

Abstract
We observed that cells lacking Rep and UvrD, two replication accessory helicases, and the recombination protein RecF are cryo-sensitive on rich medium. We isolated five mutations that suppress this Luria–Bertani (LB)-cryo-sensitivity and show that they map in the genes encoding the RNA polymerase subunits RpoB and RpoC. These rpoB (D444G, H447R and N518D) and rpoC mutants (H113R and P451L) were characterized. rpoBH447R and rpoBD444G prevent activation of the Prrn core promoter in rich medium, but only rpoBH447R also suppresses the auxotrophy of a relA spoT mutant (stringent-like phenotype). rpoCH113R suppresses the thermo-sensitivity of a greA greB mutant, suggesting that it destabilizes stalled elongation complexes. All mutations but rpoCP451L prevent R-loop formation. We propose that these rpo mutations allow replication in the absence of Rep and UvrD by destabilizing RNA Pol upon replication–transcription collisions. In a RecF+ context, they improve growth of rep uvrD cells only if DinG is present, supporting the hypothesis that Rep, UvrD and DinG facilitate progression of the replication fork across transcribed sequences. They rescue rep uvrD dinG recF cells, indicating that in a recF mutant replication forks arrested by unstable transcription complexes can restart without any of the three known replication accessory helicases Rep, UvrD and DinG.
Introduction
Replication forks are susceptible to be arrested by a variety of obstacles, including DNA-bound proteins such as RNA polymerases (RNA Pol) (Mirkin and Mirkin, 2007; Rudolph et al., 2007). DNA instability is associated with replication fork arrest (reviewed in Aguilera and Gomez-Gonzalez, 2008), and in order to limit the deleterious consequences of replication–transcription collisions, cells encode enzymes that facilitate replication through transcription units. In yeast, the Rrm3 helicase travels with the replication fork machinery and dislodges RNA Pols from tRNA and rRNA genes as well as tightly bound proteins from heterochromatin (Azvolinsky et al., 2009 and references therein). In Escherichia coli, this function was originally ascribed to the Rep helicase; first, based on the observations that in the rep mutant chromosome replication is slowed down and requires specific functions (Lane and Denhardt, 1975; Seigneur et al., 1998; Petit and Ehrlich, 2002), and second, because the purified Rep helicase is specifically capable of dislodging DNA-bound proteins (Yancey-Wrona and Matson, 1992). More recently, evidence was provided that transcribed sequences are indeed a major obstacle to replication in rapidly growing cells, and that Rep is the main, although not the only, helicase that assists replication progression across highly transcribed sequences (Guy et al., 2009; Boubakri et al., 2010).
Rep and UvrD are two 3′ to 5′ helicases that share 40% homology. The existence of a redundant function for these paralogues has been suspected since the original observation that the rep uvrD double mutant shows severe growth defects (Taucher-Scholtz et al., 1983). However, the situation turned out to be more complex and UvrD is now known to play two different roles in rep mutants, revealed by the two classes of mutations that suppress the growth defects of the rep uvrD double mutant. The first class of mutations that was identified inactivates the RecFOR recombination pathway (Petit and Ehrlich, 2002). Combined with the observation that UvrD can remove RecA from ssDNA in vitro, the rescue of rep uvrD cells by the inactivation of recombination proteins led to the proposal that UvrD is essential in rep mutants because it removes deleterious RecFOR-dependent RecA-filaments that assemble at blocked replication forks (Veaute et al., 2005; Lestini and Michel, 2008). However, the nature of the obstacles arresting replication in the first place, thus allowing RecFOR-RecA binding to forks in rep mutants, remained unknown. Moreover, the suppression of rep uvrD co-lethality by recFOR inactivation was shown to be only partial, confirming the existence of an obstacle to replication restart other than RecFOR-RecA bound to DNA in rep uvrD cells (Guy et al., 2009; Boubakri et al., 2010). Two lines of evidence identified RNA polymerases as the original cause of replication arrest in rep uvrD cells: first, mutations that map in the RNA Pol genes rpoB and rpoC were shown to suppress the growth defects of this double mutant (Guy et al., 2009; Boubakri et al., 2010); second, replication arrest sites were directly visualized in rep and rep uvrD recF mutants at inverted ribosomal operons (rrn), provided that these operons were facing replication and were highly transcribed (Boubakri et al., 2010). Rep was confirmed to be the major accessory helicase and was shown to be attracted to replication forks by a direct interaction with the replicative helicase DnaB (Guy et al., 2009). In addition to UvrD, a third player was identified, the 5′ to 3′ helicase DinG (Boubakri et al., 2010). DinG is essential for the viability of the rep uvrD recF mutant and it was proposed that, provided that recF is inactivated, DinG can clear RNA Pols from blocked replication forks in the absence of both Rep and UvrD. In addition, DinG is essential for the viability of rep and uvrD single mutants when replication collides with RNA Pol at highly expressed inverted rrn, indicating that the presence of two of these three helicases is required when replication–transcription collisions are increased. Finally, DinG has the specific function of unwinding RNA–DNA hybrids in vivo, as it is also essential for viability when replication forks are arrested by R-loops (Boubakri et al., 2010).
The described RNA Pol mutations that suppress rep uvrD growth defects are mutations that constitutively confer phenotypes akin to the induction of the stringent response (Guy et al., 2009; Boubakri et al., 2010). The stringent response is an adaptation to amino acid starvation through the induction of the alarmone ppGpp (reviewed in Potrykus and Cashel, 2008). Binding of ppGpp to RNA Pol, as well as mutations that mimic this binding, affect transcription initiation from specific promoters, which decreases the expression of ribosomal operons (rrn) and activates the expression of amino acids biosynthetic genes in relA spoT double mutants, allowing their growth in minimal medium without amino acids (Bartlett et al., 1998; 2000; Zhou and Jin, 1998; Barker et al., 2001a,b;). The so-called ‘stringent mutations’ in RNA Pol mimic the presence of ppGpp in decreasing the half-life of open complexes. Although we have not measured the half-life of transcription open complexes, we will call here ‘stringent-like’ the phenotype conferred by RNA Pol mutations that both decrease rrn expression in rich medium and allow growth of relA spoT double mutants on minimal medium. RNA Pol mutations that increase transcription of amino acid biosynthetic promoters also destabilize transcription elongation complexes (TEC) (Trautinger and Lloyd, 2002; Trautinger et al., 2005). Such mutations were thought to suppress the lethality of rep uvrD double mutants owing to the destabilization of RNA Pol–DNA complexes (Guy et al., 2009; Boubakri et al., 2010).
In this work, we report that rep uvrD recF cells grow poorly on rich medium at low temperature and we isolated five mutations suppressing this LB-cryo-sensitivity. Similarly to the mutations that suppress the growth defects of rep uvrD mutants at 37°C, the suppressor mutations isolated here in rep uvrD recF mutants at 30°C map in rpoB and rpoC. One of these mutations is close to the active site; the others are in, or close to, the primary DNA–RNA binding channel, suggesting that they affect the stability of transcription complexes on DNA. Only one of these mutations exhibits a stringent-like phenotype, showing that our assay provides a new way of isolating RNA Pol mutants that are weakly bound to DNA, in transcription initiation or elongation complexes. Furthermore, these RNA Pol mutants allow us to extend our study of helicases that assist replication progression across transcription obstacles.
