

Abstract
Acrolein is a ubiquitous environmental pollutant and an endogenous product of lipid peroxidation. It is also generated during the metabolism of several drugs and amino acids. In this study, we examined the effects of acrolein on endothelial cells. Treatment of human umbilical vein endothelial cells (HUVECs) with 2 to 10 μM acrolein led to an increase in the phosphorylation of eIF-2α within 10 to 30 min of exposure. This was followed by alternate splicing of XBP-1 mRNA and an increase in the expression of the endoplasmic reticulum (ER) chaperone genes Grp78 and Herp. Within 2–4 h of treatment, acrolein also increased the abundance and the nuclear transport of the transcription factors ATF3, AFT4, and CHOP. Acrolein-induced increase in ATF3 was prevented by treating the cells with the chemical chaperone – phenylbutryic acid (PBA). Treatment with acrolein increased phosphorylation of ERK1/2, p38, and JNK. The increase in JNK phosphorylation was prevented by PBA. Acrolein treatment led to the activation and nuclear translocation of the transcription factor NF-κB and an increase in TNF-α, IL-6 and IL-8, but not MCP-1, mRNA. Increased synthesis of cytokine genes and NF-κB activation were not observed in cells treated with PBA. These findings suggest that exposure to acrolein induces ER stress and triggers the unfolded protein response and that NF-κB activation and stimulation of cytokine production by acrolein could be attributed, in part, to ER stress. Chemical chaperones of protein-folding may be useful in treating toxicological and pathological states associated with excessive acrolein exposure or production.
Keywords: endothelial dysfunction, pollution, human, oxidative stress, aldehydes
INTRODUCTION
Humans are exposed to acrolein and related aldehydes from a variety of sources (Feron et al., 1991; Ghilarducci et al., 1995). Acrolein and other aldehydes are toxic components of automobile exhaust and smog and have been detected in high concentrations in cigarette, cotton, wood, and coal smoke. Aldehydes such as acrolein are also present in contaminated water and as natural constituents of several foods. Measurable concentration of acrolein (1–5 μg/kg) has been detected in several foods such as bread, cheese and beer. Cooking and frying further increase aldehyde concentration in foods. Several aldehydes including acrolein are also generated endogenously during peroxidation of unsaturated fatty acids in lipoproteins or membrane phospholipids (Uchida et al., 1998a). In addition, acrolein is also generated at sites of inflammation by myeloperoxidase-catalyzed oxidation of threonine (Anderson et al., 1997). Acrolein could also be generated from glucose by retro-aldol cleavage during Maillard-type reactions (Mottram et al., 2002). Consequently, high abundance of acrolein-modified proteins has been detected in tissues under several pathological conditions that are associated with high oxidative stress and inflammation such as atherosclerosis, Parkinson’s and Alzheimer’s disease and diabetes (Daimon et al., 2003; Lovell et al., 2001; Shao et al., 2005; Uchida et al., 1998b).
The α,β-unsaturated aldehydes such as acrolein, crotonaldehyde, trans-2-hexenal and 4-hydroxy trans-2-nonenal (HNE) are highly reactive and toxic. These aldehydes react readily with cellular nucleophiles and cause extensive protein and DNA modifications (Esterbauer et al., 1991). As a result, cells exposed to such aldehydes undergo glutathione depletion and changes in energy generation, respiration, calcium homeostasis, proliferation (Esterbauer et al., 1991; Uchida et al., 1998a) and ion channel conduction pathways (Bhatnagar, 1995). Extensive data indicate that aldehydes such as acrolein contribute to cardiovascular morbidity and mortality. Unsaturated aldehydes generated in oxidized LDL promote uptake of the lipoprotein by scavenger receptors and promote plaque formation and induce endothelial dysfunction (Bhatnagar, 2004). Exposures associated with pollutants containing high levels of aldehydes such as tobacco smoke (Barnoya et al., 2005; Howard et al., 1998) or traffic emission (Peters et al., 2004) are associated with an increase in chronic risk of cardiovascular disease as well as acute myocardial infarction. Indeed, the cardiovascular disease burden of such exposure far exceeds their carcinogenic risk (Bhatnagar, 2006) Nonetheless, the mechanism by which aldehydes such as acrolein induce cardiovascular dysfunction and disease remain poorly understood.
The endothelium is a vulnerable target of acrolein and related aldehydes. Acrolein induces endothelium-dependent relaxation of mesenteric arteries (Awe et al., 2006) and pre-contracted aortic rings (Tsakadze et al., 2003) and acrolein exposure in mice compromises vessel contractility (Tsakadze et al., 2003). Endothelial cells exposed to acrolein in culture show an inflammatory phenotype (Park et al., 2007), an increase in superoxide production (Jaimes et al., 2004) and eventual apoptosis (Misonou et al., 2006). However, the mechanisms of acrolein-induced endothelial toxicity remain poorly understood. Given the high propensity of acrolein and related aldehydes to modify cellular proteins, we hypothesized that acrolein interferes with protein folding or it increases the accumulation of misfolded and modified proteins leading to the induction of ER stress. The secretory pathway in the ER synthesizes, modifies and delivers active proteins where they are needed. Conditions under which the influx of nascent, unfolded proteins exceeds the folding capacity of the ER activate signaling pathways (UPR) to restore physiological function (Schroder et al., 2005; Xu et al., 2005; Zhang et al., 2004). Cytosolic perturbation outside the ER, such as inhibition of the proteasome can be propagated to the ER to cause ER stress and activate the unfolded protein response (UPR). To match demand and capacity, the folding demand is decreased by transiently inhibiting translation and transcription of secreted proteins. The clearance and degradation of misfolded proteins is increased. Accordingly, we studied the effects of acrolein on ER stress pathways and their relationship to acrolein-induced endothelial activation. Our results show that endothelial activation by acrolein was associated with robust induction of the alarm and the adaptation pathways of the UPR and ameliorating ER stress by chemical chaperones prevents the transcription of inflammatory genes such as TNF-α, IL-6 and IL-8 in endothelial cells. Collectively, these observations reveal a novel mechanism of acrolein toxicity and suggest clinically-relevant strategies for preventing pathological and toxicological states associated with excessive acrolein exposure or generation.
MATERIALS AND METHODS
Materials
Acrolein and other chemicals were purchased from Sigma (St. Louis, MO, USA). Primary antibodies against phospho-eIF-2α (Ser51), total-eIF-2α, HDAC1 (histone deacetylase 1), phospho-(Thr202/Tyr204) and total-p42/44, phospho-(Thr180/Tyr182) and total-p38, phospho-(Thr183/Tyr185) and total-JNK/SAPK (1:1000) as well as HRP-linked secondary antibodies goat anti rabbit (used at a dilution of 1:2000) or goat anti-mouse antibody (1:2500) were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against ATF3, Grp78, NF-κB-p65 (1:500) and ATF4 (1:2000) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against XBP-1 (COOH-terminus, 1:5000) from BioLegend (San Diego, CA, USA), anti-CHOP (1:1000) from Affinity BioReagents, (Golden, CO, USA) and actin (1:2000) from Sigma-Aldrich, (St. Louis, MI, USA) were used. Electrophoresis and Western blot supplies were purchased from BioRad (Hercules, CA, USA). Primers for PCR were obtained from Integrated DNA Technologies, Coralville, IA, USA.
