

Abstract
Background and purpose:
Lipid rafts and caveolae are membrane microdomains with important roles in cell survival signalling involving the Akt pathway. Cholesterol is important for the structure and function of these microdomains. The ginsenoside Rh2 exhibits anti-tumour activity. Because Rh2 is structurally similar to cholesterol, we investigated the possibility that Rh2 exerted its anti-tumour effect by modulating rafts and caveolae.
Experimental approach:
A431 cells (human epidermoid carcinoma cell line) were treated with Rh2 and the effects on cell apoptosis, raft localization and Akt activation measured. We also examined the effects of over-expression of Akt and active-Akt on Rh2-induced cell death.
Key results:
Rh2 induced apoptosis concentration- and time-dependently. Rh2 reduced the levels of rafts and caveolae in the plasma membrane and increased their internalization. Furthermore, Akt activity was decreased and consequently, Akt-dependent phosphorylation of Bad, a pro-survival protein, was decreased whereas the pro-apoptotic proteins, Bim and Bax, were increased upon Rh2 treatment. Unlike microdomain internalization induce by cholesterol depletion, Rh2-mediated internalization of rafts and caveolae was not reversed by cholesterol addition. Also, cholesterol addition did not restore Akt activation or rescue cells from Rh2-induced cell death. Rh2-induced cell death was attenuated in MDA-MB-231 cells over-expressing either wild-type or dominant-active Akt.
Conclusions and implications:
Rh2 induced internalization of rafts and caveolae, leading to Akt inactivation, and ultimately apoptosis. Because elevated levels of membrane rafts and caveolae, and Akt activation have been correlated with cancer development, internalization of these microdomains by Rh2 could potentially be used as an anti-cancer therapy.
Keywords: lipid rafts and caveolae, cholesterol, apoptosis, Akt, ginsenosides
Introduction
Lipid rafts are heterogeneous, highly dynamic, cholesterol- and sphingolipid-enriched microdomains of the plasma membranes (Jacobson et al., 2007). Caveolae, a subclass of lipid rafts, are characterized by flask-like invaginations of the plasma membrane that are distinguished from bulk lipid rafts by the presence of caveolin-1 (Patra, 2008). Rafts and caveolae contain various signalling molecules, such as receptors for insulin and epidermal growth factor (EGF), Ras, FAS/CD95 and Wnt, and thus their integrity is critical for cellular signalling to regulate cell proliferation, motility, and survival (Hancock, 2006; Patra, 2008). Rafts exist in a tightly packed liquid-ordered (Lo) state. Serving as a spacer between the hydrocarbon chains of sphingolipids, cholesterol is essential for maintaining this Lo structure of the rafts and caveolae (Hancock, 2006; Jacobson et al., 2007). Therefore, the depletion of cholesterol from the plasma membrane causes disruption of rafts and caveolae, resulting in the release of their constituents into a non-raft/caveolae membrane and leading to inappropriate cellular signalling events such as Akt inactivation, and thus deregulating cellular functions (Li et al., 2006; Patra, 2008).
Akt, also known as protein kinase B, is a serine/threonine kinase and its activation is known to block apoptosis by regulating pro- and anti-apoptotic molecules. For example, whereas some, such as glycogen synthase kinase-3β, caspase 9 and Bad are inactivated, other pro-apoptotic molecules, such as Bim and Fas ligand, are down-regulated. Akt also promotes cell survival and growth by potentiating NF-κB activity and the mammalian target of rapamycin (mTOR) (Franke, 2008). Because the phosphatidylinositol 3 kinase (PI3K)/Akt signalling pathway mediates cell proliferation, survival and motility, Akt is implicated in the oncogenesis of many cancers. Akt is activated by moving to the plasma membrane where it is phosphorylated by phosphoinositide-dependent kinases (PDKs) (Franke, 2008). Akt has been reported to partition into rafts and caveolae (Adam et al., 2007), which play a crucial role in triggering the PI3K/Akt signalling pathway by facilitating Akt recruitment to the plasma membrane (Lasserre et al., 2008). In addition, data from our laboratory and others have demonstrated that Akt activity and cell survival are dependent on cholesterol content. Lowering cellular cholesterol, either by cholesterol depletion or inhibition of synthesis, results in Akt inactivation and cell death, further indicating that the integrity of rafts and caveolae are critical for cell survival signalling involving Akt activation (Zhuang et al., 2002; Zhuang et al., 2005).
Panax ginseng has been used as a traditional medicine for the treatment of various diseases including cancers. Ginsenosides are the major pharmacologically active components of ginseng and exhibit various biological effects such as anti-inflammatory and anti-cancer effects (Yue et al., 2007). Rh2 is a ginsenoside (Figure 1) with anti-tumour effects, inhibiting cell proliferation and inducing apoptosis in many human tumour cells (Attele et al., 1999). This compound can arrest cell cycle at G1 phase by modulating cell cycle-related proteins and induce cell death through activation of death-associated signalling pathways, such as JNK and caspase as well as promote death receptor expression (Cheng et al., 2005; Ham et al., 2006). Interestingly, with their dammarane skeleton and ability to be incorporated into the plasma membrane, ginsenosides are structurally similar to cholesterol and its derivatives (Yue et al., 2007). Therefore, it is possible that the anti-tumour effect of Rh2 could be associated with alterations in rafts and caveolae.
Figure 1.
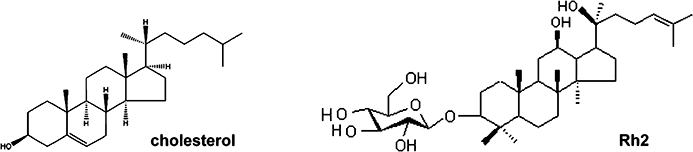
Structures of cholesterol and Rh2.
Removal of cholesterol from the membrane using methyl-β cyclodextrin (MβCD) and/or inhibition of cholesterol biosynthesis causes loss or structural disruption of rafts and caveolae on the cell surface (Li et al., 2006; Park et al., 2009). In addition, natural sterols such as plant and fungal sterols promote the formation of tightly packed, ordered lipid domains by lipids with saturated acyl chains (Tarahovsky et al., 2008). Given that cholesterol is an essential lipid component of rafts and caveolae, involved in Akt activation (Li et al., 2006; Adam et al., 2007), and that phytosterols affect formation of rafts and caveolae (Tarahovsky et al., 2008), the present study investigated whether Rh2, a ginsenoside and a phytosterol, exerted anti-tumour effects through its effect on rafts and caveolae. In this study, we have demonstrated that Rh2 induced lipid raft internalization that was not restored by cholesterol. Moreover, Rh2 caused Akt inactivation, which is critical for cell death promoted by the compound.
Methods
Cell culture
The human epidermoid carcinoma cell line, A431, human breast cancer cell line, MDA-MB-231, human prostate cancer cell line, PC-3, and human embryonic kidney cell line, HEK293 were obtained from the American Type Culture Collection (Rockville, MD, USA). Dulbecco's modified Eagle's medium (DMEM) and RPMI 1640 with L-glutamine, fetal bovine serum (FBS) and antibiotic-antimycotic (100×) were purchased from Gibco Laboratories (Grand Island, NY, USA). A431 and HEK293 cells were grown at 37°C in DMEM supplemented with 10% FBS and antibiotic-antimycotic (1×). MDA-MB-231, PC3 cells were grown in RPMI 1640 containing 10% FBS and antibiotic-antimycotic (1×). The cells were grown to approximately 70% confluence and were then serum-starved for 4 h using DMEM or RPMI containing 0.1% bovine serum albumin (BSA) prior to treatment. Cells were treated with indicated concentrations of reagents in the DMEM or RPMI containing 0.1% BSA.
Rh2 treatment
A431 cells were plated at 5 × 103 cells per 100 µL of culture medium containing 10% serum in 96-well microtiter plate, or 5 × 105 cells per 2 mL of culture medium in 60-mm tissue culture plate. The next day, cells were serum-starved in the medium containing 0.1% BSA for 4 h, and then treated with Rh2 in the same medium. Twenty millimolars Rh2 stock solution was prepared in dimethyl sulfoxide (DMSO). DMSO treatment was used as a control.