Results
rep uvrD recF mutants are cryo-sensitive
We constructed rep uvrD recF triple mutants in the presence of a conditional plasmid that carries the rep wild-type gene (pAM-rep; Lestini and Michel, 2008). This plasmid only replicates in the presence of the Lac promoter inducer, IPTG and is cured upon cell propagation in the absence of IPTG. We analysed the properties of rep uvrD recF cells cured of pAM-rep. For historical reasons the work was realized part in an AB1157 background (classically used for homologous recombination studies) and part in an MG1655 background (the more generally used, sequenced wild-type strain). In the AB1157 background, colony formation was delayed on LB at 37°C and 30°C; at 25°C only a variable subpopulation of cells formed colonies (JJC4048, Table 1). In the MG1655 background, results were similar except for a partial defect of plating efficiency on LB at 30°C (JJC5136 and JJC5166 Table 1). Finally, we constructed an Hfr strain, which allowed the co-introduction of the three rep uvrD recF mutations by conjugation, and observed that in this Hfr-PK3-PO131 background the growth defect was more pronounced than in other backgrounds, as plasmid-less rep uvrD recF cells were not recovered with the expected efficiency even on minimal medium (MM) at 37°C (not shown). In order to understand the reasons for the cryo-sensitivity of rep uvrD recF mutants, we studied five AB1157 rep uvrD recF suppressed clones that are able to form large colonies on LB at 30°C in 2 days. The rpoCΔ215–220 mutation, previously shown to restore the viability of rep uvrD cells at 37°C (Boubakri et al., 2010), and a suppressor mutation identified in the Hfr rep uvrD recF context at 37°C were included in this study.
Table 1.
rpoBsup and rpoCsup mutations suppress the LB-cryo-sensitivity of rep uvrD recF, rep uvrD recO and rep uvrD recQ.
| 37°C |
30°C |
25°C |
|||
|---|---|---|---|---|---|
| Strain | Relevant genotype | MM casa | LB | LB | LB |
| JJC4048 | rep uvrD recF | 1.3109 ± 3.7108 | 7.3108 ± 5108 | 8108 ± 2.2108 | < 107 |
| JJC4038/JJC4162 | rep uvrD recF rpoCP451L | 1.1109 ± 2.5108 | 2.2109 ± 2109 | 9.8108 ± 8.1108 | 6.4108 ± 1.1108 |
| JJC4039/JJC4170 JJC4053/JJC4340 | rep uvrD recF rpoCH113R | 1.0109 ± 1.7108 | 1.1109 ± 3.9108 | 9.3108 ± 4108 | 7.7108 ± 3.6108 |
| JJC4040/JJC4163 | rep uvrD recF rpoBH447R | 8.1108 ± 1.2108 | 8.7108 ± 4.5108 | 1.1109 ± 5.6108 | 1.1109 ± 6.2108 |
| JJC4041/JJC4164 | rep uvrD recF rpoBD444G | 8.3108 ± 2.1108 | 8.5108 ± 3.1108 | 8.2108 ± 6.7108 | 8.8108 ± 7.6108 |
| JJC4186/JJC4200 | rep uvrD recF rpoBN518D | 6.2108 ± 1108 | 8.7108 ± 2.3108 | 7.3108 ± 2.5108 | 6.3108 ± 2.1108 |
| JJC1706-S/JJC2488-S | rep uvrD recO | 1.1109 ± 7.9108 | 6.7108 ± 2.6108 | 5.4108 ± 1.1108 | < 107 |
| JJC4283 | rep uvrD recO rpoCP451L | 9.8108 ± 2.2108 | 8.6108 ± 1.9108 | 5.3108 ± 5.2108 | 6.1108 ± 1.7108 |
| JJC4344 | rep uvrD recO rpoCH113R | 1.3109 ± 5108 | 1.2109 ± 2.9108 | 1.4109 ± 1.1108 | 1.6109 ± 6.6108 |
| JJC4342 | rep uvrD recO rpoBH447R | 7.7108 ± 2.1108 | 7.3108 ± 2.5108 | 5108 ± 2108 | 5.1108 ± 1.9108 |
| JJC4324 | rep uvrD recO rpoBD444G | 1.2109 ± 8.3108 | 1.3109± 9.8108 | 1.3109 ± 1109 | 1.3109 ± 1.1109 |
| JJC4257 | rep uvrD recO rpoBN518D | 9.1108 ± 3.4108 | 6.8108 ± 1.3108 | 6.6108 ± 1.8108 | 5.6108 ± 2.5108 |
| JJC5261 | rep uvrD recO rpoCΔ215–220 | 3108 ± 1108 | 7.5108 ± 8.7107 | 6.1108 ± 1.1107 | 6.8108 ± 2.9107 |
| JJC3122-S | rep uvrD recQ | 5.8108 ± 1.3108 | 3.2107 ± 2.3107 | 3.1106 ± 3.3106 | < 107 |
| JJC4902/JJC5313 | rep uvrD recQ rpoCP451L | 1.1109 ± 2.5108 | 8.2108 ± 3.9108 | 6.6109 ± 4.6108 | 9.6108 ± 4.3108 |
| JJC4906 | rep uvrD recQ rpoCH113R | 1109 ± 4.2108 | 1.1109 ± 2.1108 | 1.2109 ± 1.3108 | 1.1109 ± 2.2108 |
| JJC4904 | rep uvrD recQ rpoBH447R | 8.9108 ± 3.4108 | 8.6108 ± 2.5108 | 7.6108 ± 1.7108 | 8.3108 ± 1.9108 |
| JJC4903 | rep uvrD recQ rpoBD444G | 6108 ± 1.4108 | 7.5108 ± 1.9108 | 7.2108 ± 1.4108 | 6.8108 ± 2.2108 |
| JJC4901/JJC5309 | rep uvrD recQ rpoBN518D | 7.2108 ± 1.5108 | 7.1108 ± 1.8108 | 5.1108 ± 1.8108 | 4.2106 ± 3.9106 |
| JJC4905 | rep uvrD recQ rpoCΔ215–220 | 7.3108 ± 5.3107 | 7.8108 ± 2.6107 | 8.5108 ± 6.6107 | 9.8108 ± 2.3108 |
| JJC5136a/JJC5166a | rep uvrD recF | 6.5108 ± 1.1108 | 2.8108 ± 1.7108 | 1.7107 ± 5.8106 | < 107 |
| JJC5152a/JJC5153a | rep uvrD recF rpoCΔ215–220 | 6.5108 ± 1.9108 | 6.3108 ± 1.5108 | 6.6108 ± 1.8108 | 5.1108 ± 8.4107 |
| JJC2451 (pEM001)-S | rep uvrD recF[pEM001] | 4.2108 ± 9107 | 2.2108 ± 1108 | < 107 | < 107 |
Context MG1655. All other strains are in an AB1157 context.
Colonies were counted after 24 h incubation (LB 37°C), 48 h incubation (MM 37°C and LB 30°C), or 3 days incubation (MM 30°C and LB 25°C). Numbers in italics indicate the formation of small colonies appearing 24 h later than wt.