Cell culture and acrolein treatment
Human umbilical vein endothelial cells (PromoCell, Heidelberg, Germany) were cultured in endothelial basal medium (EBM, Clonetics/Lonza, Walkersville, MD, USA) supplemented with human endothelial growth factor (hEGF), hydrocortisone, gentamicin/amphotericin B (GA), bovine brain extract (BBE) and 2% fetal bovine serum (FBS, EGM SingleQuots®, Clonetics/Lonza, Walkersville, MD, USA) as suggested by the supplier under standard cell culture conditions (37°C, 5% CO 2). Cells (passages 3–12), grown to sub-confluence (80–90%), were treated with acrolein at the indicated concentrations up to 2h in Hanks’ balanced salt solution (HBSS, pH7.4, 20 mM HEPES, 135 mM NaCl, 5.4 mM KCl, 1.0 mM MgCl2, 2.0 mM CaCl2, 2.0 mM NaH2PO4, 5.5 mM Glucose). For long-term incubations required to measure changes in ATF4, CHOP or Grp78 protein expression, nuclear translocation of ATF3 and NF-κB-p65-unit, CHOP, IL-6, IL-8, TNF-α and MCP-1 mRNA expression, HBSS was replaced after 2h with the complete media and the cells were incubated for an additional 2, 4 or 10h. Cells treated with thapsigargin (1 μM, TG) were used for positive control. For phenylbuytric acid (PBA; Pfaltz & Bauer, Waterbury, CT, USA) treatment, the cells were pre incubated for 16h with 10 mM PBA in media, and the cells were treated with acrolein in the presence of PBA. After treatment, the cells were washed three times with ice-cold PBS (Invitrogen, Carlsbad, CA, USA) and processed for Western or PCR analysis.
Western Blot Analysis
Total cell lysates were prepared by scraping the cells in lysis buffer containing 25 mM HEPES (pH 7.0), 1 mM EDTA, 1 mM EGTA, 1% NP-40 and 0.1 % SDS supplemented with 1:100 protease inhibitor cocktail (Protease Inhibitor Cocktail P8340, Sigma, St. Louis, MO, USA) and 1:100 phosphatase inhibitor cocktail (Halt™ Phosphatase Inhibitor Cocktail, Pierce, Rockland, IL, USA). After sonication, the cell suspension was centrifuged (14,000xg, 5 min, 4°C) and total protein in the supernatant was measured using a commercial kit (Protein-Assay, Bio-Rad, Hercules, CA, USA). To prepare nuclear extracts, cells were harvested by scraping in buffer A (pH 7.9) containing 10 mM HEPES, 10 mM KCl and 1.5 mM MgCl2 supplemented with protease and phosphatase inhibitor cocktail (1:100). After 15 min incubation on ice, the suspension was mixed by inversion with buffer B (buffer A containing 2.5% NP-40) to a final concentration of 0.25% NP-40. Following centrifugation (2000xg, 10 min, 4°C) the pellet containing the nuclei was resuspended in buffer C (pH 7.9, 20 mM HEPES, 0.45 M NaCl, 1 mM EDTA) and incubated for 15 min at 4°C. The nuclear fractions we re extracted by centrifugation at 14,000xg, for 10 min at 4°C.
Heat-denatured (5 min, 95°C) proteins, supplemented with 5x bromphenol sample buffer (312.5 mM Tris, pH 6.8, containing 10% glycerol, 11.5% SDS, and 0.1% bromphenol) were separated by SDS-PAGE and transferred to PVDF membranes. The membranes were processed by standard immuno-detection techniques according to the manufacturer guidelines. Western blots were developed using ECL® plus Western blotting detection reagents (Amersham Biosciences, Piscataway, NJ, USA) and imaged on a Typhoon 9400 variable mode imager (Amersham Biosciences, Piscataway, NJ, USA). Band intensities were quantified using Image Quant TL software (Amersham Biosciences, Piscataway, NJ, USA).
RNA isolation and PCR Analysis
Total RNA was extracted from cells using the TRIzol method (Invitrogen, Carlsbad, CA, USA). 2 μg of RNA were converted into cDNA using oligo(dT)18 primer (Integrated DNA Technologies, Coralville, IA, USA) following the manufacturers protocol (AMW Transcriptase, Promega, Madison, WT, USA). After synthesis (42°C, 60min) cDNA was heated at 94°C for 5 min and kept at −20°C until use. XBP-1 splicing was analyzed by regular PCR using primers spanning the splice junction of XBP-1 (forward primer 5′-CAGCGCTTGGGGATGGATGC-3′, reverse primer 5′-CCATGGGGAGATGTTCTGGA-3′). The cDNA was supplemented with primers and PCR-mix according to the manufacturers guidelines (PCR Master Mix, Promega, Madison, WT, USA) to a total volume of 25μl. Amplification was performed using following PCR program: 94°C -3min (denaturation), 3 0 cycles: 94°C-45 sec, 50°C-45 sec (annealing), 72°C-45 sec (extension) and 72°C-7 min (final extension). Finally, 3 μl of the PCR product were separated by electrophoresis using a 3% (w/v) agarose gel. Ethidium bromide stained gels were imaged using a Typhoon 9400 variable mode imager (Amersham Biosciences, Piscataway, NJ, USA) and band intensities were quantified using Image Quant TL software (Amersham Biosciences, Piscataway, NJ, USA).
Quantitative real-time PCR using SYBR Green/Fluorescin PCR Master Mix (Super Array, Frederick, MD, USA) was performed in an iCycler iQ™ (BioRad, Hercules, CA, USA) using the following primer sets: CHOP forward primer: 5′-GGA GCT GGA AGC CTG GTA TGA GG-3′, reverse primer: 5′-TCC CTG GTC AGG CGC TCG ATT TCC-3′; Grp78: forward primer: 5′-CGG GCA AAG ATG TCA GGA AAG-3′, reverse primer: 5′-TTC TGG ACG GGC TTC ATA GTA GAC-3′; MCP-1: forward primer: 5′-TGC TGA TAG CAG CCA CCT TCA TTC-3′, reverse primer: 5′-GAC ACT TGC TGC TGG TGA TTC TTC-3′; IL-6: forward primer: 5′-TTC TCC ACA AGC GCC TTC GGT CCA-3′, reverse primer: 5′-AGG GCT GAG ATG CCG TCG AGG ATG T A-3′; IL-8: forward primer: 5′-CCA CAC TGC GCC AAC ACA-3′, reverse primer: 5′-TCA CTG ATT CTT GGAT ACC ACA GAG A-3′; TNF-α: forward primer 5′-TGA TCC CTG ACA TCT GGA ATC TG-3′, reverse primer: 5′-GCT GGG CTC CGT GTC TCA-3′; GAPDH: forward primer 5′-CGC TCT CTG CTC CTC CTG TT-3′, reverse primer: 5′-CCA TGG TGT CTG AGC GAT GT-3′. Primers for the amplification of Herp were purchased from Superarray (Frederick, MD, USA). Quantitative RT-PCR was performed under the following parameters: 2 min for 50°C, 5 min 95°C (initial denaturation) followed by 40 amplification cycles (denaturation: 95°C for 15 sec, annealing: 53°C for 30 sec, extension: 72°C for 30 se c) and 1 min denaturation at 95°C, 1 min 55°C, and 85 cycles at 55°C for 10 sec. Relative mRNA amount were calculated by the ΔΔCt method using GAPDH as control. All primers displayed only 1 dissociation peak.
Statistical analysis
Data are presented as mean ± SE. For multiple comparisons, the data were analyzed by One-Way ANOVA followed by Tukey post-hoc testing.