Over-expression of wild type-Akt and dominant-active Akt in MDA-MB-231
Constructs for wild type-Akt and dominant-active Akt were a kind gift from Dr Jong-Sun Park (Chung-nam National University, Korea). pLL3.7 was purchased from Addgene Inc. (Cambridge, MA, USA). Its U6 promoter was digested with restriction enzymes XbaI/HpaI and then the corresponding sites were replaced by EF1α promoter/enhancer as described by Invivogen (San Diego, CA, USA) to over-express a gene of interest. The XbaI/BglII fragment digested from either pCMV5.activeAKT or pCMV5.AKT vector was inserted into AvrII/BamHI sites in pLL3.7EF1α vector. The resultant pLL3.7ECMV.AKT or pLL3.7ECMV.activeAKT vectors also co-expressed enhanced green fluorescent protein (EGFP) as a reporter gene. Lentivirus production was done with minor modifications as previously described (Rubinson et al., 2003). In brief, we co-transfected pLL3.7 vector and four packaging vectors into 293FT cells and harvested the resulting supernatant after 48 h. Titers were determined by counting GFP-expressing cells post infection of 293FT cells with serial dilutions of lentivirus.
Cell viability assay
The cells were plated at 5 × 103 cells per 100 µL of culture medium in 96-well microtiter plate, followed by treatment. The effects of the treatments on cell viability were determined with 3-(4,5-dimethylthiazol-2-yl)-5(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay (Promega, Madison, WI, USA) as described in the manufacturer's instruction. Absorbance was measured at 490 nm for MTS assay with an ELISA plate reader from MTX Lab Systems, Inc. (Vienna, VA, USA). Each experiment was performed in triplicate.
Immunoblotting
After washing with ice-cold phosphate-buffered saline (PBS; 10 mM Na2HPO4, pH 7.4, 145 mM NaCl, and 2.7 mM KCl), cells were lysed with 2× sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer [20 mM Tris, pH 8.0, 2% SDS, 2 mM dithiothreitol (DTT), 1 mM Na3VO4, 2 mM ethylenediaminetetraacetic acid (EDTA), 20% glycerol] and boiled for 5 min. Protein concentration of each sample was determined using a Micro-BCA protein assay reagent as described by the manufacturer. In all, 30 µg of total cellular protein was separated by 10% SDS-PAGE and then transferred to polyvinylidene difluoride (PVDF) membranes as described before (Kim et al., 2000). The membranes were blocked overnight at 4°C in 20 mM Tris, pH 8.0, 150 mM NaCl, and 0.05% Tween 20 (TBST) containing either 5% BSA (for immunoblotting with anti-phospho-Akt antibody) or 5% non-fat dried milk (for immunoblotting with other antibodies). The membranes were then incubated with the primary antibody for 1 h at 37°C, washed three times with TBST, incubated with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG or goat anti-rabbit IgG secondary antibodies for 1 h at 37°C, and then washed with TBST three times. The labeled proteins were visualized using the enhanced chemi-luminescence method. In the instances where the same membrane was reprobed with a different primary antibody, the membrane was incubated in a stripping buffer (62.5 mM Tris, pH 6.8, 2% SDS, and 100 mM DTT) at 70°C for 30 min, washed extensively, reblocked with 5% non-fat milk, and then reprobed with another antibody as described above. Bands were quantitated by densitometry using NIH Image J software (NIH, Bethesda, MD, USA).
Flow cytometer analysis
Cells were trypsinized and suspended in PBS containing 2.5 mM EDTA, 2.5 mM ethylene glycol tetraacetic acid and 1% BSA. For the measurement of mitochondrial membrane potential changes, cells were incubated with 20 nM DiOC6 in PBS or 2 µM JC-1 in PBS for 15 min at 37°C, followed by flow cytometer analysis (FACSCalibur; Becton Dickinson Bioscience, San Jose, CA, USA). JC-1 is a lipophillic dye and it is a sensitive indicator for mitochondrial membrane potential (Salvioli et al., 1997). At low membrane potential, JC-1 occurs as a monomer that emits green fluorescent, and at higher membrane potential JC-1 forms aggregates that emit red fluorescence. For flow cytometer analysis of sub-G1 DNA contents, cells were fixed with 70% ethanol overnight at –20°C and stained with 0.5 ng·mL–1 propidium iodide (PI) plus 0.5 mg·mL–1 RNase A. For the apoptosis assay, treated cells were harvested and incubated for 15 min at room temperature with fluorescein isothiocyanate (FITC)-conjugated annexin-V reagent (2.5 µg·mL–1) and PI (5 µg·mL–1) in binding buffer followed by flow cytometer analysis. The Data were analysed with Cell Quest Software (BD Bioscience, San Jose, CA).
Immunofluorescence and confocal microscopy imaging
Cells grown on cover slips were fixed with 2% paraformaldehyde in PBS at room temperature for 10 min, rinsed with PBS, and treated with 1.5 mg·mL–1 glycine in PBS to quench free aldehyde group. Cells were then stained either with filipin (0.05 mg·mL–1) or CTXB-Alexa555 (0.04 µg·mL–1) for plasma membrane cholesterol and GM1 staining, respectively. For caveolin-1 staining, cells were permeablized with 0.2% Triton X-100 in PBS for 5 min, blocked in 2% BSA in PBS for 1 h at room temperature. And cells were incubated with anti-primary antibodies or non-specific IgG overnight at 4°C, and then washed in PBS. Cells then were exposed to Alexa 488-conjugated or Alexa 568 conjugated secondary IgG for 1 h at room temperature. The cover slips were washed in PBS and mounted on glass slides. For nucleus staining, cells were incubated with 4′6-diamidino-2-phenylindole 2HCl (DAPI; 0.5 µg·mL–1) in PBS. After washing with PBS, cells were examined using Carl Zeiss fluorescence microscopy or inverted laser-scanning microscopy (Thornwood, NY, USA).
Purification of caveolae
For extraction of caveolae, 2 × 107 cells were washed in PBS and solubilized in 1 mL lysis buffer [75 mM NaCl, 500 uM EDTA, 12.5 mM HEPES (pH6.5), 1 mM PMSF, protease inhibitor complex] containing 1% Triton X-100 for 30 min on ice. Cells were thoroughly homogenized with a Dounce tissue grinder, and an equal volume of 80% sucrose (150 mM NaCl, 25 mM HEPES pH6.5) was mixed with the lysate. It was overlaid with 7 mL of 30% sucrose followed by 2.5 mL of 5% sucrose. The gradient was centrifuged with a SW41 rotor at 4°C for 20 h at 274 000×g (Beckman Instruments, Palo Alto, CA, USA). Eleven gradient fractions (1 mL each) were harvested from the top (fraction numbers 1–11). Twenty microlitres of fractions were mixed with 5× SDS-sample buffer, boiled for 5 min and separated by SDS-PAGE followed by immunoblotting. For detection of successful rafts and caveolae isolation, we performed dot-blotting using HRP-conjugated cholera toxin-B subunit (CTXB). Two microlitres of each fraction was dot blotted on nitrocellulose membranes and stained with HRP-conjugated CTXB that binds to GM-1, a marker of rafts and caveolae.
Data analysis
All data points represented the mean value of at least three independent experiments with triplicates for each. Statistical significance was determined by Student's t-test, with P < 0.05 taken to show significant differences between means.
Materials
Ginsenoside-Rh2 was purchased from BTGin (Chung-Nam, Korea) and dissolved in DMSO at a concentration of 20 mM and stored at –20°C. Alexa Fluor 555 conjugated-cholera toxin subunit B, Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 568 mouse IgG were from Molecular Probes (Eugene, OR, USA). Anti-Bcl-xL, anti- EGF receptor (EGFR), anti-Src, anti-caveolin-1, anti-Bax, horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG and goat anti-rabbit IgG were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-phospho-Akt (Ser473), anti-Akt, anti-phospho-extracellular signal regulated kinase (ERK)1/2, anti-phospho-Src, anti-phospho-EGFR (1068), anti-caspase-8, anti-caspase-3, anti-poly (ADP-ribose) polymerase (PARP), anti-phospho-SAPK/JNK antibodies were from Cell Signalling Technology (Beverly, MA, USA). Anti-phospho-caveolin-1 antibodies and FITC annexin V apoptosis detection kit were obtained from BD Pharmingen (San Jose, CA, USA). Anti-Bim/BOD antibody was from Stressgen (Ann Arbor, MI, USA). 3,3′-diethyloxacabocyanine iodide (DiOC6), JC-1 assay kit and DAPI from Molecular Probes. Recombinant human EGF was purchased from Upstate (Lake Placid, NY, USA). Immobilion-P PVDFmembranes (0.45 µm) were from Millipore (Bedford, MA, USA). Micro-BCA protein assay reagents and Chemiluminescent reagents were from Pierce (Thermo Fisher Scientific Inc, Rockford, IL, USA). MβCD, filipin, water-soluble cholesterol, simvastatin, PI solution were from Sigma-Aldrich.
