypoxia- and reperfusion-induced rise in [Ca2+]i and the contractile deficit. In patch clamp experiments, riluzole blocked the persistent sodium current with an IC50 of 2.7 µM, whereas the block of the transient sodium current was only apparent at concentrations above 30 µM.
Conclusions and implications:
Riluzole preferentially blocked INaP and was protective in cardiac ischaemia and reperfusion. Thus block of the persistent sodium current would be a viable method of ameliorating cardiac ischaemic and reperfusion damage.
Keywords: ischaemia, reperfusion, persistent sodium current (INaP), riluzole, cardioprotection
Introduction
Reperfusion of myocardium after a period of ischaemia (post-ischaemic myocardium) often results in an increase in cellular damage and functional impairment. For example, in the rat, post-ischaemic myocardium displays an elevated diastolic calcium to about 0.5 µM from control values of about 0.3 µM accompanied by an increased left ventricular end-diastolic pressure (LVEDP) to about 80 mmHg from a control value of about 10 mmHg and a prolonged calcium transient (Meissner and Morgan, 1995). These observations have subsequently generally been confirmed (Saini and Dhalla, 2005), although other groups have disputed whether the increased [Ca2+]i is directly related to the increased diastolic tension (Eberli et al., 2000), pointing out that the situation is complicated by changes in myofilament calcium sensitivity. Nevertheless, it is a consistent observation that post-ischaemic myocardium displays an increase in [Ca2+]i. The increase in [Ca2+]i appears to follow, and depend upon, a preceding increase in [Na+]i. The mechanistic link proposed is that an increased [Na+]i reduces the electrochemical gradient for Na+ which normally drives the sodium-calcium exchanger, NCX (Lazdunski et al., 1985), and there is now a great deal of supporting evidence for this. However, the source of the increased [Na+]i is still not clear. It is unlikely that reduction or cessation of function of the Na-KATPase alone is sufficient, and it appears that an abnormal influx of Na+ occurs (rather than merely a failure of Na+ extrusion), particularly during reperfusion. One mechanism that can produce an abnormal Na+ influx is the enhanced activity of the sodium-hydrogen exchanger, NHE1 (Xiao and Allen, 2000; Murphy and Allen, 2009). With this in mind, there has been considerable effort at developing compounds aimed at blocking NHE1 as a protective strategy against reperfusion damage, such as cariporide. In animal experiments, NHE1 blockers are reasonably effective at ameliorating reperfusion damage (Hartmann and Decking, 1999), but clinical trials with these agents have been disappointing (Theroux et al., 2000; Zeymer et al., 2001). This may be because another source of Na+ influx contributes to the rise in [Na+]i during ischaemia and/or reperfusion. Previous reports have suggested that this could be because of the persistent sodium current (INaP). This current is carried by a non-inactivating Na+ channel, also known as the TTX-sensitive Na+ channel, which is activated in hypoxia (Ju et al., 1996b) and although the amplitude of the current through this channel is small (in the order of a hundred pA), its persistence when the channel is activated permits a significant rise in [Na+]i. Moreover low doses of TTX, which block this current, have been shown to reduce the rise in [Na+]i in hypoxia as well as, and in synergy with, cariporide (Eigel and Hadley, 1999). TTX is not practical as a cardioprotective agent because of its high potency to block neuronal sodium channels and produce brain death but an agent specific for the persistent channel may have therapeutic potential.
Here we report on a potential novel cardioprotective agent, riluzole, which appears to act via a selective block of INaP. Riluzole has been shown to be protective against neuronal ischaemia in a variety of models (Bae et al., 2000; Baptiste and Fehlings, 2006; Heurteaux et al., 2006). This effect of riluzole is generally thought to be because of its ability to block glutamate release (≈10 µM) and/or postsynaptic NMDA receptors IC50≈ 18 µM] (Bryson et al., 1996; Doble, 1996; Lamanauskas and Nistri, 2008), although a variety of other mechanisms have been proposed, including activation of G-protein dependent signal transduction (Doble, 1996), inhibition of tyrosine phosphorylation (IC50≈ 0.5 µM) (Peyclit et al., 2001), activation of the tandem pore K+ channels TRAAK and TREK-1 (≈100 µM) (Duprat et al., 2000) and inhibition of neuronal sodium channels (8 mg·kg−1 i.p. in rat) (Ates et al., 2007). Although riluzole can protect the retina from neurodegeration induced by ischaemia (Izumi et al., 2003), there is, at present, no data on whether riluzole may be protective against ischaemia in tissues other than neuronal tissues. Here we show that riluzole is cardioprotective in isolated rat hearts and prevents the rise in [Ca2+]i induced in single myocytes by hypoxia and re-oxygenation. Riluzole also blocked INaP in single myocytes, with more than 10-fold greater potency than block of the transient sodium current (INaT). Furthermore, the optimal concentration for all of these effects was about the same (≈3 µM). We therefore suggest that riluzole ameliorates ischaemic and reperfusion damage as a result of its block of INaP in the heart.
Methods
All animal care and experimental procedures for these experiments conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication no. 85-23, revised 1996) and were approved by the Animal Ethics Committees of the University of Adelaide and Leeds University.
Isolated heart
Male Sprague-Dawley rats weighing 350 to 450 g were anaesthetized by I.P injection of 30 mg·kg−1 sodium pentobarbitone with 2000 units of heparin. When deeply anesthetized, the hearts were removed and perfused via the aorta in a Langendorff apparatus. Hearts were paced at 4 Hz by stimulation of the right atrium. An intraventricular saline-filled balloon was placed in the left ventricle through an incision in the left atrium for measurement of left ventricular pressure. Coronary flow was maintained at 10 mL·min−1 (except during ischaemia). The perfusate had the composition (in mM): CaCl2 1.5, NaCl 111.0, KCl 4.0, MgCl2 0.6, NaHCO3 23.9, NaH2PO4 1.2, d-glucose 12.0 and was equilibrated with 95% O2 and 5% CO2 The whole apparatus was contained in a humidified temperature controlled chamber at 37°C. Global ischaemia was induced by stopping the perfusion pump. Riluzole (Sigma Pharmaceuticals, St. Louis, MO, USA) was dissolved in a stock solution of propylene glycol (PEG; Sigma Pharmaceuticals) at 10 mM, and diluted slowly into the perfusate while stirring to achieve the final concentration. Riluzole was present in the perfusate at either 1, 3 or 10 µM (n = 4 per group) throughout the experiment, and control solutions (n = 4) contained the vehicle alone at the same concentration as the highest used in the experimental solutions. The vehicle at this concentration had no observable effect on the hearts.
In a separate series of experiments, epicardial monophasic action potentials (MAPs) were recorded from the surface of the left ventricle in isolated hearts with control perfusate, and with riluzole added to the perfusate at 3, 10 and 30 µM (n = 6).
Data was digitized at 2 KHz using ‘Powerlab’ hardware and software from AD Instruments (Sydney, Australia). Measurements of left ventricular pressure (LVP) and the differential of LVP (dLVP/dt) were made during normal perfusion, during ischaemia, and 1 min after reperfusion, by averaging the measured variables over 10 consecutive beats. Action potential duration (APD) measurements were made with the ‘peak parameters’ add-on module for Chart 5 (AD Instruments) All measurements were made without knowledge of the treatments. Data are presented as means ± standard error of the mean and statistical analysis (Student's t-test) was done in Excel (Microsoft). Significance was taken at P < 0.05.