RESULTS
Acrolein increases the phosphorylation of eIF-2α
ER stress triggers the unfolded protein response (UPR), which has distinct alarm and adaptive phases (Schroder and Kaufman, 2005; Xu, Bailly-Maitre, and Reed, 2005; Zhang and Kaufman, 2004). The alarm phase results in the activation of eIF-2α and alternative splicing of XBP-1. These pathways activate stress signaling (e.g., JNK) and stimulate host-defense responses such as NF-κB. This response also results in the activation of transcription factors ATF3 and ATF4, which in turn regulate the expression of genes that encode proteins that regulate pathways of cell survival and death (e.g., the transcription factor, CHOP). The adaptive phase results in the upregulation of ER chaperone proteins that ameliorate ER stress and extinguish UPR. This phase is to reestablish normal ER function. Accordingly, we examined the major mediators of each of these pathways. For the first series of experiments, the cells were treated in HBSS with 10 or 25 μM acrolein for 5 to 30 min, and changes in the phosphorylation of eIF-2α were measured by Western analysis. No change in cell viability, as assessed by the MTT assay, was observed under these conditions (data not shown). As shown in Fig. 1A, no increase in phospho-eIF-2α was observed 5 min after acrolein treatment; however, after 10 min, a significant increase in eIF-2α phosphorylation was observed with 25 μM acrolein. After 15 or 30 min of exposure, significant phosphorylation of eIF-2α was observed with both concentrations of acrolein tested. Using an optimal time of 30 min, we then studied the concentration-dependence of the response. As shown in Fig. 1B, treatment with as little as 2 μM acrolein induced statistically significant increase in eIF-2α phosphorylation. At a concentration of 25 μM acrolein-induced eIF-2α phosphorylation was similar in magnitude to that observed with thaspigargin (TG, used as positive control). Based on these data, we conclude that exposure to acrolein results in a dose-dependent, robust, and early increase in eIF-2α phosphorylation.
Fig. 1.

Acrolein treatment increases phosphorylation of eIF-2α: (A) Western blot analysis of lysates prepared from human umbilical vein endothelial cells (HUVECs) after 5, 10, 15, or 30 min of treatment with the indicated concentrations of acrolein. Upper panels show representative Western blots developed with anti-phospho eIF-2α and those developed with an anti-eIF-2α antibody which recognizes the non-phosphorylated and phosphorylated forms of the protein. The extent of phosphorylation was calculated by normalizing the band intensity obtained with the phospho-specific antibody with the antibody detecting total eIF-2α protein and is presented as a bar graph (n=3). (B) Western blots for phospho- and total-eIF-2α in lysates of HUVECs treated with the indicated concentrations of acrolein for 30 min. Cells treated for 30 min with 1 μM thaspigargin (TG) were used as positive controls. Group data, calculated from the band intensity of phospho- normalized to total-eIF2α was shown in a bar graph (n=5). Data are presented as mean ± SE, * p<0.05, ** p<0.01, versus untreated cells; # p<0.05 versus cells treated with the same dose of acrolein for 10 min.
Next, we investigated activation of the bZIP transcription factor XBP-1. Activation of XBP-1 is a characteristic feature of ER stress (Schroder and Kaufman, 2005). XBP-1 gets activated by splicing of the 165bp containing unspliced pre-mRNA (uXBP-1) to the 139bp mature mRNA (sXBP-1) fragment. As shown in Fig. 2A, treatment with 5–10 μM acrolein for 2h resulted in a 3.5- to 16-fold increase in the alternatively spliced form of XBP-1. The increase in sXBP-1 mRNA at 10 μM acrolein was accompanied by a significant decrease in the levels of the uXBP-1 mRNA. Interestingly, treatment with 25 μM acrolein decreased XBP-1 splicing. The increase in sXBP-1 mRNA at 10 μM acrolein corresponded with a nearly 2-fold increase in sXBP-1 protein compared with a 3.4-fold increase in the abundance of this protein in cells treated with thaspigargin (TG, Fig. 2 inset). These data indicate that treatment with acrolein results in the appearance of an alternatively spliced form of XBP-1 mRNA which is accompanied by a corresponding increase in the abundance of sXBP-1 protein.
Fig. 2.

Acrolein induces alternative splicing of XBP-1 mRNA: (A) HUVECs were treated with the indicated concentrations of acrolein for 2h and RNA in the cell lysates was amplified by PCR using primers spanning the splice junction of XBP-1. (B) Data from densitometrical analyses of both unspliced (uXBP-1, 165 bp) and spliced (sXBP-1, 139 bp) XBP-1 mRNA are presented as mean ± SE; * p<0.05 and ** p<0.01 (n=3). (C) Western Blot analyses (inset) of lysates prepared from cells treated with 10 μM acrolein or 1 μM thapsigargin (TG) for 2 h developed with a specific antibody against the spliced variant of XBP-1 (sXBP-1). Arrow indicates the 54 kDa spliced form of XBP-1. Representative fold-changes in band intensity in treated cells normalized to untreated cells are listed.
Acrolein upregulates ATF4/CHOP and ATF3
Although eIF-2α activation suppresses transcription, it selectively leads to an increase in ATF4. It is currently believed that the newly synthesized ATF4 then increases the transcription of CHOP and ATF3 (Xu, Bailly-Maitre, and Reed, 2005; Zhang and Kaufman, 2004). CHOP and ATF3 have opposing effects. While CHOP promotes apoptosis, ATF3 suppresses the expression of CHOP and prevents inflammation. To examine these pathways, the cells were treated with different concentrations of acrolein for variable times. Treatment with 10 μM acrolein led to a 3-fold increase in ATF4 protein 4h after treatment, which was comparable to the increase in AFT4 protein in thapsigargin-treated cells (Fig. 3A). In cells treated with 25 μM acrolein; however, no significant increase in ATF4 protein was observed. These observations indicate that acrolein, at low concentrations, is a potent inducer of ATF4. To examine events downstream to ATF4, changes in ATF3 (Fig. 3B) were measured in acrolein-treated cells. Treatment with 2 to 10 μM acrolein led to a significant increase in ATF3 expression. This increase was detectable in cells treated with 2 μM acrolein and was maximal at 10 μM. At higher concentrations (25 μM) acrolein failed to induce ATF3. Indeed the levels of ATF3 in cells treated with 25 μM acrolein were lower than in untreated cells. Because ATF3 is a transcription factor, we next studied its nuclear translocation. As shown in Fig. 3C, treatment with 10 μM acrolein resulted in a maximal (3-fold) increase in the nuclear abundance of ATF3. Nevertheless, 6 h after acrolein stimulation, the nuclear levels of ATF3 seem to decline, indicating that the protein is rapidly removed or degraded in the nucleus. These results indicate that an increase in ATF3 is an early and sensitive response mounted in cells exposed to low levels of acrolein.
Fig. 3.