Results
Rh2, a ginsenoside, induced apoptosis in A431 cells
Ginsenosides are the most prominent saponins of ginseng and provide most of its pharmacological effects, such as regulation of angiogenesis and anti-tumour activity (Yue et al., 2007). Among the many ginsenosides, Rh2 has been show to induce apoptosis in various cancer cells including prostate cancer and neuroblastoma cells (Yue et al., 2007). As shown in Figure 1, the chemical structure of Rh2 is similar to that of cholesterol, a critical lipid component of lipid rafts and caveolae. To test the possibility that Rh2 exerts its effect by altering the structure or function of rafts and caveolae and mimicking cholesterol, we treated A431 cells, a human epidermoid carcinoma cell line, with Rh2 and examined cell growth. As illustrated in Figure 2A and B, Rh2 inhibited cell growth in a dose- and time-dependent manner. This growth inhibition appeared to be related to apoptotic cell death, as determined by flow cytometer analysis of Annexin-V and PI-stained A431 cells as shown in Figure 2C. However, there was little apoptosis induced by Rh2 in a non-cancer cell line, HEK293cells (Figure 2C). To test whether this Rh2 effect was limited to A431, we employed another human cancer cell line, PC-3 cells. We found that Rh2 also induced apoptosis of PC-3 cells time-dependently (Figure 2C), indicating that Rh2 effect is not restricted to A431 cells. We also evaluated the apoptotic effects of Rh2 in non-adherent cells using the NK/T lymphoma cell line, Hank-1, and we found that Rh2 induced apoptosis of Hank-1, indicating that Rh2 effect is not restricted to adherent cells (data not shown).
Figure 2.
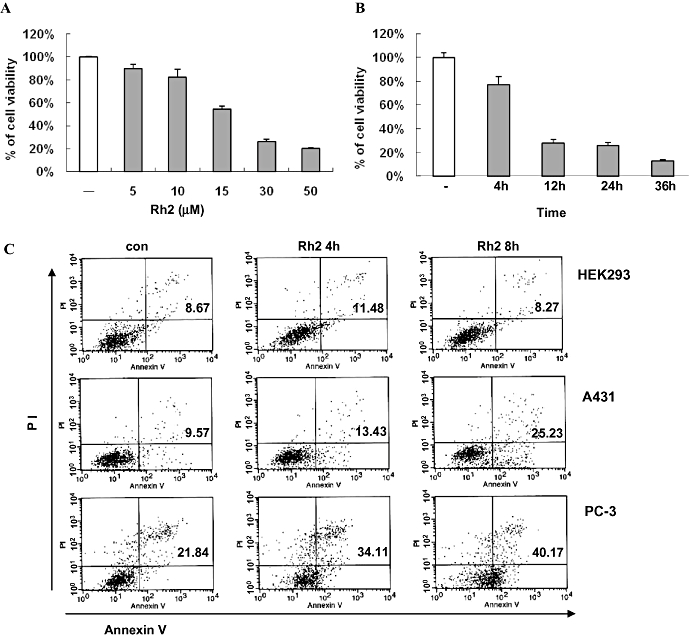
Effect of Rh2 on cell viability. (A) Serum-starved A431 cells were treated without [dimethyl sulfoxide (DMSO) only] or with various concentrations of Rh2 for 24 h and cell viability assessed by 3-(4,5-dimethylthiazol-2-yl)-5(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay. (B) Serum-starved A431 cells were treated with 30 µM Rh2 for 0–36 h and cell viability assessed by MTS assay. Error bars represent the mean ± standard deviation of three independent experiments. Similar results were observed in two independent experiments. (C) Serum-starved A431 cells and HEK293 cells were treated with 30 µM Rh2 for 0–8 h, stained with annexin V-FITC and PI, and subjected to flow cytometer analysis. Fluorescence dot blots of annexin V-positive (horizontal axis) and PI-positive (vertical axis) cells are shown. Cells that were positively stained by annexin V-FITC only (early apoptosis) and positive for both annexin V-FITC and PI (late apoptosis) were quantitated, and both subpopulations were considered as overall apoptotic cells. Similar results were observed in two independent experiments.
We further examined apoptotic features of A431 cells induced by Rh2, such as changes in cell morphology, increase in sub-G1 population, nuclei condensation by DAPI staining, and the loss of mitochondrial membrane potential by uptake of DiOC6 and JC-1, a mitochondrial specific dye. Using phase contrast microscopy, cell fragmentation was observed in the cells treated with Rh2, but not cholesterol (Figure 3A). Incubation with the lipid raft disrupting agents MβCD and filipin, which deplete and sequester cholesterol, respectively, also caused changes in cell morphology (Figure 3B) and ultimately cell death (data not shown). In addition, Rh2 treatment caused nuclei segmentation, as indicated by comparison with the positive control, staurosporine (Figure 3C). An increase in the number of cells in the sub-G1 phase (Figure 3D) and changes in the mitochondrial membrane potential (Figure 3E) were also observed. JC-1 is a fluorescent probe that is used to analyse changes of mitochondrial membrane potential via flow cytometer. JC-1 selectively enters mitochondria and changes in the ratio of green and red fluorescence indicate changes of mitochondrial membrane potential. Consistent with data in Figure 3E, there was a decrease in red fluorescence and an increase in green fluorescence upon Rh2 treatment (Figure 3F), suggesting a decrease in mitochondrial membrane potential. Collectively, these findings indicate that Rh2 treatment induced apoptosis in A431 cells.
Figure 3.
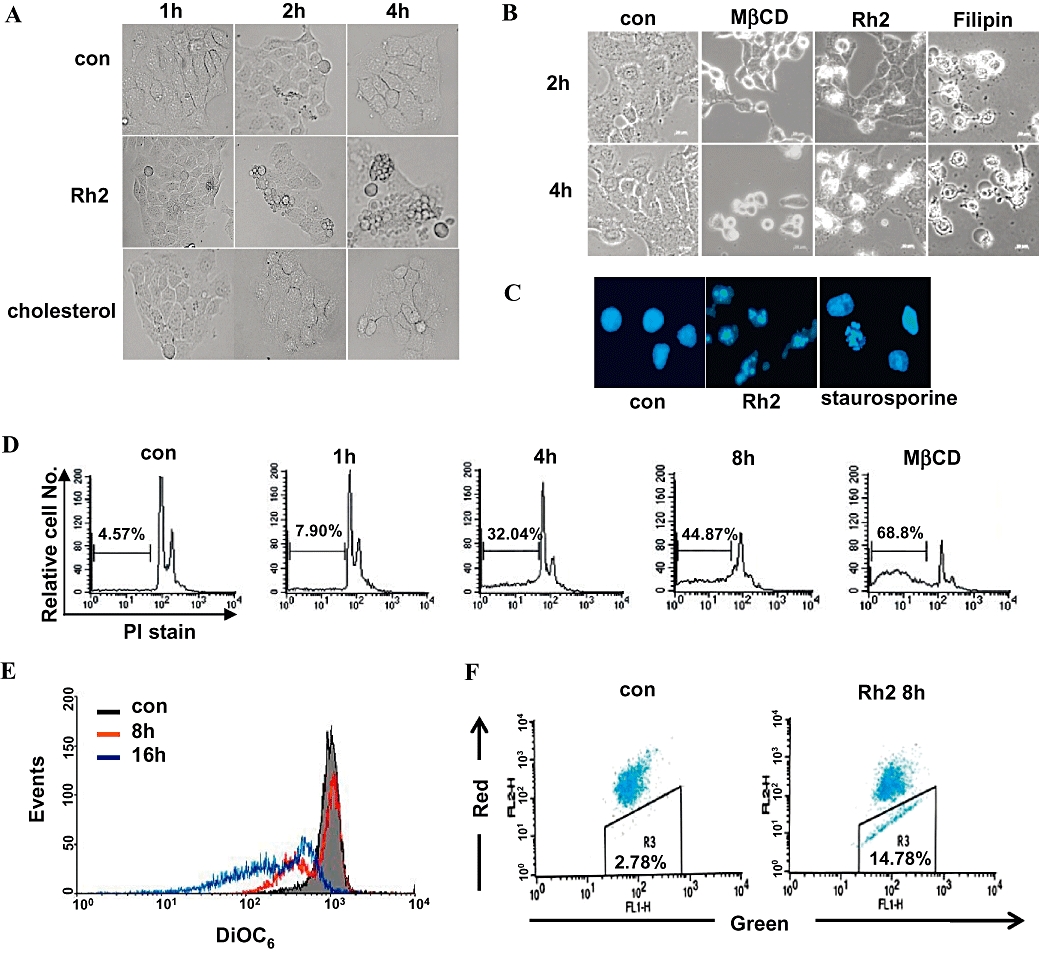
Apoptotic features of Rh2-induced cell death. (A and B) Serum-starved A431 cells were treated either without or with 30 µM Rh2, 300 µM cholesterol, 5 mM methyl-β cyclodextrin (MβCD), or 8 µg·mL–1 filipin for the indicated times, and then phase contrast images were taken. (C) Serum-starved A431 cell were treated either with 30 µM Rh2 for 8 h or 1 µM staurosporin for 4 h, stained with 4′6-diamidino-2-phenylindole 2HCl and analysed by fluorescence microscopy. (D) Serum-starved A431 cells were treated either without or with 30 µM Rh2 or with 5 mM MβCD for 0–8 h, and then cells were fixed and stained with propidium iodide (PI), followed by sub-G1 analysis (values indicate the percentage of cells with sub-G1 DNA content). (E) A431 cells were treated without or with 30 µM Rh2 for 8 and 16 h, and then incubated with DiOC6 to monitor mitochondrial membrane potential (ΔΨm) by flow cytometry. The loss of ΔΨm was equated with decreased fluorescence and shift to the left. (F) A431 cells were treated without or with 30 µM Rh2 for 8 h, and then ΔΨm was measured using the JC-1 kit by flow cytometer. The loss of ΔΨm was equated with decreased red fluorescence. These experiments were repeated three separate times with comparable results.