Measurement of [Ca2+]i and contraction in single isolated myocytes
Myocytes were isolated from adult Wistar rat hearts as described previously (McCrossan et al., 2004). The freshly isolated myocytes were incubated with 2 µM Fura 4-AM (Molecular Probes, Eugene, OR, USA) and gently shaken for 20 min at room temperature (20–24°C). Cells were then resuspended in a HEPES-based solution (see further discussion) and left in the dark for 30 min before use to allow for de-esterification of the dye. Myocytes were placed on the stage of a Nikon Eclipse microscope for simultaneous recording of cell length and [Ca2+]i. Cells were field stimulated at 2 Hz at 120% threshold voltage. For all measurements, cells were superfused continuously at 37°C with a HEPES-based solution containing (in mM): 137 NaCl; 5.4 KCl; 0.33 NaH2PO4; 0.5 MgCl2; 5 HEPES; 5.6 glucose; 1 CaCl2 at pH 7.4. Cell length was monitored using a video-edge detector (Crescent Electronics, Salt Lake City, UT, USA). For the [Ca2+]i measurement, myocytes were alternately illuminated at 340 and 380 nm, using an Optoscan monochromator (Cairn Research, Kent, UK). The fluorescent emission at 510 nm was collected by a photomultiplier tube and the ratio of emitted light in response to excitation at 340 and 380 nm was calculated to provide an index of [Ca2+]i. In order to induce hypoxia, the superfusate was bubbled with 100% N2, which was also blown over the surface of the partially closed experimental chamber to maintain a low PO2 (≈28 mmHg) in the experimental chamber. Low PO2 was confirmed by measuring samples of the perfusate using a blood gas analyser (Bayer Rapid Lab 348, Siemens Healthcare Diagnostics, Deerfield, IL, USA). Myocytes were exposed to an O2-bubbled solution for 10 min. to record pre-hypoxic values, the superfusing solution was then vigorously bubbled with 100% N2 for 20 min. (or until the cell stopped contracting), finally the solution was re-bubbled with O2 and responses monitored for 20 min (or until cell death). Cell contractions and [Ca2+]i were recorded at 5 min. intervals. The protocol was performed in the absence and the presence of 1, 3 and 10 µM riluzole (Sigma), prepared as above.
[Ca2+]i in each group for all time points was compared with analysis of variance, and the differences between groups compared with t-tests. The number of cells ‘surviving’ (i.e. contracting) was compared with an overall exact chi-squared test (3 d.f.), which was highly statistically significant (P < 0.001), followed by pairwise comparisons between the control and each concentration group using a Fisher Exact Test (two-sided).
Patch clamp experiments
Myocytes were isolated from adult rat hearts as noted earlier. When required, cells were transferred to the recording chamber on the stage of an inverted microscope. Sodium currents were recorded in whole cell patch clamp mode using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA), with borosilicate electrodes having resistances of <1 M Ω. Whole cell capacitance and series resistance compensation was achieved using the controls on the amplifier. The signal was filtered at 1 KHz and recorded at 12 bit resolution at 2 KHz. Recording was only performed if series resistance compensation of at least 90% could be achieved.
The standard external (bath) solution for INaP recordings contained (mM): NaCl, 150; HEPES, 10; KCl, 5; MgCl2, 1; CaCl2, 2; CoCl2, 5; CsCl, 5; pH adjusted to 7.4 with 5.0 M NaOH. The standard external (bath) solution for transient sodium current (INaT) recordings contained (mM): NaCl,70; HEPES, 10; KCl, 5; MgCl2, 1; CaCl2, 2; CoCl2, 5; choline, 80; CsCl, 5; pH adjusted to 7.4 with 5.0 M NaOH. The pipette solution for both INaP and INaT recording contained (mM): CsF, 120; HEPES, 10; MgCl2, 2; Na2-EGTA, 20; CaCl2, 2; pH adjusted to 7.4 with 5.0 M NaOH. Experiments were carried out at room temperature (24°C).
Riluzole, prepared as earlier, was added to the bath solution, which continuously perfused the bath (volume c. 0.5 mL) at a rate of 1 mL·min−1. Hypoxia was induced by bubbling the perfusate with 100% N2 and also blowing a gentle stream of N2 across the surface of the bath solution while maintaining the bath perfusion.
INaP was elicited by stepping the membrane potential from a holding potential of −90 to −20 mV for 250 ms. INaP was measured for each cell by averaging the current amplitude over the period from 170 to 250 ms after the voltage step and this was in turn averaged over 6 consecutive recordings taken 5 s apart. Leak correction was done by correcting the current excursion produced by a 10 ms, 20 mV pulse given 100 ms prior to the test pulse using the amplifier controls. Data were then averaged over six consecutive INaP cycles recorded 6 s apart. It was not generally possible to hold one cell long enough to produce a cumulative C-E curve for block of INaP in a single cell. Rather, the curve was constructed from as many recordings that could be obtained in different cells.
Using a similar configuration but with a lower extracellular sodium concentration in the bath solution as detailed above, INaT was generated by stepping the membrane potential to −20 mV from a holding potential of −140 mV for 200 ms. To generate activation and inactivation curves, either the holding potential or the test potential were incremented with each pulse, delivered at 1 s intervals. Data points for each curve were fitted with Boltzman distributions by least squares non-linear regression.
ECG intervals in pigs
ECGs were monitored and recorded (Data Translation) in Landrace pigs (20–35 kg, either sex). ECG intervals were measured (in seconds) by averaging five consecutive RR, PR, QRS and QTc intervals in each animal 50 min after an IP dose of 8 mg·kg−1 riluzole (n = 6) or injection of vehicle only (n = 10). This dose of riluzole was also highly effective in reducing ischaemia-induced arrhythmias and infarct size (data not shown).
Results
Riluzole protects against ischaemia-reperfusion damage in isolated hearts
Exposing isolated hearts to 30 min of global ischaemia followed by reperfusion produced a substantial functional deficit in control hearts. In each control heart there was an obvious contracture immediately upon reperfusion, as demonstrated by a pronounced increased in diastolic LVP, with very little or no pulse pressure. This was followed by a moderate recovery of pulse pressure after several minutes, but diastolic pressure failed to return to normal [Figure 1A (i)]. Adding 1 µM riluzole to the perfusate did not substantially alter the observed results. However, adding 3 µM or 10 µM riluzole to the perfusate before inducing global ischaemia virtually eliminated the contracture on reperfusion and maintained the diastolic pressure during the recovery period at pre-ischaemia levels. Moreover, in the presence of 3 or 10 µM riluzole, pulse pressure returned almost immediately upon reperfusion [Figure 1A (ii)] The times taken for pulse pressure to return following reperfusion are shown in Table 1. Although 1 µM produced some reduction in the time needed for pulse pressure to return (not significant), 3 and 10 µM riluzole almost eliminated this recovery time (P < 0.002) (Table 1).
Figure 1.