Induction of ER-stress dependent transcription factors by acrolein: (A) Induction of ATF4. Representative Western blots of lysates prepared from HUVECs harvested 4h after treatment with 10 or 25 μM acrolein or 1 μM thapsigargin (TG) as described under Materials and Methods developed using anti-ATF4 antibodies. The intensity of the ATF4 positive bands was normalized to actin as shown. (B) Induction of ATF3. Western blots of lysates of HUVECs treated for 2h with the indicated concentrations of acrolein or 1 μM thapsigargin (TG). Group data of normalized band intensity was shown in the lower panel. (C) Nuclear translocation of ATF3. Nuclear extracts were separated from HUVECs treated with 0 or 10 μM acrolein and harvested after the indicated time. The intensity of the bands obtained upon Western analysis using anti-ATF3 antibody was normalized to the nuclear protein HDAC1. (D) Induction of CHOP by acrolein. Cells were harvested 6h after treatment with 10 μM acrolein as described under Materials and Methods and the CHOP mRNA was quantified by RT-PCR using CHOP-specific primer. Representative Western blot (lower panel) of lysates from HUVEC cells harvested 6h or 12h after treatment with 0 or 10 μM Acrolein developed using an antibody against CHOP using thapsigargin (TG) treated cells (6h, 1 μM) as positive control. (E) Inhibition of acrolein-stimulated ATF3 induction by PBA. HUVECs were left untreated or treated with 10 mM PBA as described under Materials and Methods and then incubated with 10 μM acrolein. Two hours after acrolein treatment, the cells were harvested and the abundance of ATF3 protein was examined by Western analysis. The band intensity due to ATF3 was normalized to actin. Data are presented as mean ± SE. * p<0.05, ** p<0.01, ***p<0.001 compared with control, and # p<0.05 PBA treated versus untreated cells (n=3–4).
In addition to ATF3, it has been shown that ATF4 also increases the transcription of the CHOP gene (Averous et al., 2004). Indeed, we found that treatment with acrolein led to a 6.5-fold increase in CHOP mRNA, 6 h after treatment (Fig. 3D). Nonetheless, no increase in CHOP protein was detected 6h or 12h after treatment with 10 μM acrolein. Further investigations using cells treated for 8 h with 25 μM acrolein likewise showed no induction of CHOP protein (data not shown).
To examine whether the increase in ATF3 is due to ER stress and UPR, the cells were treated with the chemical chaperone, PBA, which assists protein folding. Chemical chaperones such as PBA promote normal trafficking of mutant cystic fibrosis transmembrane conductance regulator from ER to the plasma membrane (Rubenstein et al., 1997), correct aberrant aquaporin-2 trafficking in nephrogenic diabetes (Tamarappoo et al., 1998) and enhance the secretion of the mutant α1-ATZ protein in α1-trypsin deficiency (Burrows et al., 2000). As before, treatment with 10 μM acrolein results in a 3-fold increase in ATF3, however, in cells treated with PBA, no increase in ATF3 was observed (Fig. 3E). Inhibition of acrolein-induced upregulation of ATF3 by the chemical chaperone suggests that induction of this transcription factor could be attributed to the unfolded protein response and could be prevented by diminishing ER stress.
Acrolein-induced activation of MAP Kinase
Activation of MAP kinases is another characteristic feature of the UPR (Xu et al., 2005; Zhang and Kaufman, 2004). Hence, we examined changes in the phosphorylation status of each of the MAP kinases in acrolein-treated cells. Western blot analysis with phospho-p38 specific antibodies showed early and persistent increase in the phosphorylation of this kinase. The levels of phospho-p38 were increased 2-fold within 5 min of acrolein exposure. The maximal increase in p38 phosphorylation was nearly 5-fold and was evident within 15 min of exposure (Fig. 4A). The increase was sustained throughout the 60 min observation period. Both 10 and 25 μM concentrations of acrolein gave similar responses. Given that p38 was activated earlier than other mediators of ER stress, we believe that it is unlikely to be due to the UPR, which, as shown above, developed with a much slower time course. A similar rapid response was observed with ERK, which was modestly (1.2 to 2.0-fold) stimulated 5 to 15 min of acrolein exposure (Fig. 4B). The increase in ERK phosphorylation declined after 30 min to acrolein exposure and was returned to nearly basal levels 60 min after treatment. The rapid time course of this kinase suggests also that its activation by acrolein is unlikely to be mediated by ER stress or UPR. In contrast, JNK activation was much slower (Fig. 4C). Western blots showed no detectable phosphorylation of p54 or p46 after 15 min of acrolein treatment. After 30 min, treatment with 10 μM acrolein resulted in a 3.4- and 5.6-fold increase in the phosphorylation of p54 and p46, respectively. Significantly, treatment with 25 μM acrolein for 30 min did not stimulate JNK phosphorylation, although a modest increase in phosphorylation was observed after 60 min. In contrast, treatment with 10 μM acrolein resulted in a 6.6- (p54) and 7.9-fold (p46) increase in JNK phosphorylation. Hence, both the time-course and the concentration-dependence of JNK phosphorylation were consistent with it being downstream of ER stress and UPR. However, to test directly whether acrolein-induced JNK phosphorylation is due ER stress, the cells were treated with PBA. As shown in Fig. 4D, treatment with PBA completely abolished the increase in JNK phosphorylation by 10 μM acrolein. In contrast, acrolein-induced phosphorylation of p38 was not affected by treatment with PBA (Fig. 4E), supporting further the UPR independent phosphorylation of p38. Collectively, these data indicate that acrolein treatment increases the phosphorylation of p38 and ERK independent of UPR. Phosphorylation of JNK, however, appears to be a delayed response and is triggered in part by ER stress.
Fig. 4.

Acrolein activates MAP Kinases: Representative Western blots prepared from lysates of HUVECs treated with 10 or 25 μM acrolein for the indicated times. Individual MAP kinases (A) p38, (B) ERK and (C) JNK were detected using phospho protein specific antibodies. The extent of phosphorylation was quantified by normalizing the intensity of the phospho-protein band with the intensity of the total protein band. Average fold-increase in phosphorylation is listed under each lane (n=3). (D) Inhibition of acrolein-induced JNK activation by PBA. HUVECs were pretreated with PBA, left untreated or treated with 10 μM acrolein for 60 min. Representative Western blots for total and phospho-JNK are shown. (E) Acrolein-induced phosphorylation of p38 was not affected by PBA. HUVEC without or after pretreatment with PBA were treated for 30 min with 10 μM Acrolein. Representative Western blots for total and phospho p38 are shown.
Acrolein induced expression of Grp78 and Herp
The final phase of the UPR is the induction of ER chaperone proteins, which is an attempt to restore ER function and increase the protein-folding capacity of the cell (Xu et al., 2005; Zhang and Kaufman, 2004). To examine, whether a brief treatment with acrolein triggers such adaptation, changes in the expression of Grp78 and Herp were examined. Grp78 is the ER chaperone protein and a master regulator of UPR. It sequesters UPR triggers to the ER member (Xu et al., 2005; Zhang and Kaufman, 2004). Herp is an ubiquitin domain ER protein that is an essential component of ER-associated protein degradation (ERAD) (Schulze et al., 2005). The Herp gene has two ER stress response elements ERSE and ESRE-II and it is regulated by XBP-1 (Kokame et al., 2001). Upregulation of Herp is a characteristic feature of ER stress. Results obtained from RT-PCR showed a significant elevation in the expression of Grp78 and Herp mRNA in cells treated with 10 μM acrolein, within 2 h of treatment (Fig. 5A). The increase in Grp78 mRNA was accompanied by an increase in Grp78 protein (Fig. 5B). Western analysis of cells treated with 10 μM acrolein showed a 2-fold increase in Grp78 protein 6h after treatment. Taken together, these data provide clear evidence that acrolein triggers UPR that results in the increase in ER proteins.
Fig. 5.

Acrolein treatment increases the expression of ER-resident chaperones of protein folding: (A) HUVECs were either left untreated or treated with 10 μM acrolein for 2 h. Changes in Grp78 and Herp mRNA were quantified by RT-PCR (n=3). (B) HUVECs treated with 10 μM acrolein were harvested after 6 h as described under Materials and Methods and Western blots were developed from their lysates using anti-Grp78 antibodies (n=6). Data are presented as mean ± SE, * p<0.05, # p=0.05).