Because we reported previously that cholesterol depletion induces down-regulation of an anti-apoptotic member of the Bcl-2 family, Bcl-xL (Li et al., 2006), we examined whether Rh2 treatment also affects expression of these proteins. As shown in Figure 4A, the level of Bcl-xL protein remained unchanged after Rh2 treatment. However, the pro-apoptotic proteins Bax and Bim were up-regulated by Rh2 in a time-dependent manner. Based on these results and the fact that Rh2 affected the mitochondrial membrane potential, caspase activation in the presence of the compound was also investigated. As shown in Figure 4C, Rh2 treatment activated caspase-8 and caspase-3, as illustrated by increases in their active forms by immunoblotting analysis with antibodies that recognise their proforms and cleaved forms. Rh2 also induced time-dependent proteolytic cleavage of PARP, a caspase-3 substrate, as indicated by the accumulation of the 85 kDa fragment (Figure 4C).
Figure 4.
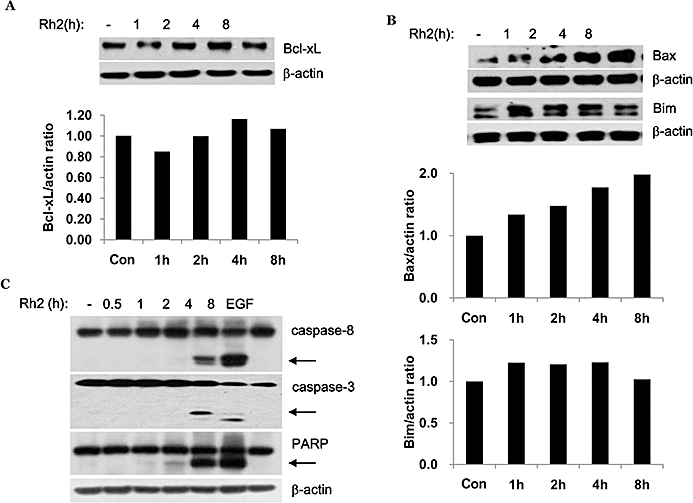
Effect of Rh2 on Bcl-xL protein levels and caspase activation. (A, B and C) Serum-starved A431 cells were treated with 30 µM Rh2 for the indicated times and cells were lysed with 2X sample buffer. Aliquots (30 µg of protein) from each treatment were subjected to immunoblotting analysis using anti-Bcl-xL, -Bax, -Bim, -caspase-8, -caspase-3, -PARP, and -β-actin antibodies (loading control). Densitometry analysis shows the band density ratios of Bcl-xL to actin, Bax to actin, and Bim to actin. These experiments were performed twice, separately, with comparable results.
Rh2 induces internalization of rafts and caveolae in A431 cells
Next, we investigated whether Rh2 treatment affected raft and caveolae levels in the plasma membrane by staining for cell surface expression of GM1, a marker for caveolae, using Alexa555-conjugated CTXB. Whereas GM1 staining was strong in the control cells, the Rh2-treated cells stained weakly in a time-dependent manner (Figure 5A). To assess whether disappearance of rafts and caveolae from the cell surface was associated with their internalisation, control and Rh2-treated cells were permeabilized and then stained intracellularly for GM1 and caveolin-1, another marker for rafts and caveolae, using Alexa555-conjugated CTXB and anti-caveolin-1 antibodies, respectively. In control cells the GM1 staining was dispersed throughout the cell, but in Rh2-treated cells, most of the staining was localised in the center (Figure 5B). We have shown previously that rafts and caveolae disruption by cholesterol depletion induced internalization of these microdomains, which could be reversed by cholesterol repletion as shown in Figure 5C as a positive control (Park et al., 2009). To test whether Rh2-induced internalization of rafts and caveolae could be reversed by cholesterol addition, cells were stained for GM1 localization after cholesterol addition to Rh2-treated cells. In MβCD-treated cells (Figure 5C), but not in those treated with Rh2 (Figure 5D), cholesterol addition was able to disperse intracellularly concentrated GM1 to the cell surface. Because caveolin-1 phosphorylation has been implicated in the endocytosis of rafts and caveolae (del Pozo et al., 2005; Park et al., 2009), we examined the phosphorylation state of this marker in Rh2-treated cells. Interestingly, caveolin-1 tyrosine phosphorylation was increased with Rh2-treatment but cholesterol addition dramatically decreased caveolin-1 phosphorylation (Figure 5E). However, Rh2 treatment did not cause any change in the distribution pattern of GM1 in the rafts and caveolae fractions (Figure 5F). These data indicate that Rh2 modified rafts and caveolae by increasing caveolin-1 phosphorylation and their internalization but not by altering the levels of GM1 within the rafts.
Figure 5.