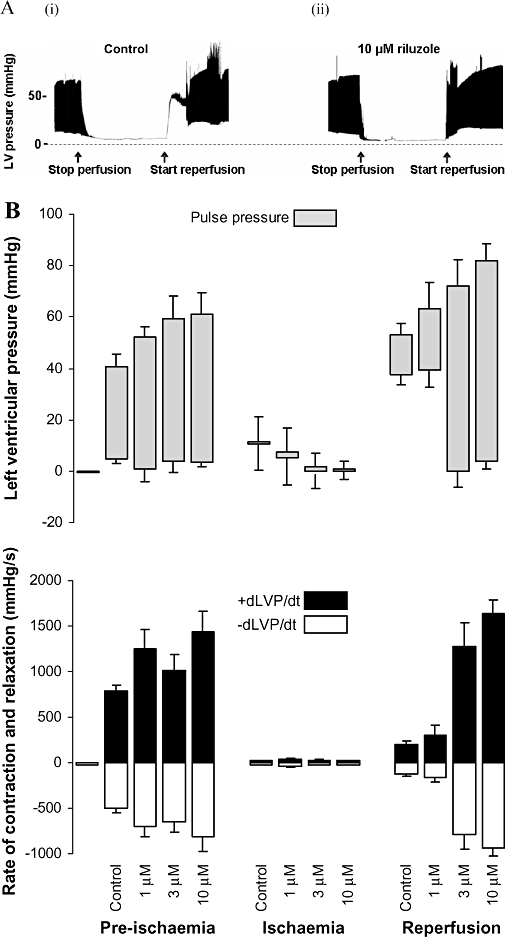
Isolated heart experiments. (A) Recordings of LVP from a rat isolated heart over time. The height of the black band illustrates the excursion from diastolic pressure to systolic pressure (individual beats cannot be seen on the time scale used). Panel (i) shows the response of an untreated heart to global ischaemia and reperfusion. The perfusion pump was stopped for 30 min at the time indicated, and restarted as indicated. Panel (ii) shows an example of the same experiment in which a different heart was perfused with 10 µM riluzole for 10 min prior to stopping the pump. (B) The experiment depicted in A was repeated in 16 hearts (4 with each concentration of riluzole). Diastolic LVP (LVPmin) and systolic LVP (LVPmax) (upper panel) and their first derivatives, rate of relaxation (−dLVP/dt) and rate of contraction (+dLVP/dt) (lower panel) were measured during normal perfusion, after 30 min of global ischaemia (just before reperfusion) and 2 min after reperfusion. Data is shown as mean ± standard error of the mean. Statistical significance is discussed in the text.
Table 1.
Time to resumption of pulse pressure after reperfusion
| Time to return of normal pulse pressure | P value | n | |
|---|---|---|---|
| Control | 8.5 ± 1.4 min | – | 4 |
| 1 µM riluzole | 5.0 ± 1.6 min | P = ns | 4 |
| 3 µM riluzole | 0.4 ± 0.0 min | P < 0.002 | 4 |
| 10 µM riluzole | 0.3 ± 0.1 min | P < 0.002 | 4 |
ns, not significant.
Figure 1B (upper panel) shows comparative LVP results (diastolic, systolic and pulse pressures) for the control, 1, 3 and 10 µM riluzole groups (n = 4 each group). Of particular interest are the changes in diastolic pressures between pre-ischaemia and reperfusion. The rises in diastolic pressure upon reperfusion in both the control and 1 µM groups were highly significant (P << 0.01) (and not significantly different from each other), whereas there were no changes in diastolic pressures for the 3 and 10 µM groups (P = 0.13 and 0.85, respectively). During the ischaemic period, diastolic pressure significantly decreased in the 3 and 10 µM groups as compared with the control group (P = 0.004 and P = 0.006, respectively), whereas with 1 µM riluzole, diastolic pressure was not significantly different (P = 0.13).
The systolic pressures immediately after reperfusion were significantly higher than during pre-ischaemia for each of the control, 1, 3 and 10 µM groups (P < 0.01 for all) (Figure 1B upper panel). Moreover, taking into account the dramatic reduction in diastolic pressure, 3 and 10 µM riluzole resulted in the heart producing a pulse pressure on reperfusion considerably larger than pre-ischaemic values.
This protective effect of riluzole was also evident in the measures of contractility (rate of increase in ventricular pressure, +dLVP/dt) and rate of relaxation (−dLVP/dt) (Figure 1B lower panel). Riluzole at all concentrations increased both rate of contraction and rate of relaxation before ischaemia when compared with control. After reperfusion, the rate of contraction and the rate of relaxation were significantly increased by 3 µM and 10 µM riluzole compared with control.
Riluzole attenuates hypoxic rise in myocyte [Ca2+]i and cell shortening
In the absence of riluzole, exposure of the cells to hypoxia produced an increase in diastolic [Ca2+]i and a reduction in the amplitude of the calcium transient [Figure 2 (iii)]. There was also an associated reduction in cell shortening [Figure 2 (i)]. Upon re-oxygenation of the cells, diastolic [Ca2+]i increased dramatically and the calcium transient and contraction were abolished [Figure 2 (iii) and (i)]. In contrast, in the presence of 3 µM riluzole, diastolic [Ca2+]i did not increase during hypoxia and the increase upon re-oxygenation was greatly attenuated [Figure 2 (iv)]. Furthermore, in the presence of 3 µM riluzole, the calcium transient [Figure 2 (iv)] and the contraction [Figure 2 (ii)] persisted throughout the hypoxia and re-oxygenation.
Figure 2.
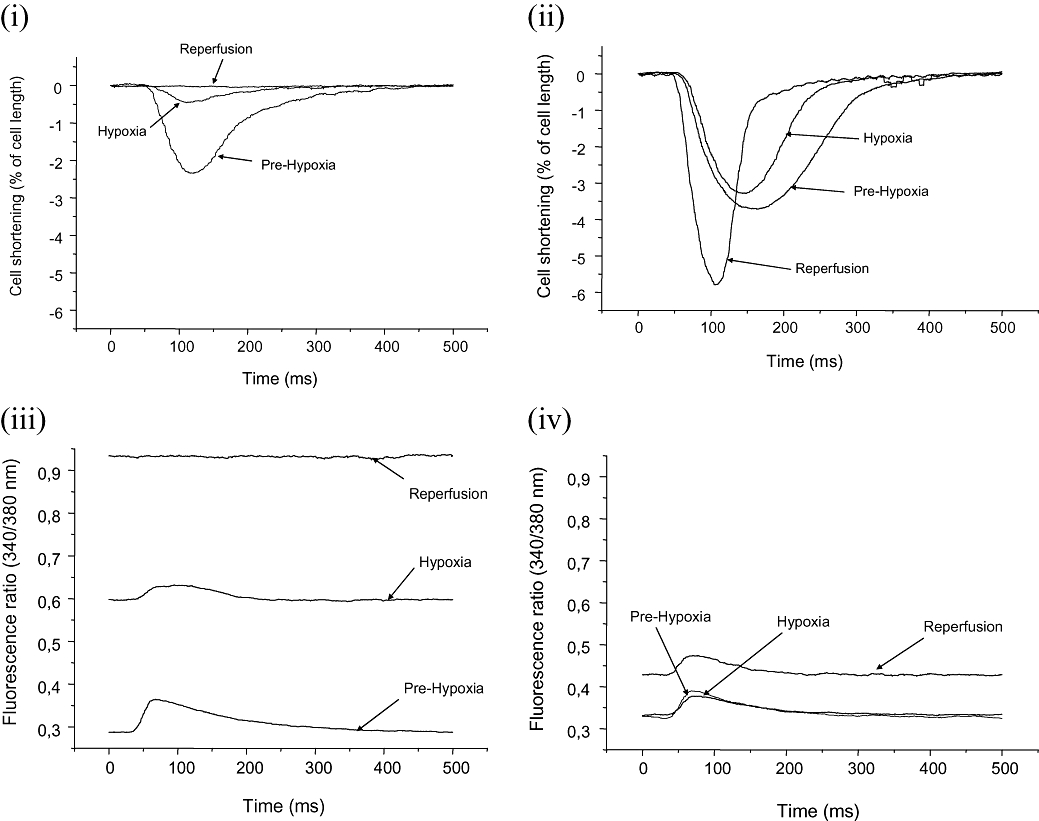
[Ca2+]i and cell shortening measurements. Traces of cell shortening (top) and [Ca2+]i (bottom) in single cells during hypoxia and re-perfusion in control [panels (i) and (iii)] and in the presence of riluzole [3 µM, panels (ii) and (iv)]. The traces were obtained during normoxia, after 20 min of hypoxia and 20 min after reoxygenation (reperfusion).