Acrolein-induced activation of NF-κB
Our results so far indicated that acrolein induces ER stress and triggers UPR; however, to assess the functional outcomes of this pathway, we examined changes in cytokine production. For this, we measured nuclear translocation of NF-κB in cells treated with acrolein. As shown in Fig. 6A, treatment with 10 μM acrolein led to a significant increase in the nuclear abundance of the NF-κB p65-unit within 30 min of stimulation. Maximal increase was observed at 1 h after treatment, whereas at 2 to 4 h NF-κB content of the nucleus returned to basal values. The increase in NF-κB-p65 translocation was diminished by pretreatment with PBA (Fig 6B), indicating that acrolein activates NF-κB by a UPR-sensitive mechanism. Activation of NF-κB by acrolein was accompanied with an increase in the transcription of genes encoding cytokines and chemokines. As shown in Fig. 7A, treatment of cells with 10 μM acrolein for 2 h resulted in a significant increase in IL-6 and TNF-α mRNA, whereas IL-8 and MCP-1 mRNA levels were not affected. In contrast, treatment with thapsigargin (TG, Fig. 7A, inset) results additively in an increase of MCP-1 and IL-8 mRNA. Treatment with acrolein led to a 6-fold increase in the transcription of IL-6 gene. The increase in IL-6 by acrolein was 2-fold higher than by thapsigargin. However, thapsigargin treatment results in an approximately 8-fold higher level of TNF-α mRNA than observed in cells treated with acrolein. In cells harvested 6h after acrolein treatment, a 3.4-fold increase in IL-8 mRNA was observed, whereas IL-6 was increased 4-fold and TNF-α 15-fold (Fig. 7B). Again, no increase in the transcription of MCP-1 mRNA was found.
Fig. 6.

Acrolein increases NF-κB activation: (A) Representative Western blots prepared from nuclear extracts of HUVECs treated with 10 μM acrolein harvested after the indicated time. Nuclear abundance of p65 was quantified using anti-NF-κB antibodies and p65 band intensity was normalized to HDAC1 band intensity in 4 independent experiments. (B) Inhibition of acrolein-stimulated NF-κB activation by PBA. HUVECs were left untreated or pretreated with 10 mM PBA and then incubated with 10 μM acrolein. One hour after acrolein treatment, the cells were harvested and the translocation of the p65 unit of NF-κB was examined by Western analysis of nuclear extracts. The band intensity of NF-κB-p65 of 3 independent experiments was normalized to HDAC1 as nuclear loading control. Data are presented as mean ± SE, * p<0.05, ** p<0.01, and ## p<0.01 PBA treated versus untreated cells.
Fig. 7.

Acrolein increases the transcription of inflammatory cytokines and chemokines by endothelial cells via ER stress. HUVECs were treated with 10 μM acrolein and harvested after 2 (A) or 6 (B) h as described in Materials and Methods. Levels of IL-6, IL-8, TNF-α and MCP-1 mRNA was quantified by RT-PCR of 3 (2h) or 6 (6h) independent experiments. Thapsigargin (1 μM, 2h) treated HUVECs were used as positive controls (A, insert) for the 2h treatment. Inhibition of the acrolein induced cytokine and chemokine transcription by PBA. HUVECs were left untreated or pretreated with 10 mM PBA and then incubated with 10 μM acrolein. Two (C) or 6 (D) hours after acrolein treatment, the cells were harvested and levels of IL-6 and TNF-α mRNA (2h) or IL-6, IL-8 and TNF-α-mRNA (6h) were examined by RT-PCR. Data from 3 to 5 independent experiments were analyzed and presented as values normalized to control. Data are presented as mean ± SE, * p<0.05, ** p<0.01, ***p<0.001, and ### p<0.01, ### p<0.001 PBA treated versus untreated cells.
To examine whether the increases in IL-6, TNF-α and IL-8 mRNA levels were dependent on ER stress and UPR, the cells were treated with the chemical chaperone PBA. As shown in Fig. 7D & E, treatment with PBA completely abolished the increase in the transcription of these genes both 2 and 6 h after acrolein treatment. Taken together, these observations suggest that acrolein causes endothelial activation characterized by NF-κB activation and increased transcription of IL-6, IL-8 and TNF-α-mRNA. These findings also support the notion that endothelial activation by acrolein is, in part, mediated by ER stress and decreasing ER stress could diminish the acrolein-induced inflammatory response of endothelial cells.
DISCUSSION
The major findings of this study are that in endothelial cells, acrolein, a ubiquitous pollutant and an endogenous by product of lipid oxidation and myeloperoxidase, activates ER stress-dependent signaling pathways (Scheme 1) and that treatment with the chemical chaperone PBA prevents acrolein-induced activation of UPR components ATF3 and JNK and decreases cytokine synthesis. These observations reveal a novel mechanism of acrolein-induced toxicity and provide a new therapeutic modality for the treatment of toxicological and pathological states associated with excessive acrolein generation and accumulation.
Scheme 1.
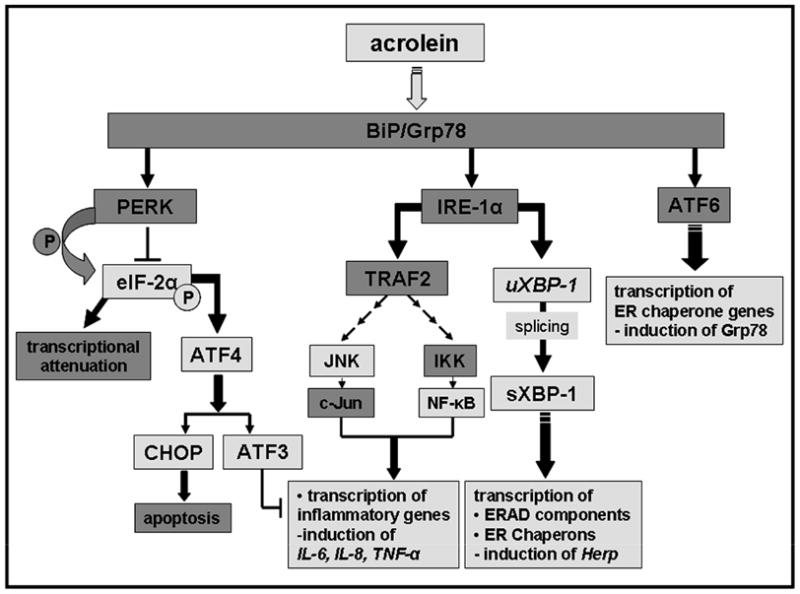
Activation of the unfolded protein response by acrolein. Specific signaling events activated by acrolein are indicated by light grey highlighted boxes.
Human exposures to acrolein are common and frequent. The concentration of S-(3-hydroxypropyl)mercapturic acid (HPMA) in the urine of healthy, non-smoking adults was 220 μg to 422 μg/L (or 0.7 to 1.9 μM) (Carmella et al., 2007; Roethig et al., 2007). This concentration of the acrolein metabolite is 100-fold higher than that of the corresponding metabolite of 4-hydroxy trans-2-nonenal (HNE) (Alary et al., 1998), which is an abundant product of lipid peroxidation. Given that only 56–67 % of acrolein is excreted in the urine and that HPMA accounts for only 60 % of the urinary metabolites of acrolein (Parent et al., 1996), the corresponding concentration of acrolein generated basally is likely to be near 1–2 μM. This concentration is likely to be 2 to 4-fold higher in smokers (Carmella et al., 2007). Undoubtedly, local and tissue-specific concentrations of acrolein at sites of generation or exposure are likely to be higher. Hence, the concentration range at which acrolein induces ER stress (2 to 10 μM) appears to be within the range of human exposures and the accumulation of acrolein associated with several pathological and toxicological states.