JJCn-S indicates that the strain JJCn was used after segregation of the Rep encoding plasmid. A fresh plasmid-less colony was used for each experiment and cured clones were not kept.
pEM001 is a plasmid that overexpresses RNase H.
Suppressors of rep uvrD recF LB-cryo-sensitivity map in rpoC and rpoB genes
Analysis of the subpopulation of Hfr plasmid-less colonies formed in 2 days at 37°C on MM supplemented with casamino acids revealed that one of them was resistant to rifampicin. RifR mutations map in rpoB, the gene coding for the β subunit of RNA Pol, which suggested that this particular rep uvrD recF clone carries an rpoB mutation that suppresses the rep uvrD recF growth defect (JJC4100, Table S1). To ascertain that the RifR mutation was responsible for the improved growth of the rep uvrD recF RifR clone, pAM-rep was reintroduced and the suppressed clone was P1 transduced with thiC::Tn10, a locus close to the rpoB rpoC genes. As expected, the Rif resistance phenotype was 90% linked with thiC::Tn10 (43/48 TetR transductants were RifS). Curing of pAM-rep showed that rep uvrD recF thiC::Tn10 clones that had remained RifR had kept the capacity to form colonies overnight at 37°C on LB whereas the thiC::Tn10 clones that had lost the RifR phenotype grew as poorly as the original Hfr rep uvrD recF mutant. This mutation is therefore necessary and sufficient for the improved viability of rep uvrD recF cells. Sequencing of the rpoB gene revealed the presence of a mutation, N518D, in the rpoB RifR cluster 1.
Overnight cultures of AB1157 rep uvrD recF cells were plated on LB at 30°C. Five colonies isolated in three independent experiments were kept for further studies (called S1, S2, and S3a, S3b, S3m). Replacement in the five suppressed strains of the rep uvrD recF region of AB1157 by that of the Hfr JJC4100, or of the rep::ApR allele by a rep::CmR allele, did not modify their growth properties (Table S1, data not shown). To determine whether the isolated mutations map in the rpoBC genes, a TetS derivative of each suppressed clones was P1 transduced with thiC::Tn10. For each mutant 7–11 out of 12 thiC::Tn10 transductants became cryo-sensitive, indicating that these five suppressor mutations are linked to thiC.
A plasmid carrying an IPTG-inducible rpoB+ gene was introduced in ApS derivatives of the suppressed clones and viability was measured in the presence of IPTG (Table S2). Expression of RpoB rendered S3m cryo-sensitive on LB, suggesting that this mutant carries a recessive rpoB mutation; sequencing rpoB showed the presence of a H447R mutation. In S3a, the control vector pUC19 could not be introduced whereas the plasmid pUC-rpoB+ transformed with a normal efficiency (not shown); nevertheless, transformants remained cryo-resistant (Table S2). Sequencing rpoB revealed the presence of a D444G mutation. These observations suggest that, in addition to suppressing the rep uvrD recF growth defect, the rpoBD444G mutation somehow prevents pUC propagation. This defect in plasmid propagation is recessive (as it is complemented in cis by the wild-type rpoB+ allele) whereas the suppression of rep uvrD recF cryo-sensitivity is dominant over the wild-type allele. In S1 and S3b expression of rpoB+ did not prevent growth at 25°C (Table S2), so we sequenced rpoC in these two mutants and found a P451L mutation (S1), and a H113R mutation (S3b). Finally, both rpoB and rpoC genes were sequenced in S2. rpoB was intact and rpoC carried the H113R mutation, which was thus obtained twice independently. To our knowledge, the three rpoBD444G, rpoCH113R and rpoCP451L mutations have not been described previously, whereas the rpoBN518D allele has been already isolated in a screen for RifR clones (Garibyan et al., 2003), and the rpoBH447R allele in a screen for mutations that increase the expression of amino acid biosynthetic genes in a relA spoT context (Trautinger and Lloyd, 2002).
The rposup mutations are necessary and sufficient for the suppressor phenotype
To determine whether the rpo mutations isolated here (called rposup thereafter) are necessary and sufficient for the suppressor phenotype, these mutations were transferred to a rep uvrD recO strain by P1 co-transduction with thiC::Tn10[RecF is known to act in conjunction with two other proteins, RecO and RecR (Kuzminov, 1999), and inactivation of either the RecF, RecO or RecR protein allows colony formation of rep uvrD cells at 37°C on MM with casamino acids (Petit and Ehrlich, 2002)]. rep uvrD recO cells harbouring the plasmid pBGts-rep were used for strain construction; this plasmid carries the wild-type rep gene and can be cured by growing cells at 42°C (Petit and Ehrlich, 2002). The phenotype of plasmid-less cells obtained after propagation at 42°C was analysed. The five rep uvrD recO rposup mutants formed about 100% colonies on LB at 37°C, 30°C and 25°C, as the original rep uvrD recF rposup cells (Table 1). We conclude that the rposup mutations are necessary and sufficient to restore full viability to rep uvrD recF (recO) cells at low temperatures.
RecQ acts in concert with RecFOR to promote RecA binding to blocked forks in the rep uvrD mutant (Lestini and Michel, 2008). We observed that the rep uvrD recQ mutant is more sensitive to LB than rep uvrD recF (recO) cells (Table 1), suggesting that the RecFOR proteins still bind to arrested forks in a rep uvrD recQ mutant. The rposup alleles suppressed the LB-sensitivity of rep uvrD recQ cells, with the notable exception of the RifR (rpoBN518D) mutation that suppressed only at 37°C (Table 1). The residual cryo-LB-sensitivity of the rep uvrD recQ rpoBN518D indicates that RecFOR bind blocked replication forks in the absence of RecQ in this particular RNA Pol mutant, and that this mutation is a less efficient suppressor than the others at low temperature, in agreement with its original isolation at 37°C. Nevertheless, all other rposup mutations and the rpoCΔ215–220 mutation suppress the residual growth defects of rep uvrD recQ cells.
Suppression of the LB-cryo-sensitivity of rep uvrD recF does not correlate with a stringent-like phenotype of these RNA Pol mutants
The rpoCΔ215–220 mutation alters the kinetic properties of transcription complexes, reducing rRNA transcription and increasing transcription from some amino acid biosynthetic genes (Bartlett et al., 1998). On the other hand, it behaves as most of the mutations isolated here as it suppresses the growth defects of rep uvrD recF, recO and recQ cells at all temperatures (Boubakri et al., 2010) (Table 1). In addition, one of the mutations isolated here (rpoBH447R) was also previously isolated in a screen for mutations that confer a stringent-like phenotype (Trautinger and Lloyd, 2002). These observations prompted us to test whether the rposup mutations isolated here prevent the stimulation of the rrnB core promoter after a shift to rich medium. The rposup mutations were co-transduced with thiC::Tn10 into a strain carrying a P1rrnB-lacZ fusion, which was used to compare the expression of the P1rrnB core promoter in MM and in LB (Bartlett et al., 1998). As expected, in wild-type cells the expression of lacZ from P1rrnB promoter was higher in LB than in MM, whereas P1rrnB activity remained low in both media in the presence of the rpoCΔ215–220 or rpoBH447R mutations (Fig. 1). This experiment revealed that rpoBD444G also reduces rRNA expression in LB (Fig. 1). In contrast, P1rrnB expression remained higher in LB than in minimal medium in the presence of the rpoBN518D, rpoCP451L and rpoCH113R alleles as in wild-type cells, showing that none of these three mutations affects the activity of rRNA promoter.