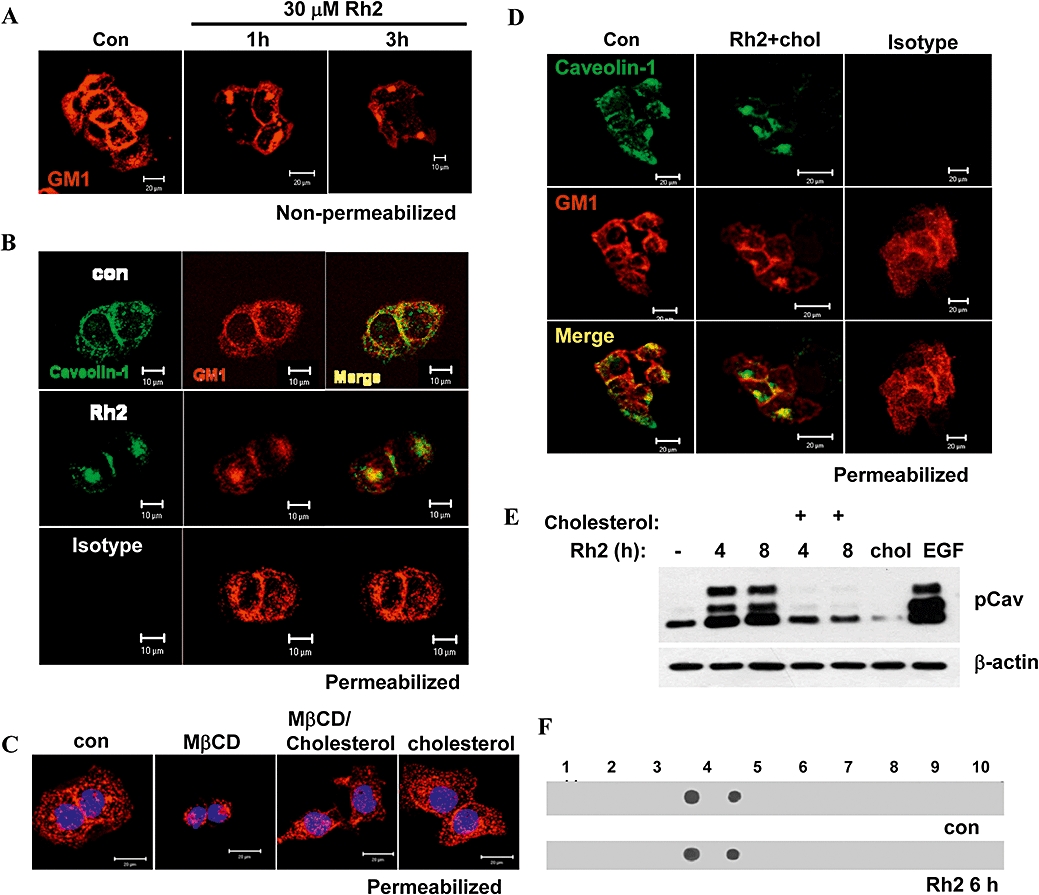
Effect of Rh2 on internalization of rafts and caveolae. Serum-starved A431 cells were treated without or with 30 µM Rh2 for indicated times and cells were stained with CTXB-Alexa555 for cell surface GM1 staining, followed by confocal microscope analysis. (B) Serum-starved A431 cells were treated with 30 µM Rh2 for 4 h and cells were permeabilized, stained with CTXB-Alexa555 and anti-caveolin-1 antibody, and then subjected to confocal microscope analysis (isotype IgG was used as a negative control for anti-caveolin-1 staining). Scale bar, 10 µm. (C) Serum-starved A431 cells were treated with either 5 mM MβCD for 3 h or with 1 mM cholesterol for 1 h. Some methyl-β cyclodextrin (MβCD)-treated cells for 2 h were incubated with 1 mM cholesterol for additional 1 h. Cells were permeabilized and stained with CTXB-Alexa555 and DAPI, followed by confocal microscopy. Scale bar, 20 µm (D) Serum-starved A431 cells were treated with 30 µM Rh2 for 3 h and treated with 300 µM cholesterol for another 1 h, and processed as described in B. Scale bar, 20 µm (E) Serum-starved A431 cells were treated without or with 30 µM Rh2 for indicated times and some MβCD-treated cells were incubated with 300 µM cholesterol for 1 h and cells. Aliquots (30 µg of protein) from each treatment were subjected to immunoblotting analysis using anti-phospho caveolin-1 (pCav) or β-actin antibodies. (F) A431 cells were treated without or with 30 µM Rh2 for 6 h and homogenized in a 2X lysis buffer containing 1% Triton X-100 as described. After sucrose gradient centrifugation, fractions were collected and subjected to dot blotting analysis using HRP-conjugated cholera toxin B-subunit that binds to GM1. Similar results were obtained in three different experiments.
Rh2-induced cell death is associated with Akt inactivation
Rafts and caveolae are important for Akt activation (Adam et al., 2007; Lasserre et al., 2008), and our previous report has demonstrated that internalization of rafts and caveolae by cholesterol depletion impaired Akt activation, which is critical for cell survival (Li et al., 2006; Park et al., 2009). Because Rh2 induced internalization of these microdomains, it is possible that Akt inactivation was involved in Rh2-induced cell death. As shown in Figure 6A, basal Akt activation decreased within 4 h and EGF was used as a positive control for Akt activation. EGF receptor activation initiates various signalling pathways including Akt and ERK1/2 for cell survival and proliferation (Pike, 2004). To test whether Rh2 could impair Akt pathway selectively, Rh2-pretreated cells were treated with EGF. EGF stimulation of the Rh2-pretreated cells resulted in EGFR activation and ERK activation as assessed by their phosphorylation, but not Akt activation, which is similar to the actions of MβCD (Figure 6B). Akt activation is known to increase cell survival by phosphorylation of a pro-apoptotic protein, Bad (Youle and Strasser, 2008). Because Akt activation was down-regulated in Rh2-treated cells, we examined Bad phosphorylation. As shown in Figure 6C, Bad phosphorylation was decreased with Rh2 treatment in a time-dependent manner. These results indicate that raft integrity is critical for Akt activation. To further determine whether Akt inactivation was critical for Rh2-induced cell death, the breast cancer cell line MDA-MB-231 (par) was infected with lentiviral vectors expressing either wild-type (WT-Akt) or dominant-active Akt (active-Akt). MDA-MB-231 cells possess lower levels of Akt than A431 cells, making the effects of Akt overexpression easier to study. Akt activity from each cell line was verified by immunoblotting using phospho-Akt antibodies as shown in Figure 7A. Morphological changes in cells treated with or without Rh2 were examined by light microscopy. Compared with cells infected with an empty vector (par), over-expression of WT-Akt or active Akt attenuated Rh2-induced morphological changes (Figure 7B). Consistent with this observation, Rh2-induced cell death was also diminished in cells expressing WT-Akt or active Akt (Figure 7C) even though Rh2 decreased Akt activation in all three cell lines (Figure 7D). To test whether Rh2-induced cell death could be reversed by cholesterol, Rh2-pretreated cells were treated with cholesterol, and then cell viability was assessed by MTS assays. Cholesterol addition had little effect on cell viability of the Rh2-treated cells when measured by MTS assay (Figure 8A) and flow cytometer analysis of annexin-V and PI-stained cells (Figure 8C). Consistent with the results in Figure 7A, cholesterol addition did not restore Akt activation (Figure 8B) despite the slight activation of Akt at 4 h in the Rh2-treated cells. These data suggest that Rh2 treatment down-regulates Akt activity, which was not reversible by cholesterol addition. Taken all together, our data indicate that Rh2 induced cell death by irreversible internalization of rafts and caveolae, thereby sequestering these membrane microdomains intracellularly and causing Akt inactivation.
Figure 6.
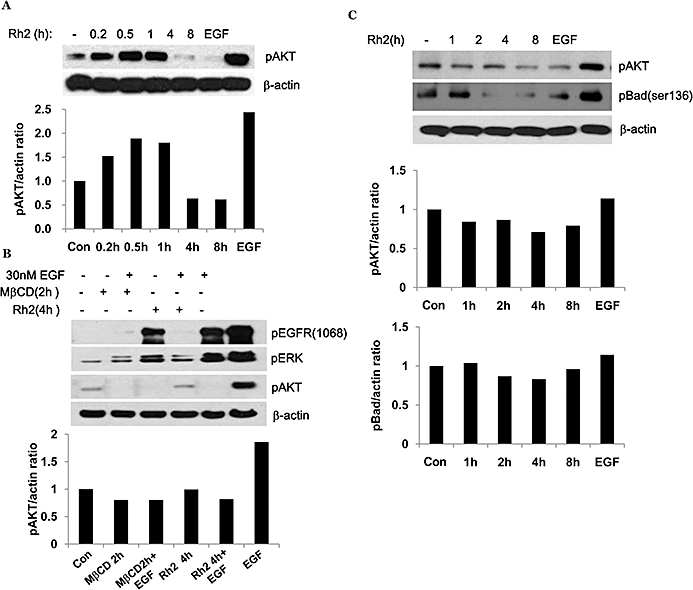
Effect of Rh2 on Akt activation. (A and C) Serum-starved A431 cells were treated either with 30 µM Rh2 for indicated times or with 30 nM EGF for 5 min as a positive control for Akt activation. 30 µg of protein from each treatment were subjected to immunoblotting analysis using anti-phospho Akt, anti-phospho-Bad, or anti-β-actin antibodies. (B) Serum-starved A431 cells were treated either with 30 µM Rh2 for 4 h or with 5 mM methyl-β cyclodextrin (MβCD) for 2 h and then treated with 30 nM EGF for 15 min. 30 µg of protein from each treatment were subjected to immunoblotting analysis using anti-phospho EGFR (1068), -phospho-Akt, -phospho ERK1 and 2, or anti-β-actin antibodies. Densitometry analysis shows the band density ratios of pAkt to actin and pBad to actin. These experiments were performed twice with comparable results.
Figure 7.