Acrolein exposures profoundly affect the endothelium. Acute exposure to acrolein induces endothelium-dependent vasodilation (Awe et al., 2006; Tsakadze et al., 2003), whereas, chronic acrolein exposure lowers contractile response to phenylephrine (Tsakadze et al., 2003). These effects may be related perhaps to activation of eNOS by acrolein (Tsakadze et al., 2003) by increasing intracellular calcium (Misonou et al., 2006). Endogenously generated acrolein could similarly affect the endothelium. Acrolein generated in oxidized lipoprotein or by myeloperoxidase is likely to accumulate in highly vascular regions and thus contribute to endothelial dysfunction. In this regard, the observations reported here demonstrate for the first time that acrolein increases the production of IL-6, TNF-α and IL-8 in endothelial cells. These findings reveal new proinflammatory and atherogenic aspects of acrolein toxicity and they raise the possibility that endogenously generated acrolein could contribute endothelial injury and inflammation. Because an increase in the production of cytokines such as TNF-α, IL-6 and IL-8 is a significant feature of atherogenesis (Daugherty et al., 2005; Tedgui et al., 2006), the generation of acrolein in arterial lesions and other sites of vascular inflammation could contribute to or promote atherogenesis. Likewise, acrolein, which is the major electrophilic component of tobacco smoke, may be partly responsible for the ability of tobacco smoke to increase NF-κB activation and monocyte adhesion to endothelial cells and to impair acetylcholine-induced arterial relaxation (Orosz et al., 2007).
Acrolein-induced increase in cytokine production was accompanied by NF-κB activation. Although increased transcriptional activation of genes encoding cytokines is frequently associated with NF-κB activation, it has been previously reported that exposure to acrolein decreases NF-κB activation in lung adenocarcinoma cells (Horton et al., 1999) and that acrolein inhibits NF-κB activation after mitogenic stimulation of T-cells (Lambert et al., 2007). The paradoxical anti-inflammatory effect of acrolein was ascribed to direct modification of NF-κB (Horton et al., 1999; Lambert et al., 2007). In contrast with these observations, we found transient increase in NF-κB activation, 0.5–1 h after treatment, after which the NF-κB returned to baseline. Hence, under the conditions used in the present study, acrolein led to an increase in NF-κB activation, thereby contributing to an inflammatory phenotype. Although multiple mechanisms can account for NF-κB activation, the results presented here suggest that ER stress may be an important mechanism for NF-κB activation as well as the increase in cytokine production seen in cells treated with acrolein.
Multiple lines of evidence suggest that acrolein is a potent inducer of ER stress. Results of our experiment show that treatment with acrolein led to the activation of multiple signaling pathways that are associated with the induction of ER stress and stimulation of the UPR. Hence, our results showing that acrolein-treated cells show an increase in eIF-2α phosphorylation, XBP-1 splicing, the induction of the ER stress responsive transcription factors - ATF3, ATF4, and CHOP, and the increase in the synthesis of ER protein – Grp78 and Herp, clearly demonstrate that acrolein triggers both the alarm and the adaptive phases of the response. Significantly, several of the adaptive responses (e.g. increases in XBP-1 splicing, ATF3, and ATF4) observed at lower concentrations of acrolein were abolished at high concentrations. While current data does not provide clear evidence why this may be the case, we speculate that high concentrations of acrolein (> 10 μM) could inhibit adaptive changes by either directly alkylating cell proteins that mediate UPR or by triggering other responses such as autophagy. Indeed our recent observations show that 25 μM acrolein activates autophagic responses (Hill et al., 2008) which could attenuate UPR by ER degradation. Alternately, low doses of acrolein activate UPR, while higher concentrations could inhibit UPR mediators by direct alkylation. A similar mechanism has been proposed to explain the effects of acrolein on caspases. Acrolein increases the cleavage of pro-caspase 3, but inhibits its activity, perhaps by direct alkylation of its active site cysteines. Other caspases are similarly affected as well. As a result, low doses of acrolein increase apoptosis, whereas apoptosis is inhibited at higher concentrations (Tanel et al., 2005). In our experiments, however, no cell death was observed and UPR responses were suppressed at 25 μM acrolein without activation of the apoptotic mediator CHOP, indicating that acrolein could prevent adaptive responses without triggering cell death. Similar results have been reported by Wu and coworkers (Wu et al., 2006) who found that HO-1 was induced in glutathione-depleted endothelial cells at 10 μM acrolein, but that the levels of HO-1 were reduced at 25 μM acrolein, indicating that at high concentrations of acrolein, the adaptive responses seen at lower concentrations is abolished or overwhelmed. Hence, high concentrations of acrolein are likely to be particularly cytotoxic because they prevent adaptive responses.
The ER stress-triggered UPR include two major branches; one that is specific to ER stress (mediated by IRE1/XBP-1 and AFT6) and the other that is shared by other cellular stress (the PERK-eIF-2α pathway) (Ma et al., 2004). Although not all mediators were examined, our results indicate that acrolein activates both these branches. The increase in XBP-1 splicing and JNK activation by acrolein is consistent with activation of ER stress and UPR-specific responses. In addition, the increases in eIF-2α phosphorylation, ATF3, ATF4, and CHOP suggest that the shared PERK pathway is also activated. That these pathways are activated specifically in response to ER stress is reinforced by the observation that treatment with the chemical chaperone PBA prevented JNK and NF-κB activation and the induction of ATF3, TNF-α, IL-6, and IL-8. The increase in the transcription of Herp and Grp78 genes in acrolein-treated cells further suggest that the ATF6-dependent, ER-stress specific pathway is also activated. Collectively, these findings provide strong evidence supporting the notion that acrolein treatment induces ER stress, which in turn leads to the activation of the major pathways of UPR. Nevertheless, activation of other processes (e.g., p38) seemed to be independent of ER stress. Hence, overall it appears that exposure to acrolein induces a variety of responses, of which ER stress appears to be a significant and critical component and that UPR trigger by ER stress induces inflammatory responses in the endothelium.
The mechanisms by which acrolein induces ER stress remain unknown. Acrolein increases intracellular calcium (Hyvelin et al., 2000; Misonou et al., 2006), induces covalent protein modifications (Burcham et al., 2006; Luo et al., 2007; Uchida et al., 1998b), depletes intracellular glutathione (Biswal et al., 2002; Ghilarducci and Tjeerdema, 1995; Patel et al., 1993) and increases the cellular generation of reactive oxygen species (Adams, Jr. et al., 1993; Li et al., 2008). Any of these changes could in principle contribute to ER stress or could act synergistically to disrupt proper protein folding and trigger UPR. Clearly, further studies are required to deconstruct specific mechanisms and to assess the contribution of each of these processes to acrolein-mediated ER stress. Nevertheless, data presented here reveal for the first time that acrolein is a potent inducer of ER stress. In addition, the study also suggest that remedial or preventive therapy with chemical chaperone may be a useful strategy for prevent pathologic or toxicologic changes in the endothelium induced by acrolein. Chemical chaperones have excellent in vivo safety profiles and have been approved by USFDA for human use (Ozcan et al., 2006) and hence their use represents a potentially beneficial modality for decreasing acrolein toxicity.