Fig. 1.
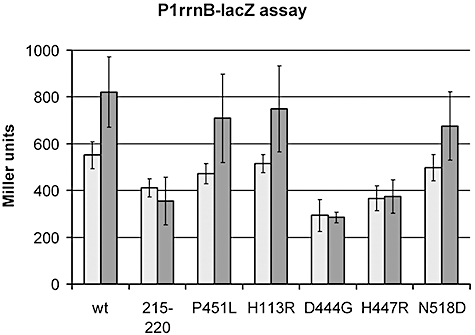
Two of the five rposup RNA Pol mutations affect rrn expression in LB. β-galactosidase assays were performed on strains carrying a P1rrnB-lacZ fusion and an rposup mutation. The height of the histograms indicates β-galactosidase Miller Units, vertical bars indicate standard deviations. Wt stands for wild-type, Δ215–220 is the control rpoCΔ215–220 mutation. P451L and H113R are rpoC mutations, D444G, H447R and N518D are rpoB mutations. Light grey: cells grown in MM; dark grey: cells grown in LB.
RelA and SpoT proteins are implicated in ppGpp alarmone synthesis. relA spoT double mutants do not induce the stringent response and are thus unable to grow on MM unless amino acids are provided. RNA Pol mutations that mimic the stringent response are classically isolated as suppressing the auxotrophy of relA spoT mutants. Although this has not been actually demonstrated, it was suggested that the inability of relA spoT mutants to grow on MM and the ability of the rpoBC suppressor mutations to suppress this defect results from effects of the mutations on transcription of amino acid biosynthetic operons (Paul et al., 2005; see Rutherford et al., 2009, for discussion). In order to analyse the capacity of the rposup mutations to suppress the auxotrophy of relA spoT double mutants, we constructed rposupΔrelA::KanR mutants (strains JJC4553 to JJC4559 Table S1) and P1-transduced them with a ΔspoT::CmR null mutation, plating half of the transduction mixture on LB and half on MM. All rposupΔrelA::KanR mutants could be transduced by the ΔspoT::CmR null mutation on LB while only one mutant, rpoBH447RΔrelA::KanR, provided transductants on MM (Table S3). As expected from this result, none of the rposupΔrelA::KanRΔspoT::CmR mutants obtained on LB could grow on MM except for the rpoBH447RΔrelA::KanRΔspoT::CmR strain (Table S3). The growth defect on MM was specific for a relA spoT context, because none of the RNA Pol mutations prevented growth on MM in RelA+ SpoT+ cells (see plating efficiencies on MM in Figs 3 and 4). This result shows that only the rpoBH447R mutation prevents the increase of rrn expression in rich medium and allows growth of a relA spoT mutant on MM. The rpoBD444G mutation only affects rrn expression and the other rposup mutations exhibit none of these stringent-like phenotypes. This result shows that four of the five mutations isolated in this study would not be obtained in a classical screen for mutations that restore growth of a relA spoT mutant on MM. Although they do not confer a stringent-like phenotype, the rposup mutations bypass the need for accessory replication helicases, suggesting that they affect the stability of transcription complexes without affecting their kinetic properties on rrn and amino acid biosynthetic gene promoters. The possible instability of the mutant RNA Pol–DNA complexes was tested by two different genetic approaches.
Fig. 3.
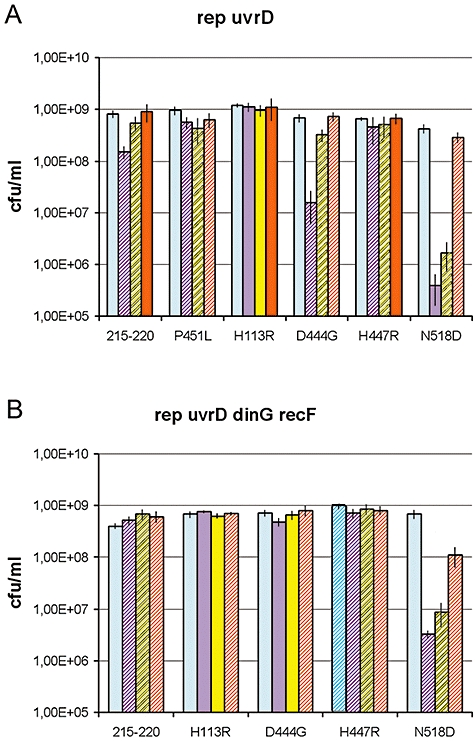
A. rposup RNA Pol mutations restore growth of rep uvrD cells. The height of the histograms indicates the number of colony-forming units (cfu) per ml, vertical bars indicate standard deviations. Rpo wild-type rep uvrD cells are not shown because they are lethal in all these conditions (plating on MM with casamino acids or on LB, at 37°C or 30°C). Mutants are in the AB1157 context, similar results were previously published for the rep uvrD rpoCΔ215–220 mutant at 37°C in the MG1655 context (Boubakri et al., 2010). Δ215–220 and H113R are rpoC mutations, D444G, H447R and N518D are rpoB mutations. Light blue: MM 30°C (plating efficiencies on MM 37°C are not shown and were similar to those at 30°C); purple: LB 25°C; yellow: LB 30°C; orange: LB 37°C. Full boxes: colonies formed in 24 h (37°C LB), 48 h (30°C LB), or 3 days (30°C MM, 25°C LB). Hatched boxes: colonies appearing 24 h later than these normal times. B. Three rposup RNA Pol mutations restore growth of rep uvrD dinG recF cells at all temperatures. Rpo wild-type and rpoCP451L cells are not shown because they are lethal under these conditions (plating on MM or on LB, at 37°C or 30°C). Mutants are in the MG1655 context, similar results were obtained in the AB1157 context (not shown). Results for the rep uvrD dinG recF rpoCΔ215–220 mutant at 37°C were previously published (Boubakri et al., 2010), and were reproduced here as a control. Plating efficiencies on MM 37°C are not shown and were similar to those at 30°C.
Fig. 4.