Compiled time course data showed that riluzole produced a concentration-dependent attenuation of the rise in diastolic [Ca2+]i seen in the control group during hypoxia (such that with 3 and 10 µM, diastolic [Ca2+]i rose very little) and a striking concentration-dependent attenuation of the rise in diastolic [Ca2+]i which occurred in the control group on re-oxygenation (Figure 3A).
Figure 3.
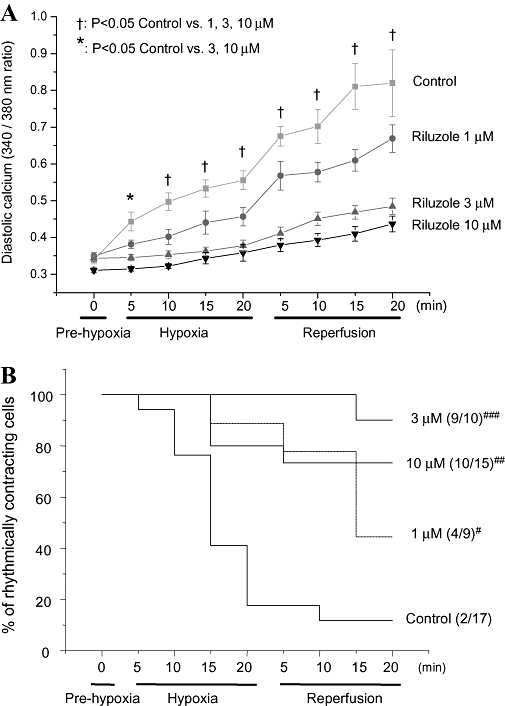
[Ca2+]i and cell shortening-pooled data. (A) Pooled data on the time course of the change in 340/380 fluorescence ratio during the experiment. The x-axis is continuous – the horizontal bars indicate the experimental protocol, with the time in each condition. ‘n’ (number of cells) for each condition and for control, 1, 3 and 10 µM were: Prehypoxia, 17, 9, 10, 15; hypoxia 9, 8, 10, 13; reperfusion 7, 7, 10, 13. Data was analysed with two-way analysis of variance; the symbols indicate the statistical significance as indicated. (B) The proportion of cells rhythmically contracting during the time course of the experiment for each of the concentrations of riluzole. The numbers of cell contracting at 20 min reperfusion was compared using an overall exact chi-squared test (3 d.f.: P < 0.001), then pairwise comparisons between the control and each concentration group using a Fisher exact test (two-sided). Significance is indicated by # (=0.138), ## (=0.003) and ### (<0.001).
Time course data on the proportion of cells contracting at each time point showed that in control solution many cells ceased contracting during hypoxia and only 2/17 cells were still contracting at the end of the reoxygenation period. Riluzole greatly enhanced the number of cells surviving and maintaining contractility; in 1 µM riluzole 4/9 (not significant compared with control) were still contracting after 20 min reoxygenation, in 3 µM 9/10 (P = 0.003 compared with control) were, and in 10 µM 10/15 were (P = 0.001 compared with control) (Figure 3B).
Riluzole block of INaT and INaP: patch clamp data
Block of INaT
Riluzole produced no observable block of peak INaT amplitude at concentrations up to 10 µM. Thirty µM and 100 µM (the highest possible given the solubility of riluzole) produced a small block, of the order of 10% (Figure 4A). Riluzole produced a concentration-dependent positive shift of the activation voltage for INaT (Table 2 and Figure 4B). There was also a concentration-dependent negative shift in the inactivation curve (Table 3 and Figure 4C). Note that no effects on inactivation or activation voltage dependence were apparent at 3 µM, the concentration for 50% block of INaP (see further discussion).
Figure 4.
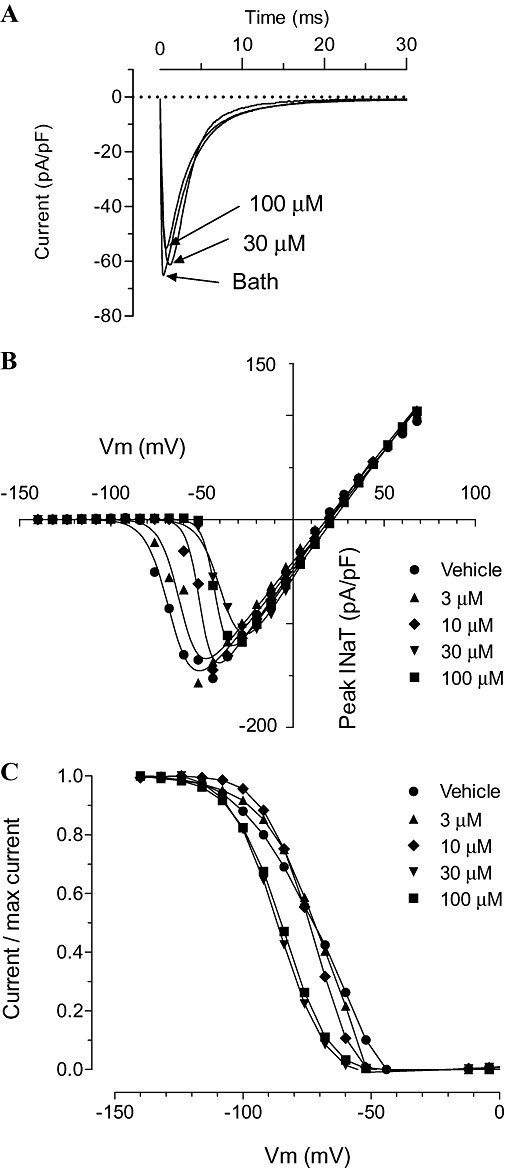
Patch clamp experiments: effects of riluzole on INaT. (A) An example of recordings of INaT in a single cell in control (bath) solutions and in the presence of 30 and 100 µM riluzole. INaT was evoked by a membrane voltage step from −140 to −20 mV lasting 200 ms, at 2 s intervals. (B) Averaged I–V curves for INaT recording in control solution, 3, 10, 30 and 100 µM riluzole. INaT was evoked by a voltage step from −140 mV to a test potential lasting 200 ms, which was incremented from −140 mV to +70 mV at 2 s intervals (n = 4 for each curve). The lines show the best fit of the equation y = (Vm – Erev) * {(Gmax)/[1 + exp(V50 – Vm)/slope)]} The parameters for each of the fits are given in Table 2. (C) Averaged inactivation curves for INaT in control solution, 3, 10, 30 and 100 µM riluzole. INaT was evoked by a step from a holding potential to −20 mV for 200 ms, at 2 s intervals the holding potential being incremented from −140 to 70 mV after 4 iterations at each step (n = 4 for each curve). Data is normalized to the maximum current for each group. Lines show the best fits to the data of the equation: y = 1/[1 + exp(V50 – Vm)/slope] The parameters derived from these fits are shown in Table 3.
Table 2.