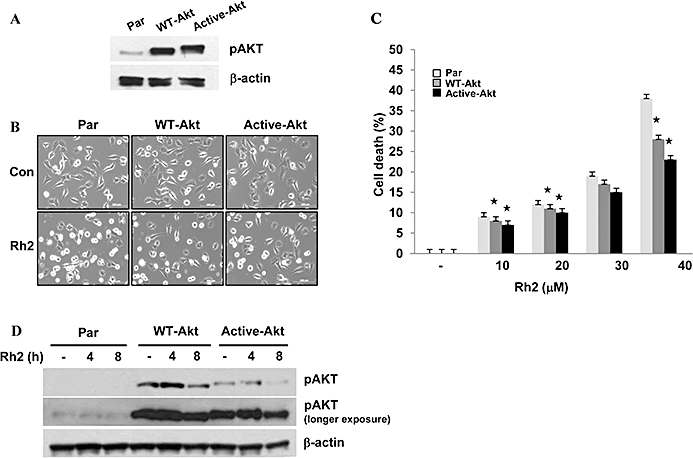
Effect of Akt activity on Rh2-induced cell death. (A) MBA-MB-231 cells were infected with lentivirus encoding either vector only (Par), wild type Akt (wt-Akt), or dominant-active Akt (active-Akt). Akt activity was assessed by immunoblotting analysis using anti-phospho and anti-β-actin antibodies. (B) MBA-MB-231-Par, -wt-Akt, -active-Akt cells were serum starved for 4 h and then treated with 30 µM Rh2 for 6 h and phase contrast images were taken. (C and D) MB-231-Par, -wt-Akt, -active-Akt cells were serum starved for 4 h and then treated with 30 µM Rh2 for 6 h (C) or indicated times (D) and cell viability was measured by 3-(4,5-dimethylthiazol-2-yl)-5(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium assay (C) or Akt activity was analysed by immunoblotting using phospho-Akt antibody (D). Densitometry analysis shows the band density ratios of pAkt to actin. Values represent the mean ± standard deviation of three independent experiments. Similar results were obtained in three different experiments.  , P < 0.01 versus methyl-β cyclodextrin-treated parental cells.
, P < 0.01 versus methyl-β cyclodextrin-treated parental cells.
Figure 8.
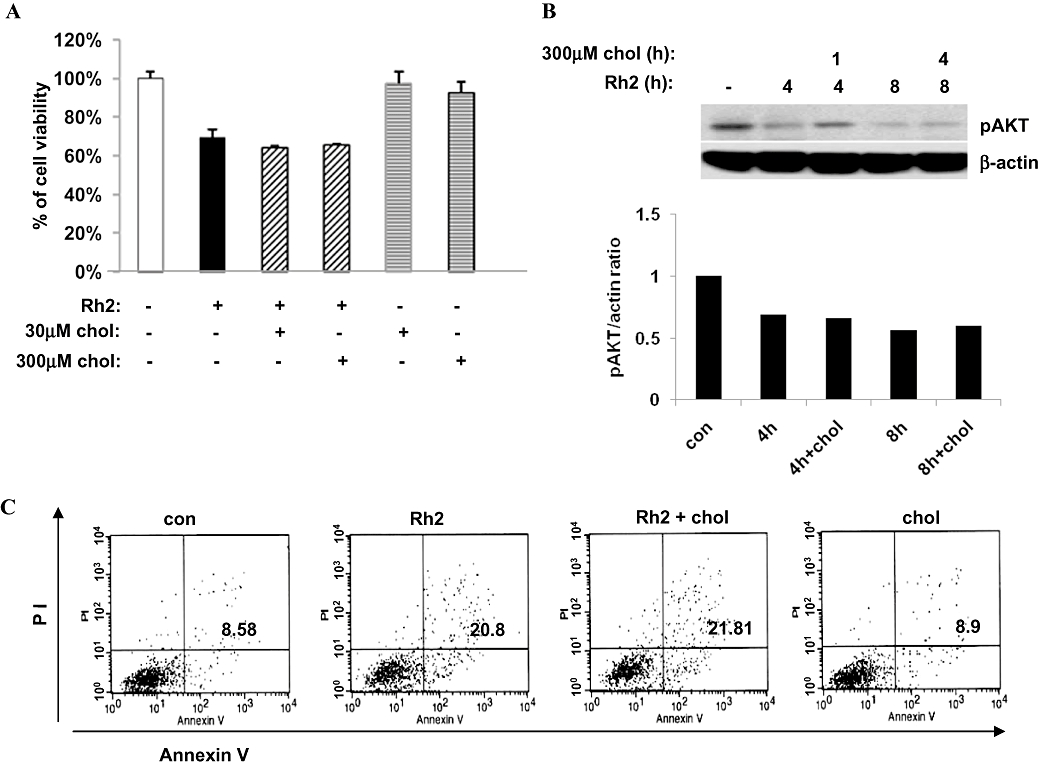
Effect of cholesterol addition on Rh2-induced cell death. (A) Serum-starved A431 cells were treated either with 30 µM Rh2, 30 µM cholesterol, or 300 µM cholesterol. Some Rh2-treated cells for 4 h were incubated with either 30 µM cholesterol or 300 µM cholesterol for additional 4 h. Cell viability was measured by 3-(4,5-dimethylthiazol-2-yl)-5(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium assay. (B) Serum-starved A431 cells were treated either with 30 µM Rh2 for 4 h or 8 h. Some Rh2-treated cells were with 300 µM cholesterol for 1 h or 4 h and processed for immunoblotting analysis using anti-phospho-Akt and anti-β-actin antibodies. (C) A431 cells were treated as described in Figure 8A and subjected to flow cytometer analysis of annexin V-FITC and propidium iodide (PI)-stained cells. Annexin V-FITC only (early apoptosis) and positive for both annexin V-FITC and PI (late apoptosis) were quantitated, and both subpopulations were considered as overall apoptotic cells. These experiments were performed twice with comparable results.
Discussion and conclusions
The specialized membrane domains, known as lipid rafts and caveolae, contain many proteins activated by receptor tyrosine kinases such as the EGF receptor, and other proteins involved in cell transformation, tumour development and metastasis, including CD44, Src and caveolin-1 (Patra, 2008). Cholesterol is a major lipid component of rafts and caveolae. Cholesterol depletion therefore results in disruption and internalization of rafts and caveolae, thereby inhibiting cellular signalling pathways, mediated by rafts and caveolae, and inducing apoptosis (Li et al., 2006; Park et al., 2009). Based on this knowledge, one can postulate that a reagent capable of modulating the integrity and/or localization of rafts and caveolae would also affect cell survival. In the present study, we have demonstrated that the ginsenoside Rh2, which is structurally similar to cholesterol, induced internalization of rafts and caveolae and Akt inactivation, leading to apoptosis.
Ginsenosides are active components of ginseng and are responsible for most of the pharmacological effects of ginseng extracts, such as anti-inflammatory and anti-tumour activities. The ginsenosides are glycosides containing an aglycone (protopanaxadiol or protopanaxatriol) with a dammarane skeleton (steroid-like skeleton), which is also found in cholesterol (Figure 1). The hydroxyl (-OH) group of ginsenosides interact with polar groups of plasma membrane phospholipids and the β-OH group of cholesterol whereas the hydrophobic steroid backbone associates with the hydrophobic side chains of fatty acids and cholesterol (Yue et al., 2007). Among them, Rh2 is known to possess a potent anti-tumour activity including cell growth inhibition and apoptosis induction in various cancer cell lines, such as prostate, ovarian and cervical cancer cell lines (Tode et al., 1993; Ham et al., 2003; Yue et al., 2007). In our study, Rh2 appeared to not affect the viability of a normal cell line, HEK293, when compared with the cancer cell lines, A431 and PC-3 (Figure 2C). In addition, it was reported that a daily oral administration of Rh2 showed anti-tumour effect in nude mice bearing HRA human ovarian cancer xenograft with no sign of toxicity (Nakata et al., 1998). Furthermore, recent pre-clinical evaluation of Rh2 revealed that Rh2 is a stable compound that can be formulated for oral gavage (Musende et al., 2009). As to in vivo efficacy and toxicity, treatment with Rh2 was achieved at a dose that was well tolerated by the animals. In addition, Rh2 exhibits its anti-tumour effect when used to treat established tumours derived following subcutaneous injection of PC-3 cells (Musende et al., 2009).