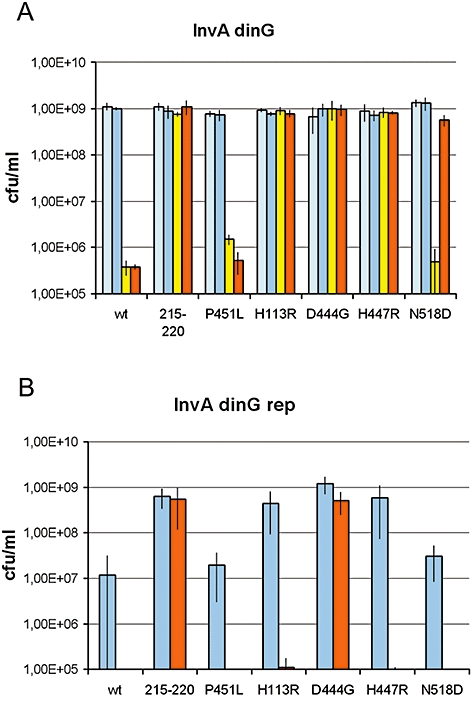
A. Three rposup RNA Pol mutations restore growth of InvA dinG cells at all temperatures. The height of the histograms indicates the number of cfu per ml, vertical bars indicate standard deviations. Results for the InvA dinG rpoCΔ215–220 mutant at 37°C were previously published (Boubakri et al., 2010), and were reproduced here as a control. Δ215–220, P451L and H113R are rpoC mutations, D444G, H447R and N518D are rpoB mutations (MG1655 context). Symbols are as in Fig. 3, dark blue are plating efficiencies on MM at 37°C. B. Three rposup mutations restores growth of InvA dinG rep cells at 37°C on MM, only rpoBD444G also restores viability on LB. Results for the InvA rep dinG rpoCΔ215–220 mutant were previously published (Boubakri et al., 2010), and were reproduced here as a control.
rposup mutations improve the resistance to UV irradiation of a ruv mutant and the rpoCH113R mutation rescues a greA greB double mutant
RuvABC is a recombination complex that acts at the last step of homologous recombination by resolving recombination intermediates called Holliday junctions (Kuzminov, 1999). ruv mutants are hypersensitive to UV irradiation and rpoB or rpoC mutations that exhibit a stringent-like phenotype partially relieve this hypersensitivity. It was proposed that they increase the intrinsic instability of RNA Pol–DNA complexes when RNA Pol is blocked by a DNA lesion (Trautinger and Lloyd, 2002). A ruvABC deletion was introduced in all rposup mutants and the UV resistance of the resulting strains was measured. With the exception of the rpoCH113R allele, which was poorly viable in a ruv mutant context and yielded variable results, all mutations improved the UV resistance of the ruvABC mutant (Fig. 2A). This result supports the idea that the mutations isolated here affect the stability of RNA Pol–DNA complexes, either at promoters or in TEC.
Fig. 2.
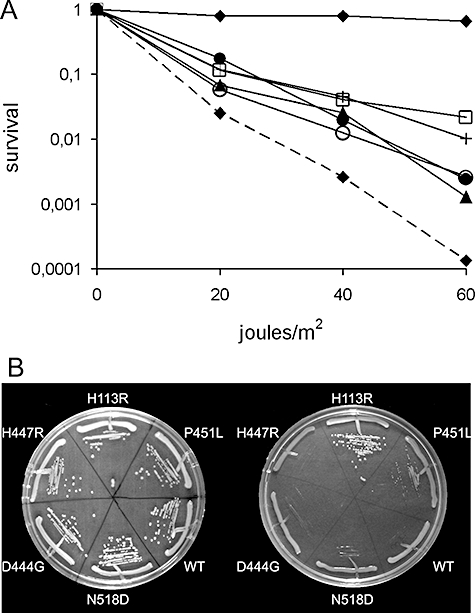
A. Four rposup RNA Pol mutations improve growth of UV-irradiated ruvABC mutants. Survival of UV-irradiated cells, results are the average of three to six independent determinations. Wild-type cells (JJC40) diamonds full line, ruvABC (JJC754) diamond dashed line, rpoCΔ215–220ruvABC (JJC4886) crosses, rpoCP451LruvABC (JJC4548) open square, rpoBH447R ruvABC (JJC4832) closed circle, rpoBD444G ruvABC (JJC4549) open circles, rpoBN518D ruvABC (JJC4547) triangles. B. The rpoCH113R mutation restores the viability of the ΔgreA::CmRΔgreB::KanR double mutant at 42°C. ΔgreA::CmRΔgreB::KanRrposup strains were streaked on LB Cm plates at 30°C (left) and at 42°C (right). Top sector, greA greB rpoCH113R (JJC4923) the only thermoresistant greA greB double mutant. Turning in the clockwise direction from this mutant: greA greB rpoCP451L (JJC5456), greA greB Rpo wt (JJC5455), greA greB rpoBN518D (JJC4818), greA greB rpoBD444G (JJC4820), greA greB rpoBH447R (JJC4821).
When RNA Pol encounters a block during elongation and backtracks, the transcription factors GreA and GreB suppress pausing by stimulating the intrinsic nucleolytic activity of RNA Pol (reviewed in Borukhov et al., 2005). greA greB double mutants are non-viable at high temperature, presumably because prolonged RNA polymerase pausing prevents replication and/or transcription. A mutation isolated through its stringent-like phenotype was previously reported to suppress the thermo-sensitivity of greA greB mutants (Trautinger and Lloyd, 2002). greA greB rposup mutants were constructed and tested for growth at 42°C. Only the rpoCH113R mutation allowed the growth of greA greB mutant at 42°C (Fig. 2B). This result presumably reflects the destabilization of backtracked RNA Pol by the rpoCH113R, but this level or type of destabilization is not essential for the growth of rep uvrD recF cells at 30°C as it is observed for only one of the suppressor mutations. This result also indicates that mutations that do not affect rrn or amino acid biosynthetic gene expression can nevertheless decrease the stability of stalled TEC enough to rescue a greA greB mutant. Altogether, we infer from the phenotypes conferred by the RNA Pol mutations described here that these mutations compromise the stability of transcription complexes; the consequences of this putative destabilization were further investigated in backgrounds that lack different accessory helicases.
Several rposup mutants rescue rep uvrD mutants in a RecF+ context
If the viability of rep uvrD recF rposup mutants results from less replication arrest, or from facilitated replication restart, the inactivation of recF might not be needed for viability. Actually, certain RNA Pol mutations including the rpoCΔ215–220 allele were reported to suppress the growth defect of rep uvrD cells in a RecF+ context at 37°C (Guy et al., 2009; Boubakri et al., 2010). To construct rep uvrD rposup mutants, rep and uvrD were introduced in rposup single mutants containing pGBts-rep, and/or the rposup mutation was co-transduced with thiC::Tn10 to a rep uvrD (pAM-rep) mutant. The viability of plasmid-less clones was measured after curing cells by propagation at 42°C (pGBts-rep) or in the absence of IPTG (pAM-rep) (Fig. 3A). The rpoCH113R mutation allowed formation or rep uvrD colonies on LB at all temperatures. The rpoCP451L mutant yielded slow-growing colonies that were heterogeneous in size at low temperature. Two of the mutations that affect rrn expression (rpoCΔ215–220, rpoBH447R) restored rep uvrD cells viability on LB, but colony formation was delayed at low temperature and also slightly decreased for rpoCΔ215–220 at 25°C. In the rpoBD444G context, colony formation on LB was delayed at all temperatures and significantly decreased at 25°C. Finally, the rpoBN518D mutation had only a partial effect at 37°C and did not allow colony formation at low temperatures. These observations indicate that in one rep uvrD rposup mutant (rpoCH113R) RecFOR does not bind replication forks at any temperature. In the other rep uvrD rposup mutants, although RecFOR is not lethal any more as in rep uvrD Rpo+ cells, it remains partly deleterious, slowing down and/or preventing growth, mainly at low temperature. Therefore, the modifications of RNA Pol activity caused by the different mutations determine the accessibility of replication forks to RecFOR recombination proteins.