Parameters from fits to I–V curves for INaT
| Bath | 3 µM | 10 µM | 30 µM | 100 µM | |
|---|---|---|---|---|---|
| Erev | 20.23 | 18.43 | 19.97 | 20.77 | 22.06 |
| Gmax | 2.22 | 2.20 | 2.43 | 2.69 | 2.35 |
| V50 | −66.44 | −61.02 | −50.65 | −37.67 | −42.26 |
| Slope | 6.4 | 5.5 | 3.7 | 6.1 | 3.5 |
| Standard error | |||||
| Erev | 0.845 | 1.125 | 0.743 | 3.054 | 0.356 |
| Gmax | 0.054 | 0.073 | 0.062 | 0.029 | 0.028 |
| V50 | 0.795 | 1.037 | 0.467 | 1.092 | 0.294 |
| Slope | 0.629 | 0.836 | 0.406 | 0.596 | 0.257 |
| Goodness of fit | |||||
| R2 | 0.9908 | 0.9816 | 0.9997 | 0.9904 | 0.9977 |
Equation fitted: y = (Vm – Erev) * [(Gmax)/(1 + exp(V50 – Vm)/slope] where Vm, membrane potential; V50, membrane potential at which activation is 50% maximum; Gmax, maximum conductance; Erev, reversal potential for Na+.
Table 3.
Inactivation parameters for INaT
| Bath | 3 µM | 10 µM | 30 µM | 100 µM | |
|---|---|---|---|---|---|
| V50 | −73.41 | −72.51 | −74.73 | −87.1 | −85.37 |
| Slope | 11.4 | 8.9 | 7.5 | 8.1 | 8.7 |
| Standard error | |||||
| V50 | 0.737 | 0.594 | 0.388 | 0.501 | 0.229 |
| Slope | 0.64 | 0.5158 | 0.3375 | 0.4293 | 0.1979 |
| Goodness of fit | |||||
| R2 | 0.997 | 0.9973 | 0.9987 | 0.9978 | 0.9996 |
Equation fitted: y = 1/(1 + exp(V50 – Vm)/slope), where Vm, membrane potential; V50, membrane potential at which activation is 50% maximum.
Block of INaP
Consistent with previously reported results (Ju et al., 1996b), INaP was greatly enhanced by exposure of the myocytes to hypoxia. An example for one cell is shown in Figure 5A, in which exposure to hypoxia increased INaP from −47 to −157 pA. Figure 5A also shows that exposure of the myocytes under hypoxic conditions to the highest concentration of vehicle (PEG) used in the experiments produced no perceptible change in the current.
Figure 5.
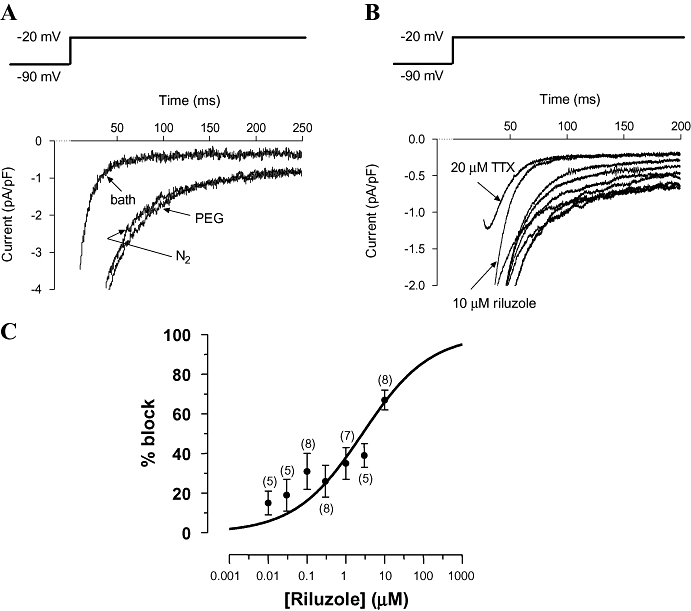
Patch clamp experiments: effects of riluzole on INaP. (A) High gain recording of the sodium current evoked by a membrane voltage step from −90 to −20 mV for 250 ms, at 2 s intervals. The current peaks (INaT) are truncated by the amplifier. Currents are shown in one cell while perfused with normoxic bath solution, hypoxic bath solution and hypoxic bath solution containing vehicle (propylene glycol) equivalent to that used to solubilize 100 µM riluzole (cell capacitance = 167 pF). (B) High gain recording of the sodium current evoked as in A in a cell perfused with hypoxic bath solution with vehicle alone, or riluzole at a concentration of 0.01, 0.03, 0.1, 0.3, 1.0, 3.0 and 10 µM (traces from bottom to top, respectively, 10 µM trace labelled). The topmost trace was recorded in the presence of 20 µM TTX (labelled) (cell capacitance = 287 pF). (C) Compiled data for the block of INaP evoked as in A. Data points show mean and standard error of the mean for % block of INaP in hypoxic solution at each concentration of riluzole; numbers in brackets indicate the number of rats (n) for each data point (multiple cell recordings from a single rat were averaged and counted as one recording). The line shows the best fit of the Hill equation with the Hill coefficient fixed at 0.5: IC50 was 2.7 µM (95% confidence interval 1.0 to 7.1 µM). [note that 100 µM (75% block) was the limit of solubility of riluzole, so a maximum block could not be determined].
Riluzole blocked the hypoxia-enhanced INaP in a concentration-dependent manner over the concentration range 0.1 to 10 µM. An example is shown for one cell in Figure 5B. In this example, 10 µM riluzole produced a similar degree of block of INaP as did 20 µM TTX. In contrast, the block of INaT was much less with 10 µM riluzole than with 20 µM TTX.
Compiled data on the block of INaP by concentrations of riluzole over the range 0.01 µM to 10 µM were fitted with a concentration-response curve with a Hill coefficient of 0.5 and an IC50 of 2.7 µM (95% confidence interval 1.0 to 7.1 µM) (Figure 5C). As a comparison, the relative block required to negate the effects of hypoxia (i.e. to reduce INaP to its normoxic value) was about 38% on Figure 5C, corresponding to a riluzole concentration of between 1 and 3 µM.
Other electrophysiological effects: rat isolated heart MAP and ECG intervals in pigs
In normoxic rat isolated hearts (n = 6), the duration of epicardial MAPs at APD20, APD50 and APD80 was not significantly altered by addition of riluzole at concentrations of 3, 10 and 30 µM to the perfusate (Figure 6). In pigs, riluzole produced no change in ECG intervals 50 min after a dose of 8 mg·kg−1 IP (Table 4, n = 6 for riluzole group, n = 10 for control)
Figure 6.
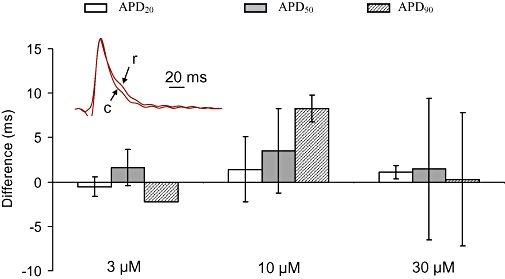
Effect of riluzole on monophasic action potentials (MAPs) in rat normoxic heart. Change in the duration of epicardial MAPs induced in normoxic isolated hearts (n = 6) by riluzole at 3, 10 and 30 µM, compared with control (zero riluzole). Bars show change (as a percentage of control value) in APD20, APD50 and APD80. Error bars show standard error of the mean. None of the changes were significant. The inset shows superimposed examples of MAPs from one heart recorded before (arrowed ‘c’) and during perfusion with 30 µM riluzole (arrowed ‘r’).