Because Rh2 is structurally similar to cholesterol and can interact with membrane lipids (Yue et al., 2007), it is possible that Rh2 exerts its anti-tumour effect by modulating rafts and caveolae, thereby altering rafts and caveolae-dependent signalling events such as Akt activation. The present study demonstrated that Rh2 treatment resulted in internalization and thus redistribution of rafts and caveolae from the plasma membrane to the cytoplasm. Cholesterol reduction, either by depletion or inhibition of synthesis, results in cell death caused by Akt inactivation in human prostate cancer cells in vitro and in vivo (Zhuang et al., 2002; Zhuang et al., 2005), as well as in the human cervical cancer cell line, A431 (Li et al., 2006). In addition, Lasserre et al. (2008) illustrated the fundamental role of sphingolipid and cholesterol in nanodomain formation by inhibiting synthesis of sphingolipid and cholesterol using the inhibitors, myriocin and zaragozic acid, respectively. They also showed that raft nanodomains play a crucial role in Akt recruitment to the plasma membrane, as well as its activation (Lasserre et al., 2008). Therefore, it is possible that internalization of rafts and caveolae induced by Rh2 leads to Akt inactivation. In addition, Rh2 treatment blocked EGF-stimulated Akt activation whereas it did not affect EGF-mediated EGFR phosphorylation and ERK1/2 activation (Figure 6), indicating a selective effect on Akt of Rh2 treatment.
Akt activation mediates cell survival signalling. For example, Akt-mediated Bad phosphorylation at Ser 136 promotes cell survival by inhibiting its interaction with anti-apoptotic Bcl2 family members like Bcl-xL to prevent cytochrome C release. In addition, Akt activation down-regulates a pro-apoptotic protein Bim via phosphorylation and subsequent degradation (McCubrey et al., 2007; Giannoni et al., 2008). Consistent with these reports, Rh2-induced Akt inactivation resulted in a decrease in Bad phosphorylation and Bim up-regulation, both of which are pro-apoptoic signalling events (Figures 4 and 6). Another pro-apoptotic protein, Bax, was also up-regulated (Figure 4) as seen in human blastoma (Kim and Jin, 2004). Bax up-regulation could increase the Bax/Bcl-2 ratio, stimulating the release of cytochrome C from mitochondria (McCubrey et al., 2007).
We cannot exclude the involvement of other pathways in Rh2-mediated apoptosis in A431 cells. It has been reported that Rh2 induces mitochondrial depolarization and apoptosis via reactive oxygen species (ROS) and JNK-1 (Ham et al., 2006). We also observed that Rh2 increased ROS production and induced JNK-1 activation in A431 cells (Supporting Information Figure S1). Because Fas is present in the lipid rafts (Hancock, 2006; Patra, 2008) and because Rh2 induces ligand-independent Fas activation in HeLa cells (Yi et al., 2009), we examined whether Fas was activated in A431 cells. There was some complex formation between Fas and FADD in the Rh2-treated cells (Supporting Information Figure S2), indicating that Fas activation might be partially responsible for Rh2-induced cell death in A431 cells. Although Akt inhibition is known to activate the Fas-mediated apoptotic pathway via FLIP down-regulation (Uriarte et al., 2005), this was unlikely in our system as there was little change in FLIP protein levels with Rh2 treatment (Supporting Information Figure S2).
Cholesterol addition could reverse cholesterol depletion-induced effects via rerouting rafts and caveolae to the cell membrane and Akt reactivation (Park et al., 2009 and Figure 5). In contrast, exposure of Rh2-treated cells to cholesterol could not restore the localization of rafts and caveolae or activate Akt (Figures 5 and 8). Consistent with this lack of effect, cholesterol addition did not delay Rh2-induced cell death (Figure 8). It is likely that the internalization of rafts and caveolae induced by Rh2 is irreversible, even in the presence of cholesterol. This could be because trafficking of rafts and caveolae in the Rh2-treated cells is different from that of cholesterol-depleted cells. In HeLa cells, cholesterol overloading overcomes Rh2-induced raft disruption and apoptosis (Yi et al., 2009), which is in contrast to our results. This difference could be a result of either differences in cell type or treatment. We treated cells with Rh2 to disrupt rafts first and then treated with cholesterol to test whether or not the disrupted rafts could be restored by cholesterol addition. However, Yi's group treated cells with Rh2 and excess cholesterol simultaneously, or pre-treated the cells with excess cholesterol first and then administered Rh2. Rh2 may not disrupt rafts when cells are overloaded with cholesterol.
Lipids, lipid derivatives, and phytosterols are known to incorporate into the plasma membrane and regulate cell viability. For example, incorporation of 7-ketocholesterol into lipid rafts induces apoptosis of smooth muscle cells via Akt dephosphorylation. However, α-tocopherol pre-treatment prevents 7-ketocholesterol incorporation into the rafts and thus preserves Akt activation and cell survival (Royer et al., 2009). Studies have demonstrated that docosahexaenoic acid (DHA) is incorporated into the plasma membrane and decreases the EGFRs associated with rafts and caveolae (Schley et al., 2007). Because Rh2 is also known to incorporate into the plasma membrane and bind to cholesterol (Yue et al., 2007), Rh2 possibly incorporates into rafts and caveolae domains so that it can alter rafts and caveolae-mediated signalling events such as Akt activation. Interestingly, Rh2 appeared to alter the localization of rafts and caveolae, but not their level overall because there was little change in the total levels of rafts and caveolae, as determined by GM1, when these microdomains were purified from Rh2-treated cells (Figure 5). Similarly, it has been reported that 7-ketocholesterol did not affect the distribution of sphingolipids/cholesterol within the lipid raft fractions, although it did incorporate into rafts and inactivated Akt (Royer et al., 2009).
In summary, we have demonstrated that Rh2 causes internalization of rafts and caveolae and Akt inactivation, which can lead to cell death. Because the presence of rafts and caveolae on the cell surface is critical for PI3K/Akt activation, impairment of Akt activation and cell death in response to Rh2 treatment are consistent with this concept. Since rafts and caveolae are linked to cancer development, targeting these microdomains has great potential as a strategy for cancer eradication. Therefore, the present study demonstrates that Rh2 can modulate rafts and caveolae and influence cell viability regardless of cholesterol addition, which is a major component of rafts and caveolae. These data provide a biological basis for the potential therapeutic applications of Rh2 in cancer therapies that target rafts and caveolae.
Acknowledgments
This work was supported by a research grant from the National Cancer Center in Korea.
Glossary
Abbreviations:
- CTXB
cholera toxin-B subunit
- MβCD
methyl-β cyclodextrin
- PARP
poly (ADP-ribose) polymerase
- PI3K
phosphatidylinositol 3 kinase
Conflicts of interest
The authors state no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Appendix S1 Induction of apoptosis by Rh2, an active compound in ginseng via lipid rafts/caveolae internalization and Akt inactivation.
Figure S1 Effects of Rh2 on ROS production. A, Serumstarved A431 cells were treated without (control) or with 30 μM Rh2 for 4 h and cells were loaded with 20 μM 2′,7′-dichlorofluorescein (DCF; a fluorescent dye). Intracellular ROS levels were analysed by flow cytometer. B, Serum-starved A431 cells were treated with 30 μM Rh2 for the indicated times and cells were lysed with 2X sample buffer. Aliquots (30 μg of protein) from each treatment were subjected to immunoblotting analysis using anti-phospho-JNK1/2 and anti-b-actin antibodies (loading control). These experiments were performed two separate times with comparable results.