Acknowledgments
This work was supported in part by NIH grants ES11860, HL55477, HL59378, and HL65618 and a grant from Philip Morris.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Adams JD, Jr, Klaidman LK. Acrolein-induced oxygen radical formation. Free Radic Biol Med. 1993;15:187–193. doi: 10.1016/0891-5849(93)90058-3. [DOI] [PubMed] [Google Scholar]
- 2.Alary J, Debrauwer L, Fernandez Y, Cravedi JP, Rao D, Bories G. 1,4-Dihydroxynonene mercapturic acid, the major end metabolite of exogenous 4-hydroxy-2-nonenal, is a physiological component of rat and human urine. Chem Res Toxicol. 1998;11:130–135. doi: 10.1021/tx970139w. [DOI] [PubMed] [Google Scholar]
- 3.Anderson MM, Hazen SL, Hsu FF, Heinecke JW. Human neutrophils employ the myeloperoxidase-hydrogen peroxide-chloride system to convert hydroxy-amino acids into glycolaldehyde, 2-hydroxypropanal, and acrolein. A mechanism for the generation of highly reactive alpha-hydroxy and alpha, beta-unsaturated aldehydes by phagocytes at sites of inflammation. J Clin Invest. 1997;99:424–432. doi: 10.1172/JCI119176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Averous J, Bruhat A, Jousse C, Carraro V, Thiel G, Fafournoux P. Induction of CHOP expression by amino acid limitation requires both ATF4 expression and ATF2 phosphorylation. J Biol Chem. 2004;279:5288–5297. doi: 10.1074/jbc.M311862200. [DOI] [PubMed] [Google Scholar]
- 5.Awe SO, Adeagbo AS, D’Souza SE, Bhatnagar A, Conklin DJ. Acrolein induces vasodilatation of rodent mesenteric bed via an EDHF-dependent mechanism. Toxicol Appl Pharmacol. 2006;217:266–276. doi: 10.1016/j.taap.2006.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnoya J, Glantz SA. Cardiovascular effects of secondhand smoke: nearly as large as smoking. Circulation. 2005;111:2684–2698. doi: 10.1161/CIRCULATIONAHA.104.492215. [DOI] [PubMed] [Google Scholar]
- 7.Bhatnagar A. Electrophysiological effects of 4-hydroxynonenal, an aldehydic product of lipid peroxidation, on isolated rat ventricular myocytes. Circ Res. 1995;76:293–304. doi: 10.1161/01.res.76.2.293. [DOI] [PubMed] [Google Scholar]
- 8.Bhatnagar A. Cardiovascular pathophysiology of environmental pollutants. Am J Physiol Heart Circ Physiol. 2004;286:H479–H485. doi: 10.1152/ajpheart.00817.2003. [DOI] [PubMed] [Google Scholar]
- 9.Bhatnagar A. Environmental cardiology: studying mechanistic links between pollution and heart disease. Circ Res. 2006;99:692–705. doi: 10.1161/01.RES.0000243586.99701.cf. [DOI] [PubMed] [Google Scholar]
- 10.Biswal S, cquaah-Mensah G, Datta K, Wu X, Kehrer JP. Inhibition of cell proliferation and AP-1 activity by acrolein in human A549 lung adenocarcinoma cells due to thiol imbalance and covalent modifications. Chem Res Toxicol. 2002;15:180–186. doi: 10.1021/tx015552p. [DOI] [PubMed] [Google Scholar]
- 11.Burcham PC, Pyke SM. Hydralazine inhibits rapid acrolein-induced protein oligomerization: role of aldehyde scavenging and adduct trapping in cross-link blocking and cytoprotection. Mol Pharmacol. 2006;69:1056–1065. doi: 10.1124/mol.105.018168. [DOI] [PubMed] [Google Scholar]
- 12.Burrows JA, Willis LK, Perlmutter DH. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: A potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency. Proc Natl Acad Sci USA. 2000;97:1796–1801. doi: 10.1073/pnas.97.4.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carmella SG, Chen M, Zhang Y, Zhang S, Hatsukami DK, Hecht SS. Quantitation of acrolein-derived (3-hydroxypropyl)mercapturic acid in human urine by liquid chromatography-atmospheric pressure chemical ionization tandem mass spectrometry: effects of cigarette smoking. Chem Res Toxicol. 2007;20:986–990. doi: 10.1021/tx700075y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daimon M, Sugiyama K, Kameda W, Saitoh T, Oizumi T, Hirata A, Yamaguchi H, Ohnuma H, Igarashi M, Kato T. Increased urinary levels of pentosidine, pyrraline and acrolein adduct in type 2 diabetes. Endocr J. 2003;50:61–67. doi: 10.1507/endocrj.50.61. [DOI] [PubMed] [Google Scholar]
- 15.Daugherty A, Webb NR, Rateri DL, King VL. Thematic review series: The immune system and atherogenesis. Cytokine regulation of macrophage functions in atherogenesis. J Lipid Res. 2005;46:1812–1822. doi: 10.1194/jlr.R500009-JLR200. [DOI] [PubMed] [Google Scholar]
- 16.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 17.Feron VJ, Til HP, de VF, Woutersen RA, Cassee FR, van Bladeren PJ. Aldehydes: occurrence, carcinogenic potential, mechanism of action and risk assessment. Mutat Res. 1991;259:363–385. doi: 10.1016/0165-1218(91)90128-9. [DOI] [PubMed] [Google Scholar]
- 18.Ghilarducci DP, Tjeerdema RS. Fate and effects of acrolein. Rev Environ Contam Toxicol. 1995;144:95–146. doi: 10.1007/978-1-4612-2550-8_2. [DOI] [PubMed] [Google Scholar]
- 19.Hill BG, Haberzettl P, Ahmed Y, Srivastava S, Bhatnagar A. Unsaturated lipid peroxidation-derived aldehydes activate autophagy in vascular smooth-muscle cells. Biochem J. 2008;410:525–534. doi: 10.1042/BJ20071063. [DOI] [PubMed] [Google Scholar]
- 20.Horton ND, Biswal SS, Corrigan LL, Bratta J, Kehrer JP. Acrolein causes inhibitor kappaB-independent decreases in nuclear factor kappaB activation in human lung adenocarcinoma (A549) cells. J Biol Chem. 1999;274:9200–9206. doi: 10.1074/jbc.274.14.9200. [DOI] [PubMed] [Google Scholar]
- 21.Howard G, Wagenknecht LE, Burke GL, ez-Roux A, Evans GW, McGovern P, Nieto FJ, Tell GS. Cigarette smoking and progression of atherosclerosis: The Atherosclerosis Risk in Communities (ARIC) Study. JAMA. 1998;279:119–124. doi: 10.1001/jama.279.2.119. [DOI] [PubMed] [Google Scholar]
- 22.Hyvelin JM, Roux E, Prevost MC, Savineau JP, Marthan R. Cellular mechanisms of acrolein-induced alteration in calcium signaling in airway smooth muscle. Toxicol Appl Pharmacol. 2000;164:176–183. doi: 10.1006/taap.1999.8879. [DOI] [PubMed] [Google Scholar]
- 23.Jaimes EA, DeMaster EG, Tian RX, Raij L. Stable compounds of cigarette smoke induce endothelial superoxide anion production via NADPH oxidase activation. Arterioscler Thromb Vasc Biol. 2004;24:1031–1036. doi: 10.1161/01.ATV.0000127083.88549.58. [DOI] [PubMed] [Google Scholar]
- 24.Kokame K, Kato H, Miyata T. Identification of ERSE-II, a new cis-acting element responsible for the ATF6-dependent mammalian unfolded protein response. J Biol Chem. 2001;276:9199–9205. doi: 10.1074/jbc.M010486200. [DOI] [PubMed] [Google Scholar]