Table 4.
Effect of riluzole (8 mg·kg−1) on ECG intervals in conscious pigs
|
R–R |
P–R |
QRS |
QTc |
|||||
|---|---|---|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | |
| Control | 0.824 | 0.065 | 0.119 | 0.012 | 0.067 | 0.004 | 0.584 | 0.022 |
| Riluzole | 0.725 | 0.052 | 0.112 | 0.005 | 0.068 | 0.002 | 0.541 | 0.007 |
| P | 0.259 | 0.568 | 0.853 | 0.103 | ||||
SEM, standard error of the mean.
Discussion
There is now a considerable body of evidence to show that the key event in the sequence triggered by hypoxia or ischaemia in the heart is a rise in [Na+]i (Shryock and Belardinelli, 2008). The mechanisms underlying this rise in [Na+]i are not clear. As noted above, one well described mechanism, the inhibition of NHE1 during ischaemia and the subsequent large H+ extrusion and Na+ influx during reperfusion, falls short of fully explaining the rise in Na+ and the ensuing cellular damage from ischaemia and reperfusion. Studies have shown that the rise in [Na+]i during ischaemia can be attenuated by blockers of NHE1, such as cariporide: (Ten Hove et al., 2005). However, in animal models, block of NHE does not completely prevent the rise in Na+ or provide complete protection from damage, a result reflected in the disappointing results with NHE1 blockers in clinical trials (Theroux et al., 2000; Zeymer et al., 2001). Animal experiments have also shown that reduction in [Na+]i and protection from reperfusion damage can equally and synergistically be provided by blocking voltage-dependent sodium channels (Eigel and Hadley, 1999; ten Hove et al., 2007).
As the transient, voltage-dependent, sodium channels rapidly inactivate, they cannot be the source of a sustained Na+ influx during ischaemia or reperfusion. Rather, the influx appears to occur through a population of Na+ channels that do not inactivate, and which give rise to a ‘persistent’ sodium current, INaP (Saint et al., 1992; Baetz et al., 2003). This current has been shown to be greatly enhanced by hypoxia (Ju et al., 1996b; Hammarstrom and Gage, 2002) and it would seem that block of this current should provide protection against ischaemia/reperfusion damage. Indeed, over 10 years ago, Le Grand et al. showed that low concentrations of TTX (320 nM, a concentration that produces a relatively selective block of the INaP over INaT) can alleviate the contractile dysfunction caused by ischaemia in guinea pig isolated heart (Le Grand et al., 1995). More recently, the potential for agents that block INaP as a cardioprotective agents has also been discussed by others (Belardinelli et al., 2006; Saint, 2008). A working hypothesis that has been proposed, on the basis of the effects of TTX, amiloride and zoniporide, is that the Na+ influx during ischaemia is predominantly caused by flux through INaP, while the large rise in [Na+]i upon reperfusion is caused by the activity of NHE1. Hence, block of INaP should present a useful strategy to protect against ischaemic damage, and at least one agent, ranolazine, is in late development with this proposed mechanism of action (Belardinelli et al., 2006), although ranolazine also has other possible mechanisms of action.
This lack of selective blockers has presented difficulties in exploiting block of INaP as a strategy. Some agents do exhibit some selectivity, such as lidocaine and TTX (Ju et al., 1992; Ju et al., 1994) and ranolazine (Antzelevitch et al., 2004), however, the difference in potency for block of INaP and INaT with these agents is small and the block of INaT is potentially lethal. Although newer agents are being developed to have an increased selectivity for block of INaP (Vacher et al., 2009; Vie et al., 2009), it is difficult to develop these agents because the molecular mechanisms producing INaP are not well understood. The cardiac isoform of (transient) voltage-gated sodium channels (Nav1.5) is quite different from the neuronal (Nav1.1, Nav1.2, Nav1.3, Nav1.6, Nav1.7, Nav1.8 and Nav1.9) and skeletal muscle (Nav1.4) isoforms, and the cardiac isoform displays a quite different pharmacological profile from the others (channel nomenclature follows Alexander et al., 2009). In parallel with this, it is not known whether INaP in other tissues has the same pharmacology as that seen in the heart.
There is data to show that, in the heart, INaP has a different pharmacological profile from INaT (Ju et al., 1992; Ju et al., 1996a), a difference that argues for it being caused by a distinct, as yet unknown, sodium channel isoform. However, a different conclusion arises from the observation that expression of human cardiac α-subunit of the Na+ channel (hH1) in HEK293 cells results in a sodium current having both transient and persistent components, the persistent component being enhanced by hypoxia (Hammarstrom and Gage, 2002). This suggests that INaP arises from a subset of the (homogeneous) channel population entering a different gating state, with the enhancement of INaP by hypoxia being caused by modulation of channel gating. In this case, the apparent different pharmacology of the channel must arise from the kinetics of block of the channel in different gating states. The difficulty in finding a blocker of cardiac INaP is therefore compounded not only by finding a blocker which is selective for INaP over INaT, but one which is also selective for the cardiac isoform.
We report here that riluzole, which has been suggested to block neuronal INaP, does indeed block INaP in cardiac myocytes. Moreover, riluzole blocks INaP in a concentration-dependent manner between 0.01 and 10 µM, these concentrations being similar to those used for neuronal treatment and already having been shown to not be harmful in humans (Le Liboux et al., 1999). We also show that riluzole has minimal effect on INaT even at 10-fold higher concentrations than those required to block INaP.
It has been suggested that small, non-inactivating currents can arise because of the overlap of the inactivation and activation voltage dependence- giving rise to ‘window currents’ at certain membrane potentials. We showed here that riluzole shifts the voltage dependence of activation and inactivation of INaT in ways that reduce the overlap of the inactivation and activation curves of INaT, which would tend to reduce a window current, consistent with a block of INaP. However, this occurred only at higher concentrations- no shift was seen at 3 µM, which produces a substantial block of INaP. Furthermore, it has been shown previously that INaP is not a window current (Saint et al., 1992). In our experiments reported here, we elicited INaP by a voltage step to −20 mV, well beyond the region where overlap of inactivation and activation curves would be expected to produce a window current.
In the isolated heart, 3 and 10 µM riluzole offered substantial cardioprotection by significantly reducing the degree of contracture during ischaemia as well as the degree of contracture and the time to the return of pulse pressure upon reperfusion. Riluzole also reduced the diastolic pressure and increased the systolic pressure post reperfusion thus significantly and beneficially improving myocardial recovery. In addition, riluzole increased the rate of ventricular relaxation and the rate of ventricular contractility thus further enhancing myocardial recovery. Of note was the observation that riluzole produced positive intropic and lusitropic effects when added to the heart before ischaemia; the systolic pressures, +dLVP/dt and –dLVP/dt, were significantly increased for all concentrations (Figure 1B– pre-ischaemia values). The inotropic effect was also apparent in the isolated cell experiments (Figure 2), where an increase in contractility appeared to occur without an obvious increase in the calcium transient. As we have shown that riluzole did not affect the MAP, this suggests that perhaps it can act to sensitize the contractile proteins to calcium. However, the lack of correlation between this effect and the protective effect (e.g. at 1 µM there is an obvious inotropic effect, but no significant protection) indicates that it is not responsible for the protective effect. We have not investigated this inotropic and lusitropic effect further in this study.