Rposup mutations rescue rep uvrD recF dinG mutants but not rep uvrD dinG mutants
The viability of the rep uvrD recF mutant relies on the presence of a third helicase called DinG (Boubakri et al., 2010). We previously reported that the rpoCΔ215–220 mutation suppresses the lethality of rep uvrD recF dinG mutants at 37°C on MM and on LB, but not that of rep uvrD dinG mutants, indicating that it facilitates replication restart in the absence of all three accessory helicases only if RecF does not poison arrested forks (Boubakri et al., 2010). We tested whether the rposup mutations also suppress rep uvrD recF dinG lethality by constructing rep uvrD recF dinG rposup mutants in two ways, first a dinG deletion was introduced in the original rep uvrD recF rposup clones (JJC4043 to JJC4047, context AB1157, Table S1), and second the rposup mutations were introduced in a rep uvrD recF dinG mutant (JJC5405 to JJC5426, context MG1655, Table S1). Results were similar in both backgrounds (data not shown and Fig. 3B). rpoCP451L was the only mutation that did not suppress the lethality of rep uvrD recF dinG cells as no plasmid-less cells could be obtained (not shown). The best suppressor mutation was again the rpoCH113R, which restored 100% rep uvrD dinG recF plating efficiency at 37°C (although colony formation was delayed) and at low temperature (Fig. 3B). The three mutations rpoCΔ215–220, rpoBH447R and rpoBD444G also restored 100% plating efficiency but colony formation was delayed on LB at 37°C for rpoBD444G, on LB at all temperatures for rpoCΔ215–220, and in all growth conditions for rpoBH447R. Finally, rpoBN518D allowed normal colony formation on MM but formed only about 10% of heterogeneous slow-growing colonies on LB at 37°C and did not suppress the rep uvrD recF dinG lethality on LB at low temperature.
It was previously reported that rpoCΔ215–220 only rescues a rep uvrD dinG mutant that lacks RecF (Boubakri et al., 2010). To address this question for the rposup mutants, rep uvrD dinG rposup RecF+ mutants were constructed, first by introducing the dinG deletion in the original rep uvrD rposup mutants previously made RecF+ (JJC5253 to JJC5257 context AB1157 and JJC5258 context JJC4100, Table S1) and second by introducing successively all three helicase deletions in the rposup mutants (JJC4911 to JJC4918, JJC5310 and JJC5311, context AB1157, Table S1). All strains yielded similar results: the rposup mutations did not suppress the lethality of rep uvrD dinG mutants in a RecF+ context, as either no plasmid-less colonies could be recovered after growth under non-permissive conditions for plasmid replication (rpoBN518D), or only a few plasmid-less colonies were obtained that could not be propagated and eventually acquired additional suppressive mutations, possibly because the uvrD context is mutator (data not shown). In conclusion, in the absence of all three helicases the rposup mutations restore cell viability only if recF is inactivated, as previously observed for the rpoCΔ215–220 mutation in a MG1655 background at 37°C (Boubakri et al., 2010). This result indicates that, at least in the absence of the three Rep, UvrD and DinG helicases, replication forks are still arrested by the encounter of the rposup mutated RNA polymerases, allowing RecFOR to gain access to DNA. This result also indicates that DinG is responsible of the viability of rep uvrD rposup DinG+ mutants shown in Fig. 3A, as the inactivation of dinG in this context is lethal.
To further test the effects of the rposup mutations on replication–transcription collisions, we introduced these mutations in cells where such collisions are increased by a chromosome rearrangement.
Rescue of helicase mutants that carry an inverted rrn operon depends on the rpo mutation
Inversion of an rrn operon creates a region of increased head-on collisions between replication and transcription. Such inversions render the dinG mutant sensitive to rich medium because of R-loop formation, and strongly impair growth of the rep dinG double mutant, even on MM, because of DNA Pol–RNA Pol collisions (Boubakri et al., 2010) (Fig. 4A). In a strain that carries an inverted rrnA operon (InvA) the rpoCΔ215–220 mutation suppresses the growth defects of both dinG and dinG rep mutants at 37°C, on MM and on LB, presumably by decreasing the transcription efficiency of rrnA (Boubakri et al., 2010) (Fig. 4A).
We tested whether the rposup mutations decrease the formation of R-loops in inverted rrn by introducing these mutations in InvA dinG cells and measuring plating efficiencies of the resulting combination of mutations on LB. rpoCΔ215–220, previously tested at 37°C only, was tested here at 30°C. The rpoCH113R mutation and the three mutations that affect rrn expression (rpoBH447RrpoBD444G and rpoCΔ215–220) restored full viability of the InvA dinG mutant at 37°C and 30°C (Fig. 4A). Therefore, they decrease R-loop formation in this context at both temperatures. In contrast the RifR mutation (rpoBN518D) allowed InvA dinG colony formation on LB at 37°C but not at 30°C, and the rpoCP451L mutations had no effect. The phenotype of this last mutant suggests that suppression of the rep uvrD recF LB-cryo-sensitivity by rposup mutations is independent of R-loop removal. To ascertain directly that the LB-cryo-sensitivity of the rep uvrD recF mutant does not result from R-loop formation, a plasmid that overexpresses RnaseH (pEM001) was introduced in this mutant. As shown in Table 1, this plasmid did not improve plating efficiency. In conclusion, most of the rposup mutations suppress or decrease R-loop formation in InvA cells, allowing InvA dinG viability, but conversely suppression of R-loops is neither necessary nor sufficient to improve growth of rep uvrD recF cells at 30°C on LB.
rposup mutations were also introduced in an InvA dinG rep mutant, where replication is arrested in the inverted rrnA by collisions with RNA Pol (Boubakri et al., 2010). As previously reported the mutation rpoCΔ215–220 restored a normal plating efficiency on MM and LB (Boubakri et al., 2010), and as expected the rpoBD444G mutation, which decreases Prrn efficiency (Fig. 1), exhibited a similar phenotype (Fig. 4B). Surprisingly, the rpoBH447R mutation improved colony formation only on MM. It is conceivable that the decrease of rrn expression measured in Fig. 1 with a promoter deprived of its FIS-binding sites is compensated by the presence of these sites at inverted rrnA (Bartlett et al., 2000). Finally, rpoCH113R also improved viability on MM whereas the rpoCP451L and rpoBN518D mutations had no effect. Altogether, these results indicate that in rich medium only two mutations, rpoCΔ215–220 and rpoBD444G, destabilize RNA Pol enough to allow replication across an inverted rrn in the absence of Rep and DinG, i.e. when only the UvrD helicase is active. Among the suppressor mutations that do not affect rrn expression, rpoCH113R is the only one that rescues an InvA dinG rep mutant, and only on MM.