Figure S2 Effects of Rh2 on FLIP protein levels and DISC formation. A, Serum-starved A431 cells were treated with 30 μM Rh2 for the indicated times and lysed with IPA buffer. The cell extracts (500 μg of protein) were subjected to immunoprecipitation (IP) of FAS and immunocomplexes were analysed by immunoblotting using anti-FADD antibody. B, Serum-starved A431 cells were treated with 30 μM Rh2 for the indicated times and cell lysates were subjected to immunoblotting analysis using anti-FLIP, -FAS, -FADD and -beta;-actin antibodies (loading control). These experiments were performed two separate times with comparable results.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Adam RM, Mukhopadhyay NK, Kim J, Di Vizio D, Cinar B, Boucher K, et al. Cholesterol sensitivity of endogenous and myristoylated Akt. Cancer Res. 2007;67:6238–6246. doi: 10.1158/0008-5472.CAN-07-0288. [DOI] [PubMed] [Google Scholar]
- Attele AS, Wu JA, Yuan CS. Ginseng pharmacology: multiple constituents and multiple actions. Biochem Pharmacol. 1999;58:1685–1693. doi: 10.1016/s0006-2952(99)00212-9. [DOI] [PubMed] [Google Scholar]
- Cheng CC, Yang SM, Huang CY, Chen JC, Chang WM, Hsu SL. Molecular mechanisms of ginsenoside Rh2-mediated G1 growth arrest and apoptosis in human lung adenocarcinoma A549 cells. Cancer Chemother Pharmacol. 2005;55:531–540. doi: 10.1007/s00280-004-0919-6. [DOI] [PubMed] [Google Scholar]
- del Pozo MA, Balasubramanian N, Alderson NB, Kiosses WB, Grande-García A, Anderson RG, et al. Phospho-caveolin-1 mediates integrin-regulated membrane domain internalization. Nat Cell Biol. 2005;7:901–908. doi: 10.1038/ncb1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke TF. PI3K/Akt: getting it right matters. Oncogene. 2008;27:6473–6488. doi: 10.1038/onc.2008.313. [DOI] [PubMed] [Google Scholar]
- Giannoni E, Buricchi F, Grimaldi G, Parri M, Cialdai F, Taddei ML, et al. Redox regulation of anoikis: reactive oxygen species as essential mediators of cell survival. Cell Death Differ. 2008;15:867–878. doi: 10.1038/cdd.2008.3. [DOI] [PubMed] [Google Scholar]
- Ham YM, Choi JS, Chun KH, Joo SH, Lee SK. The c-Jun N-terminal kinase 1 activity is differentially regulated by specific mechanisms during apoptosis. J Biol Chem. 2003;278:50330–50337. doi: 10.1074/jbc.M302997200. [DOI] [PubMed] [Google Scholar]
- Ham YM, Lim JH, Na HK, Choi JS, Park BD, Yim H, et al. Ginsenoside-Rh2-induced mitochondrial depolarization and apoptosis are associated with reactive oxygen species- and Ca2+-mediated c-Jun NH2-terminal kinase 1 activation in HeLa cells. J Pharmacol Exp Ther. 2006;319:1276–1285. doi: 10.1124/jpet.106.109926. [DOI] [PubMed] [Google Scholar]
- Hancock JF. Lipid rafts: contentious only from simplistic standpoints. Nat Rev Mol Cell Biol. 2006;7:456–462. doi: 10.1038/nrm1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K, Mouritsen OG, Anderson RG. Lipid rafts: at a crossroad between cell biology and physics. Nat Cell Biol. 2007;9:7–14. doi: 10.1038/ncb0107-7. [DOI] [PubMed] [Google Scholar]
- Kim YS, Jin SH. Ginsenoside Rh2 induces apoptosis via activation of caspase-1 and -3 and up-regulation of Bax in human neuroblastoma. Arch Pharm Res. 2004;27:834–839. doi: 10.1007/BF02980175. [DOI] [PubMed] [Google Scholar]
- Kim YN, Wiepz GJ, Guadarrama AG, Bertics PJ. Epidermal growth factor-stimulated tyrosine phosphorylation of caveolin-1. Enhanced caveolin-1 tyrosine phosphorylation following aberrant epidermal growth factor receptor status. J Biol Chem. 2000;275:7481–7491. doi: 10.1074/jbc.275.11.7481. [DOI] [PubMed] [Google Scholar]
- Lasserre R, Guo XJ, Conchonaud F, Hamon Y, Hawchar O, Bernard AM, et al. Raft nanodomains contribute to Akt/PKB plasma membrane recruitment and activation. Nat Chem Biol. 2008;4:538–547. doi: 10.1038/nchembio.103. [DOI] [PubMed] [Google Scholar]
- Li YC, Park MJ, Ye SK, Kim CW, Kim YN. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am J Pathol. 2006;168:1107–1118. doi: 10.2353/ajpath.2006.050959. quiz 1404-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musende AG, Eberding A, Wood C, Adomat H, Fazli L, Hurtado-Coll A, et al. Pre-clinical evaluation of Rh2 in PC-3 human xenograft model for prostate cancer in vivo: formulation, pharmacokinetics, biodistribution and efficacy. Cancer Chemother Pharmacol. 2009;64:1085–1095. doi: 10.1007/s00280-009-0965-1. [DOI] [PubMed] [Google Scholar]
- Nakata H, Kikuchi Y, Tode T, Hirata J, Kita T, Ishii K, et al. Inhibitory effects of ginsenoside Rh2 on tumor growth in nude mice bearing human ovarian cancer cells. Jpn J Cancer Res. 1998;89:733–740. doi: 10.1111/j.1349-7006.1998.tb03278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EK, Park MJ, Lee SH, Li YC, Kim J, Lee JS, et al. Cholesterol depletion induces anoikis-like apoptosis via FAK down-regulation and caveolae internalization. J Pathol. 2009;218:337–349. doi: 10.1002/path.2531. [DOI] [PubMed] [Google Scholar]
- Patra SK. Dissecting lipid raft facilitated cell signaling pathways in cancer. Biochim Biophys Acta. 2008;1785:182–206. doi: 10.1016/j.bbcan.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Pike LJ. Lipid rafts: heterogeneity on the high seas. Biochem J. 2004;378:281–292. doi: 10.1042/BJ20031672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer MC, Lemaire-Ewing S, Desrumaux C, Monier S, Pais de Barros JP, Athias A, et al. 7-ketocholesterol incorporation into sphingolipid/cholesterol-enriched {lipid raft} domains is impaired by vitamin e: a specific role for alpha-tocopherol with consequences on cell death. J Biol Chem. 2009;284:15826–15834. doi: 10.1074/jbc.M808641200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, et al. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- Salvioli S, Ardizzoni A, Franceschi C, Cossarizza A. JC-1, but not DiOC(6)(3) or rhodamine 123, is a reliable fluorescent probe to assess Delta Psi changes in intact cells: implications for studies on mitochondrial functionality during apoptosis. FEBS LETTERS. 1997;411:77–82. doi: 10.1016/s0014-5793(97)00669-8. [DOI] [PubMed] [Google Scholar]
- Schley PD, Brindley DN, Field CJ. (n-3) PUFA alter raft lipid composition and decrease epidermal growth factor receptor levels in lipid rafts of human breast cancer cells. J Nutr. 2007;137:548–533. doi: 10.1093/jn/137.3.548. [DOI] [PubMed] [Google Scholar]
- Tarahovsky YS, Muzafarov EN, Kim YA. Rafts making and rafts braking: how plant flavonoids may control membrane heterogeneity. Mol Cell Biochem. 2008;314:65–71. doi: 10.1007/s11010-008-9766-9. [DOI] [PubMed] [Google Scholar]
- Tode T, Kikuchi Y, Kita T, Hirata J, Imaizumi E, Nagata I. Inhibitory effects by oral administration of ginsenoside Rh2 on the growth of human ovarian cancer cells in nude mice. J Cancer Res Clin Oncol. 1993;120:24–26. doi: 10.1007/BF01200720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uriarte SM, Joshi-Barve S, Song Z, Sahoo R, Gobejishvili L, Jala VR, et al. Akt inhibition upregulates FasL, downregulates c-FLIPs and induces caspase-8-dependent cell death in Jurkat T lymphocytes. Cell Death Differ. 2005;12:233–242. doi: 10.1038/sj.cdd.4401549. [DOI] [PubMed] [Google Scholar]
- Yi JS, Choo HJ, Cho BR, Kim HM, Kim YN, Ham YM, et al. Ginsenoside Rh2 induces ligand-independent Fas activation via lipid raft disruption. Biochem Biophys Res Commun. 2009;24:154–159. doi: 10.1016/j.bbrc.2009.05.028. [DOI] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Yue PY, Mak NK, Cheng YK, Leung KW, Ng TB, Fan DT, et al. Pharmacogenomics and the Yin/Yang actions of ginseng: anti-tumor, angiomodulating and steroid-like activities of ginsenosides. Chin Med. 2007;2:6. doi: 10.1186/1749-8546-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang L, Kim J, Adam RM, Solomon KR, Freeman MR. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J Clin Invest. 2005;115:959–968. doi: 10.1172/JCI200519935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang L, Lin J, Lu ML, Solomon KR, Freeman MR. Cholesterol-rich lipid rafts mediate akt-regulated survival in prostate cancer cells. Cancer Res. 2002;62:2227–2231. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.