We found that riluzole blocked the hypoxic component of INaP without affecting INaT, prevented [Ca2+]i accumulation and the cessation of cell shortening in response to hypoxia and re-oxygenation, and provided substantial cardioprotection from ischaemia and ischaemia-reperfusion, all with a similar IC50 of around 3 µM. We therefore propose that riluzole may be an effective anti-ischaemic and cardioprotective medication for use in myocardial hypoxia, ischaemia and ischaemia-reperfusion settings.
Limitations of study
Because of the difficulty in ensuring good pH control in the isolated cell experiments, because of the technical difficulty in bubbling the solutions while on the microscope stage and the movement artefacts so produced, experiments in the isolated cells were carried out in HEPES-buffered solutions. Hence, the bicarbonate-Na+ co-transporter would not be active in these cells, and this would be expected to reduce the sodium influx into the cells during the period of intracellular acidosis. This would tend to reduce the severity of ischaemic damage in these cells. These experiments also used hypoxia, rather than ischaemia, the latter not being appropriate to isolated cell measurements. Hence, the composition of the ‘extracellular’ (bath) solution will not change materially during the experiment, whereas in isolated heart during stop flow, leakage of ions and metabolites from the cells can lead to changes in extracellular composition. Nevertheless, the isolated cells showed large fluctuations in [Ca2+]i and a contractile deficit in hypoxia and ischaemia, and riluzole was effective in reducing both, consistent with results from the isolated hearts.
Potential therapeutic applications
Given that riluzole shows some degree of selectivity for block of INaP over INaT, it may have promise as an anti-ischaemic agent in clinical applications. Obvious applications would be post-myocardial infarction, at reperfusion, or as a protective agent in cardioplegia situations. As riluzole is already approved for use in humans, one can surmise that toxicity (at least acute toxicity) at the normal clinical dose of 100 mg·day−1 will not present a problem. Cardiovascular side effects are very rare at the clinically approved doses (Groeneveld et al., 2003) and the fact that we see no change in the MAP in rat hearts and no change in ECG intervals in pigs reinforces this lack of cardiac adverse effects. However, it should be noted that riluzole blocks INaP in many types of neurones, and obviously crosses the blood brain barrier, so it is possible that neuronal effects may limit the dose if it is used in a cardiac setting. Rilutek prescribing information also notes: ‘Due to its blockade of glutamatergic neurotransmission, riluzole also exhibits myorelaxant and sedative properties in animal models at doses of 30 mg·kg−1 (about 20 times the recommended human daily dose) and anticonvulsant properties at a dose of 2.5 mg·kg−1 (about 2 times the recommended human daily dose)’. Hence it seems possible that riluzole may present a novel opportunity as a cardioprotective agent. In addition, the data suggests that development of more specific blockers of INaP should be pursued with a view to improving this potential novel class of therapeutic agents.
Glossary
Abbreviations:
- dLVP/dt
deriviative of left ventricular pressure with respect to time
- INaP
persistent sodium current
- INaT
transient sodium current;
- LVEPD
left ventricular end-diastolic pressure
- LVP
left ventricular pressure
- NCX
sodium-calcium exchanger
- NHE1
sodium-hydrogen exchanger (isoform 1)
Conflict of interest
David Saint and Steven Weiss are shareholders in Neoviva Pty. Ltd., an Australian company pursuing the development of cardioprotective therapies.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C, Belardinelli L, Zygmunt AC, Burashnikov A, Di Diego JM, Fish JM, et al. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation. 2004;110:904–910. doi: 10.1161/01.CIR.0000139333.83620.5D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ates O, Cayli SR, Gurses I, Turkoz Y, Tarim O, Cakir CO, et al. Comparative neuroprotective effect of sodium channel blockers after experimental spinal cord injury. J Clin Neurosci. 2007;14:658–665. doi: 10.1016/j.jocn.2006.03.023. [DOI] [PubMed] [Google Scholar]
- Bae HJ, Lee YS, Kang DW, Koo JS, Yoon BW, Roh JK. Neuroprotective effect of low dose riluzole in gerbil model of transient global ischemia. Neurosci Lett. 2000;294:29–32. doi: 10.1016/s0304-3940(00)01536-6. [DOI] [PubMed] [Google Scholar]
- Baetz D, Bernard M, Pinet C, Tamareille S, Chattou S, El Banani H, et al. Different pathways for sodium entry in cardiac cells during ischemia and early reperfusion. Mol Cell Biochem. 2003;242:115–120. [PubMed] [Google Scholar]
- Baptiste DC, Fehlings MG. Pharmacological approaches to repair the injured spinal cord. J Neurotrauma. 2006;23:318–334. doi: 10.1089/neu.2006.23.318. [DOI] [PubMed] [Google Scholar]
- Belardinelli L, Shryock JC, Fraser H. Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart. 2006;92(Suppl 4):iv6–iv14. doi: 10.1136/hrt.2005.078790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryson HM, Fulton B, Benfield P. Riluzole. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in amyotrophic lateral sclerosis. Drugs. 1996;52:549–563. doi: 10.2165/00003495-199652040-00010. [DOI] [PubMed] [Google Scholar]
- Doble A. The pharmacology and mechanism of action of riluzole. Neurology. 1996;47:S233–S241. doi: 10.1212/wnl.47.6_suppl_4.233s. [DOI] [PubMed] [Google Scholar]
- Duprat F, Lesage F, Patel AJ, Fink M, Romey G, Lazdunski M. The neuroprotective agent riluzole activates the two P domain K(+) channels TREK-1 and TRAAK. Mol Pharmacol. 2000;57:906–912. [PubMed] [Google Scholar]
- Eberli FR, Stromer H, Ferrell MA, Varma N, Morgan JP, Neubauer S, et al. Lack of direct role for calcium in ischemic diastolic dysfunction in isolated hearts. Circulation. 2000;102:2643–2649. doi: 10.1161/01.cir.102.21.2643. [DOI] [PubMed] [Google Scholar]
- Eigel BN, Hadley RW. Contribution of the Na(+) channel and Na(+)/H(+) exchanger to the anoxic rise of [Na(+)] in ventricular myocytes. Am J Physiol. 1999;277:H1817–H1822. doi: 10.1152/ajpheart.1999.277.5.H1817. [DOI] [PubMed] [Google Scholar]
- Groeneveld GJ, Van Kan HJ, Kalmijn S, Veldink JH, Guchelaar HJ, Wokke JH, et al. Riluzole serum concentrations in patients with ALS: associations with side effects and symptoms. Neurology. 2003;61:1141–1143. doi: 10.1212/01.wnl.0000090459.76784.49. [DOI] [PubMed] [Google Scholar]
- Hammarstrom AK, Gage PW. Hypoxia and persistent sodium current. Eur Biophys J. 2002;31:323–330. doi: 10.1007/s00249-002-0218-2. [DOI] [PubMed] [Google Scholar]
- Hartmann M, Decking UK. Blocking Na(+)-H+ exchange by cariporide reduces Na(+)-overload in ischemia and is cardioprotective. J Mol Cell Cardiol. 1999;31:1985–1995. doi: 10.1006/jmcc.1999.1029. [DOI] [PubMed] [Google Scholar]