- 25.Lambert C, Li J, Jonscher K, Yang TC, Reigan P, Quintana M, Harvey J, Freed BM. Acrolein inhibits cytokine gene expression by alkylating cysteine and arginine residues in the NF-kappaB1 DNA binding domain. J Biol Chem. 2007;282:19666–19675. doi: 10.1074/jbc.M611527200. [DOI] [PubMed] [Google Scholar]
- 26.Li L, Jiang L, Geng C, Cao J, Zhong L. The role of oxidative stress in acrolein-induced DNA damage in HepG2 cells. Free Radic Res. 2008;42:354–361. doi: 10.1080/10715760802008114. [DOI] [PubMed] [Google Scholar]
- 27.Lovell MA, Xie C, Markesbery WR. Acrolein is increased in Alzheimer’s disease brain and is toxic to primary hippocampal cultures. Neurobiol Aging. 2001;22:187–194. doi: 10.1016/s0197-4580(00)00235-9. [DOI] [PubMed] [Google Scholar]
- 28.Luo J, Hill BG, Gu Y, Cai J, Srivastava S, Bhatnagar A, Prabhu SD. Mechanisms of acrolein-induced myocardial dysfunction: implications for environmental and endogenous aldehyde exposure. Am J Physiol Heart Circ Physiol. 2007;293:H3673–H3684. doi: 10.1152/ajpheart.00284.2007. [DOI] [PubMed] [Google Scholar]
- 29.Ma Y, Hendershot LM. Herp is dually regulated by both the endoplasmic reticulum stress-specific branch of the unfolded protein response and a branch that is shared with other cellular stress pathways. J Biol Chem. 2004;279:13792–13799. doi: 10.1074/jbc.M313724200. [DOI] [PubMed] [Google Scholar]
- 30.Misonou Y, Asahi M, Yokoe S, Miyoshi E, Taniguchi N. Acrolein produces nitric oxide through the elevation of intracellular calcium levels to induce apoptosis in human umbilical vein endothelial cells: implications for smoke angiopathy. Nitric Oxide. 2006;14:180–187. doi: 10.1016/j.niox.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 31.Mottram DS, Wedzicha BL, Dodson AT. Acrylamide is formed in the Maillard reaction. Nature. 2002;419:448–449. doi: 10.1038/419448a. [DOI] [PubMed] [Google Scholar]
- 32.Orosz Z, Csiszar A, Labinskyy N, Smith K, Kaminski PM, Ferdinandy P, Wolin MS, Rivera A, Ungvari Z. Cigarette smoke-induced proinflammatory alterations in the endothelial phenotype: role of NAD(P)H oxidase activation. Am J Physiol Heart Circ Physiol. 2007;292:H130–H139. doi: 10.1152/ajpheart.00599.2006. [DOI] [PubMed] [Google Scholar]
- 33.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parent RA, Caravello HE, Sharp DE. Metabolism and distribution of [2,3-14C]acrolein in Sprague-Dawley rats. J Appl Toxicol. 1996;16:449–457. doi: 10.1002/(SICI)1099-1263(199609)16:5<449::AID-JAT369>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 35.Park YS, Kim J, Misonou Y, Takamiya R, Takahashi M, Freeman MR, Taniguchi N. Acrolein induces cyclooxygenase-2 and prostaglandin production in human umbilical vein endothelial cells: roles of p38 MAP kinase. Arterioscler Thromb Vasc Biol. 2007;27:1319–1325. doi: 10.1161/ATVBAHA.106.132837. [DOI] [PubMed] [Google Scholar]
- 36.Patel JM, Block ER. Acrolein-induced injury to cultured pulmonary artery endothelial cells. Toxicol Appl Pharmacol. 1993;122:46–53. doi: 10.1006/taap.1993.1170. [DOI] [PubMed] [Google Scholar]
- 37.Peters A, von KS, Heier M, Trentinaglia I, Hormann A, Wichmann HE, Lowel H. Exposure to traffic and the onset of myocardial infarction. N Engl J Med. 2004;351:1721–1730. doi: 10.1056/NEJMoa040203. [DOI] [PubMed] [Google Scholar]
- 38.Roethig HJ, Zedler BK, Kinser RD, Feng S, Nelson BL, Liang Q. Short-term clinical exposure evaluation of a second-generation electrically heated cigarette smoking system. J Clin Pharmacol. 2007;47:518–530. doi: 10.1177/0091270006297686. [DOI] [PubMed] [Google Scholar]
- 39.Rubenstein RC, Egan ME, Zeitlin PL. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J Clin Invest. 1997;100:2457–2465. doi: 10.1172/JCI119788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 41.Schulze A, Standera S, Buerger E, Kikkert M, van VS, Wiertz E, Koning F, Kloetzel PM, Seeger M. The ubiquitin-domain protein HERP forms a complex with components of the endoplasmic reticulum associated degradation pathway. J Mol Biol. 2005;354:1021–1027. doi: 10.1016/j.jmb.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 42.Shao B, Fu X, McDonald TO, Green PS, Uchida K, O’Brien KD, Oram JF, Heinecke JW. Acrolein impairs ATP binding cassette transporter A1-dependent cholesterol export from cells through site-specific modification of apolipoprotein A-I. J Biol Chem. 2005;280:36386–36396. doi: 10.1074/jbc.M508169200. [DOI] [PubMed] [Google Scholar]
- 43.Tamarappoo BK, Verkman AS. Defective aquaporin-2 trafficking in nephrogenic diabetes insipidus and correction by chemical chaperones. J Clin Invest. 1998;101:2257–2267. doi: 10.1172/JCI2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanel A, verill-Bates DA. The aldehyde acrolein induces apoptosis via activation of the mitochondrial pathway. Biochim Biophys Acta. 2005;1743:255–267. doi: 10.1016/j.bbamcr.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 45.Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev. 2006;86:515–581. doi: 10.1152/physrev.00024.2005. [DOI] [PubMed] [Google Scholar]
- 46.Tsakadze NL, Srivastava S, Awe SO, Adeagbo AS, Bhatnagar A, D’Souza SE. Acrolein-induced vasomotor responses of rat aorta. Am J Physiol Heart Circ Physiol. 2003;285:H727–H734. doi: 10.1152/ajpheart.00269.2003. [DOI] [PubMed] [Google Scholar]
- 47.Uchida K, Kanematsu M, Morimitsu Y, Osawa T, Noguchi N, Niki E. Acrolein is a product of lipid peroxidation reaction. Formation of free acrolein and its conjugate with lysine residues in oxidized low density lipoproteins. J Biol Chem. 1998a;273:16058–16066. doi: 10.1074/jbc.273.26.16058. [DOI] [PubMed] [Google Scholar]
- 48.Uchida K, Kanematsu M, Sakai K, Matsuda T, Hattori N, Mizuno Y, Suzuki D, Miyata T, Noguchi N, Niki E, Osawa T. Protein-bound acrolein: potential markers for oxidative stress. Proc Natl Acad Sci USA. 1998b;95:4882–4887. doi: 10.1073/pnas.95.9.4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu CC, Hsieh CW, Lai PH, Lin JB, Liu YC, Wung BS. Upregulation of endothelial heme oxygenase-1 expression through the activation of the JNK pathway by sublethal concentrations of acrolein. Toxicol Appl Pharmacol. 2006;214:244–252. doi: 10.1016/j.taap.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 50.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang K, Kaufman RJ. Signaling the unfolded protein response from the endoplasmic reticulum. J Biol Chem. 2004;279:25935–25938. doi: 10.1074/jbc.R400008200. [DOI] [PubMed] [Google Scholar]