A consequence of replication arrest in the rep mutant is a reaction called replication fork reversal, in which the two ends of the newly synthesized strands at a blocked replication fork anneal to form a DNA double-strand end and a Holliday junction. This reaction renders RecBC (the recombination enzyme specific for DNA double-strand ends) essential for viability (Seigneur et al., 1998; Michel et al., 2007). Interestingly, although this study and other recent studies suggest that in the rep mutant replication forks are arrested by transcribed sequences, none of the mutations isolated here rescued the viability of rep recB cells, as no plasmid-less colonies could be recovered after propagation of rep recB rposup (pAM-rep) mutants in the absence of IPTG (data not shown, strains JJC5532 to JJC5536 in Table S1). This indicates that the rpoB mutation does not prevent replication fork reversal in a rep mutant, and that, even when the RNA Pol is mutated and weakly bound to DNA, its removal from DNA by UvrD and/or DinG takes place within the context of restarting reverted replication forks.
Discussion
We show here that transcription is a stronger obstacle to replication at lower temperatures than at 37°C and we isolated mutations that suppress the rich medium cryo-sensitivity of rep uvrD recF cells. The genetic properties of these mutants suggest that the rep uvrD and rep uvrD recF strains provide a new way of isolating mutations that decrease the stability of RNA Pol on DNA, at promoters and/or in TEC. Furthermore, the analysis of these RNA Pol mutants reinforces the notion that Rep, UvrD and DinG have redundant functions in E. coli to facilitate the progression of replication forks across transcribed sequences.
Isolation and properties of new RNA Pol alleles
The atomic structure of Thermus thermophilus and Termus aquaticus RNA Pol have led to structural models of TEC (Zhang et al., 1999; Korzheva et al., 2000; Vassylyev et al., 2002, 2007a,b). The similarity of these RNA Pols with the E. coli enzyme allows us to map the residues affected in our rpoB and rpoC mutants on these structures. The position of the mutated amino acids on a representation of a wild-type transcription complex is shown in Fig. 5. The three rpoB mutations, D444G, H447R and N518D (D324, H327 and T398 in T. thermophilus and T. aquaticus) lie in the major channel for the DNA/RNA hybrid, in, or close to the β-lobe 1, in a region previously described to be important for the stability of RNA Pol–DNA complexes (Trautinger and Lloyd, 2002; Rutherford et al., 2009). The rpoCH113R mutation (H90 in T. thermophilus and H101 in T. aquaticus) is on the other side of the major channel and although the mutated residue is buried in the enzyme, it may affect interactions of RpoC with the hybrid DNA-RNA. rpoCP451L (P719 in T. thermophilus and P730 in T. aquaticus) is in a different location in the complex, in a highly conserved region around the active site. This mutation is adjacent to a mutation isolated as a ΔdksA suppressor (H450R) and, as the RNA Pol cofactor DksA is also involved in the stability of transcription complexes, mutations in this region could affect the enzyme in another way to those which flank the major channel (Rutherford et al., 2009). In spite of the proximity of this mutation to the active site, we did not detect any deleterious effect of the rpoCP451L allele on E. coli growth (data not shown). Biochemical analysis of these mutant enzymes would tell whether the in vivo instability of transcription complexes is related to an increased propensity to pause and/or to terminate transcription.
Fig. 5.
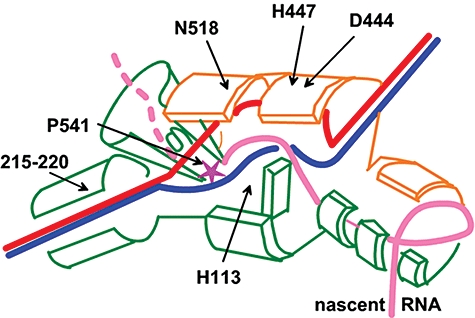
Schematic representation of the RpoB and RpoC subunits of RNA Pol showing the position of the rposup mutations. In orange RpoB, in green RpoC. Blue and red lines represent the template and non-template DNA-strands, respectively, the pink line represents the neo-synthesized RNA (the putative backtracked RNA is shown in dashed pink line). Positions of the rposup mutations are indicated (adapted with permission from Nudler, 2009).
The phenotypes of the isolated RNA Pol mutations do not allow their classification in specific groups but allow us to rank them according to the advantage that they confer to cells that lack Rep, UvrD and/or DinG helicases (summarized in Table 2). The best suppressor of growth defects in the absence of these helicases is the rpoCH113R mutation, which was isolated twice independently. This RNA Pol mutant is the only one that forms TEC unstable enough to suppress the temperature sensitivity of a greA greB double mutant. It confers LB resistance to rep uvrD, rep uvrD dinG recF and InvA dinG cells at all temperatures, and improves growth of InvA dinG rep cells at 37°C on MM. Of all mutants isolated here, the RpoCH113R RNA Pol is probably the one that forms the weakest complex on DNA. It is also the only one that slows down the growth of otherwise wild-type cells (data not shown), and could not be reliably combined with a ruvABC deletion. Slightly less efficient than rpoCH113R, the mutations rpoBH447R and rpoBD444G confer an intermediate phenotype, as the previously described rpoCΔ215–220 allele. The destabilization of transcription complexes caused by these mutations, which is deduced from their stringent-like phenotype and from the increased UV resistance conferred to ruv mutants, allows rep uvrD dinG recF and rep uvrD cells to grow on LB. They rescue InvA dinG and InvA rep dinG mutants, with the exception of the LB sensitivity of InvA rep dinG rpoBH447R cells (discussed below). Finally, the two less efficient suppressors are rpoCP451L and rpoBN518D. rpoCP451L is the only mutation that is close to the active site, while rpoBN518D was isolated at 37°C. The only sign that these mutated RNA Pols might be weakly bound to DNA, beside the rescue of rep uvrD recF cells, is the increase of UV resistance in ruv mutants. In their presence, rep uvrD cells form slow-growing colonies only at 37°C, and InvA dinG rep cells remain sick on MM. Furthermore, rpoCP451L fails to rescue InvA dinG cells on LB, while rpoBN518D only rescues it at 37°C and does not rescue rep uvrD recQ cells at 30°C.
Table 2.
Summary of the phenotypes conferred by the different rpoB/C mutations.
| P1rrnB in LB | relA spoT on MM | greA greB at 42°C | rep uvrD RecF+ (LB) | rep uvrD dinG recF (LB) | InvA dinG (LB) | InvA dinG rep (LB) | |
|---|---|---|---|---|---|---|---|
| rpoBC+ | high | − | − | − | − | − | − |
| rpoCH113R | high | − | + | + | + | + | −(MM+) |
| rpoCΔ215–220 | low | +a | − | +(∼cryoS) | +(delayed) | + | + |
| rpoBD444G | low | − | − | delayed and cryoS | + | + | + |
| rpoBH447R | low | + | − | +(delayed at low T°) | +(delayed) | + | −(MM+) |
| rpoCP451L | high | − | − | +(delayed) | − | − | − |
| rpoBN518D | high | − | − | delayed and cryoS | cryoS | cryoS | − |
In addition to the phenotypes indicated here, all mutations rescue a rep uvrD recF (recO) mutant on LB at all temperatures, all rescue a rep uvrD recQ mutant on LB (except rpoBN518D at low temperature), all improve the UV resistance of a ruvABC mutant (except rpoCH113R which could not be reliably combined with the ruvABC mutation) and none rescue the lethality of a rep uvrD dinG triple mutant.