- Heurteaux C, Laigle C, Blondeau N, Jarretou G, Lazdunski M. Alpha-linolenic acid and riluzole treatment confer cerebral protection and improve survival after focal brain ischemia. Neuroscience. 2006;137:241–251. doi: 10.1016/j.neuroscience.2005.08.083. [DOI] [PubMed] [Google Scholar]
- ten Hove M, Jansen MA, Nederhoff MG, Van Echteld CJ. Combined blockade of the Na+ channel and the Na+/H+ exchanger virtually prevents ischemic Na+ overload in rat hearts. Mol Cell Biochem. 2007;297:101–110. doi: 10.1007/s11010-006-9334-0. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Hammerman SB, Kirby CO, Benz AM, Olney JW, Zorumski CF. Involvement of glutamate in ischemic neurodegeneration in isolated retina. Vis Neurosci. 2003;20:97–107. doi: 10.1017/s0952523803202017. [DOI] [PubMed] [Google Scholar]
- Ju Y, Gage PW, Saint DA. Tetrodotoxin-sensitive inactivation-resistant sodium channels in pacemaker cells influence heart rate. Pflugers Arch. 1996a;431:868–875. doi: 10.1007/s004240050079. [DOI] [PubMed] [Google Scholar]
- Ju YK, Saint DA, Gage PW. Effects of lignocaine and quinidine on the persistent sodium current in rat ventricular myocytes. Br J Pharmacol. 1992;107:311–316. doi: 10.1111/j.1476-5381.1992.tb12743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju YK, Saint DA, Gage PW. Inactivation-resistant channels underlying the persistent sodium current in rat ventricular myocytes. Proc Biol Sci. 1994;256:163–168. doi: 10.1098/rspb.1994.0065. [DOI] [PubMed] [Google Scholar]
- Ju YK, Saint DA, Gage PW. Hypoxia increases persistent sodium current in rat ventricular myocytes. J Physiol. 1996b;497(Pt 2):337–347. doi: 10.1113/jphysiol.1996.sp021772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamanauskas N, Nistri A. Riluzole blocks persistent Na+ and Ca2+ currents and modulates release of glutamate via presynaptic NMDA receptors on neonatal rat hypoglossal motoneurons in vitro. Eur J Neurosci. 2008;27:2501–2514. doi: 10.1111/j.1460-9568.2008.06211.x. [DOI] [PubMed] [Google Scholar]
- Lazdunski M, Frelin C, Vigne P. The sodium/hydrogen exchange system in cardiac cells: its biochemical and pharmacological properties and its role in regulating internal concentrations of sodium and internal pH. J Mol Cell Cardiol. 1985;17:1029–1042. doi: 10.1016/s0022-2828(85)80119-x. [DOI] [PubMed] [Google Scholar]
- Le Grand B, Vie B, Talmant JM, Coraboeuf E, John GW. Alleviation of contractile dysfunction in ischemic hearts by slowly inactivating Na+ current blockers. Am J Physiol. 1995;269:H533–H540. doi: 10.1152/ajpheart.1995.269.2.H533. [DOI] [PubMed] [Google Scholar]
- Le Liboux A, Cachia JP, Kirkesseli S, Gautier JY, Guimart C, Montay G, et al. A comparison of the pharmacokinetics and tolerability of riluzole after repeat dose administration in healthy elderly and young volunteers. J Clin Pharmacol. 1999;39:480–486. [PubMed] [Google Scholar]
- McCrossan ZA, Billeter R, White E. Transmural changes in size, contractile and electrical properties of SHR left ventricular myocytes during compensated hypertrophy. Cardiovasc Res. 2004;63:283–292. doi: 10.1016/j.cardiores.2004.04.013. [DOI] [PubMed] [Google Scholar]
- Meissner A, Morgan JP. Contractile dysfunction and abnormal Ca2+ modulation during postischemic reperfusion in rat heart. Am J Physiol. 1995;268:H100–H111. doi: 10.1152/ajpheart.1995.268.1.H100. [DOI] [PubMed] [Google Scholar]
- Murphy E, Allen DG. Why did the NHE inhibitor clinical trials fail? J Mol Cell Cardiol. 2009;46:137–141. doi: 10.1016/j.yjmcc.2008.09.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyclit A, Keita H, Juvin P, Derkinderen P, Jardinaud F, Rouelle D, et al. Effects of riluzole on N-methyl-D-aspartate-induced tyrosine phosphorylation in the rat hippocampus. Brain Res. 2001;903:222–225. doi: 10.1016/s0006-8993(01)02429-5. [DOI] [PubMed] [Google Scholar]
- Saini HK, Dhalla NS. Defective calcium handling in cardiomyocytes isolated from hearts subjected to ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2005;288:H2260–H2270. doi: 10.1152/ajpheart.01153.2004. [DOI] [PubMed] [Google Scholar]
- Saint DA. The cardiac persistent sodium current: an appealing therapeutic target? Br J Pharmacol. 2008;153:1133–1142. doi: 10.1038/sj.bjp.0707492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint DA, Ju YK, Gage PW. A persistent sodium current in rat ventricular myocytes. J Physiol. 1992;453:219–231. doi: 10.1113/jphysiol.1992.sp019225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shryock JC, Belardinelli L. Inhibition of late sodium current to reduce electrical and mechanical dysfunction of ischaemic myocardium. Br J Pharmacol. 2008;153:1128–1132. doi: 10.1038/sj.bjp.0707522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten Hove M, Nederhoff MG, Van Echteld CJ. Relative contributions of Na+/H+ exchange and Na+/HCO3- cotransport to ischemic Nai+ overload in isolated rat hearts. Am J Physiol Heart Circ Physiol. 2005;288:H287–H292. doi: 10.1152/ajpheart.01102.2003. [DOI] [PubMed] [Google Scholar]
- Theroux P, Chaitman BR, Danchin N, Erhardt L, Meinertz T, Schroeder JS, et al. Inhibition of the sodium-hydrogen exchanger with cariporide to prevent myocardial infarction in high-risk ischemic situations. Main results of the GUARDIAN trial. Guard during ischemia against necrosis (GUARDIAN) Investigators. Circulation. 2000;102:3032–3038. doi: 10.1161/01.cir.102.25.3032. [DOI] [PubMed] [Google Scholar]
- Vacher B, Pignier C, Letienne R, Verscheure Y, Le Grand B. F 15845 inhibits persistent sodium current in the heart and prevents angina in animal models. Br J Pharmacol. 2009;156:214–225. doi: 10.1111/j.1476-5381.2008.00062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vie B, Sablayrolles S, Letienne R, Vacher B, Darmellah A, Bernard M, Feuvray D, Le Grand B. 3-(R)-[3-(2-methoxyphenylthio-2-(S)-methylpropyl]amino-3,4-dihydro-2H-1,5- benzoxathiepine bromhydrate (F 15845) prevents ischemia-induced heart remodeling by reduction of the intracellular Na+ overload. J Pharmacol Exp Ther. 2009;330:696–703. doi: 10.1124/jpet.109.153122. [DOI] [PubMed] [Google Scholar]
- Xiao XH, Allen DG. Activity of the Na(+)/H(+) exchanger is critical to reperfusion damage and preconditioning in the isolated rat heart. Cardiovasc Res. 2000;48:244–253. doi: 10.1016/s0008-6363(00)00166-8. [DOI] [PubMed] [Google Scholar]
- Zeymer U, Suryapranata H, Monassier JP, Opolski G, Davies J, Rasmanis G, et al. The Na(+)/H(+) exchange inhibitor eniporide as an adjunct to early reperfusion therapy for acute myocardial infarction. Results of the evaluation of the safety and cardioprotective effects of eniporide in acute myocardial infarction (ESCAMI) trial. J Am Coll Cardiol. 2001;38:1644–1650. doi: 10.1016/s0735-1097(01)01608-4. [DOI] [PubMed] [Google Scholar]