Several mutations that mimic the effects of ppGpp have been isolated by different means, most often because they activate amino acid biosynthetic genes in a relA spoT double mutant, enabling it to grow on minimal medium (Bartlett et al., 1998; Zhou and Jin, 1998; Trautinger and Lloyd, 2002; Broccoli et al., 2004; Szalewska-Palasz et al., 2007). Some of these mutations decrease expression from the rrn promoter regardless of the presence of the upstream FIS-binding sites (Szalewska-Palasz et al., 2007), while the FIS sites compensate for the rpoCΔ215–220 mutation effects on the rrnB promoter (Bartlett et al., 2000). One mutation conferring a stringent-like phenotype was shown to reduce R-loop formation in a non-translated sequence by causing premature transcription arrest in vivo (Broccoli et al., 2004), and two others were shown to decrease the stability of TEC in vitro (Trautinger et al., 2005). Therefore, the rpoCΔ215–220, rpoBD444G and rpoBH447R mutations may be affected for the stability of transcription initiation complexes, or for the stability of TEC, or for both. Because the FIS sites compensate for the rpoCΔ215–220 mutation effects on the rrnB promoter, this mutation is likely to rescue helicase mutants carrying an inverted rrn by destabilizing TEC, as proposed in Boubakri et al., (2010). rpoBD444G may rescue InvA dinG rep cells because the promoter remains weakly expressed on LB in the presence of the FIS Sites, or because TEC are unstable. The RpoBH447R RNA Pol may fail to rescue InvA dinG rep cells because its defects are compensated for by the presence of the FIS sites and it forms more stable TEC than the RpoCΔ215–220 RNA Pol during rrn transcription. This observation supports the idea that there is no necessary correlation between the instability of TEC and the instability of the open complexes on rrn promoters. Furthermore, we isolated here a mutant (rpoBD444G) that affects transcription initiation at rrn promoters without affecting the expression of amino acids biosynthesis genes in a relA spoT context. To our knowledge, such a mutant was not reported before. Finally, rescue of a greA greB mutant at high temperature was previously described for a RNA Pol mutation isolated as increasing amino acids biosynthetic genes expression (Trautinger and Lloyd, 2002) whereas here it is observed for a RNA Pol mutant that does not affect these genes, nor rrn expression (rpoCH113R).
We used strains carrying a chromosome inversion to measure the consequences of a weaker stability of transcription complexes when the rate of replication arrest is increased either by the formation of R-loops, or by encountering oppositely oriented highly active RNA Pol in inverted rrn. With the exception of rpoCP451L, all RNA Pol mutations isolated here prevent R-loop formation. This means that RNA Pols that form unstable transcription complexes are less prone than the wild-type enzyme to R-loop formation. Conversely, only one (rpoBD444G) could prevent the lethality associated with the encounter of a series of RNA Pol transcribing a ribosomal operon in the direction opposite to replication. This means that even RNA Pols weakly bound to DNA will arrest replication forks when the latter collide with an oppositely oriented highly transcribed operon.
How does E. coli deal with replication–transcription collisions?
The general scheme that emerges from the present study and from previous studies of rep, dinG and uvrD single and multiple mutants is that Rep is the first factor that facilitates replication through transcribed sequences in vivo (Lane and Denhardt, 1975; Seigneur et al., 1998; Petit and Ehrlich, 2002; Guy et al., 2009; Boubakri et al., 2010). In the absence of Rep, UvrD becomes essential for E. coli viability and mutations in RNA Pol suppress this co-lethality, which points to UvrD as the main back-up to Rep for RNA Pol removal (Guy et al., 2009; Boubakri et al., 2010; this work). Two lines of evidence point to DinG as the other helicase that acts as a back-up. The first is our previous report that even in the absence of RecF, the combination of the rep, uvrD and dinG mutations is lethal in E. coli expressing the wild-type RNA Pol (Boubakri et al., 2010). This suggests that DinG removes wild-type RNA Pols from replication forks when Rep, UvrD and RecF are absent. The second, is our present observation that DinG is required for suppression of the rep uvrD co-lethality by most of the rposup mutations studied here, indicating that because they are unstable on DNA, the mutant RNA Pol can be removed from replication forks by DinG regardless of the presence of RecF. However, these mutated RNA Pols do not suppress the need for RecBC in a rep mutant, indicating that DinG and UvrD act in the context of restarting reversed replication forks. These helicases may recognize some features of reversed or of the restarting replication forks that are not shared by the original replication fork, whereas Rep may act within the context of the original replication fork that encounters the obstacle. It should be noted that the rposup mutations do not prevent spontaneous replication arrest in the E. coli chromosome because they did not relieve the growth defects of a priA mutant, which lacks the main replication restart protein PriA (strains JJC5540 and JJC5541 in Table S1, data not shown).
At low temperature, even in the presence of DinG, rep uvrD recF cells grow poorly on LB, indicating that DinG has a limited ability to remove the wild-type RNA Pol from replication forks under these growth conditions. Either transcription is a stronger obstacle to replication at low temperature, for example because transcriptional complexes are more stable, or for some reason DinG is less active. Provided that RecF is absent, several RNA Pol mutations isolated here bypass not only the need for Rep and UvrD but also the need for DinG (rep uvrD dinG recF rposup are viable, Fig. 3B). Removal of the mutant RNA Pol may then be catalysed either by some yet unknown function or by the replisome itself. Actually, in a purified system in vitro the wild-type RNA Pol initiation complex can be dislodged by the replisome (Pomerantz and O'Donnell, 2010). However, as RecF remains lethal in a rep uvrD dinG rposup mutant, replication forks are still arrested in these rposup cells that lack the three helicases, which is another indication that the dislodging of RNA Pol takes place during replication restart.
Our study of RNA Pol mutants that form unstable complexes on DNA provided conditions where viability is dependent on DinG only when RecF is present (the rep uvrD rposup combinations of mutations become LB-sensitive upon DinG inactivation only in a RecF+ context). This observation suggests that DinG counteracts a deleterious action of RecF; either DinG could remove RecFOR/RecA from DNA, or it could prevent RecFOR binding. It should be noted that UvrD can act both ways, it removes RecA from DNA or prevents RecFOR binding, depending on the cause of replication arrest (Lestini and Michel, 2007). The hypothetical removal of RecFOR and/or RecA from DNA by DinG needs to be tested in vitro; nevertheless, we do not favour this hypothesis because it is difficult to explain why DinG would remove RecF only in certain RNA Pol mutants. We favour the hypothesis that DinG and RecFOR compete for blocked replication forks in rep uvrD cells. Because DinG is unable to remove the wild-type RNA Pol, rep uvrD cells are killed by RecFOR. Similarly, the rep uvrD rpoN518D mutant is non-viable on LB at low temperature because under these conditions DinG cannot dislodge this mutated RNA Pol from DNA, letting RecFOR bind to arrested replication forks. Conversely, DinG is capable of removing all other RNA Pol mutants isolated here before RecFOR binds, and consequently the viability of the rep uvrD mutant becomes independent of the recF context in these RNA Pol mutants.
Experimental procedures
Strains and plasmids
The strain backgrounds are MG1655, or JJC40, which is an hsdR Thr+ Pro+ derivative of AB1157 (leu-6 thi-1, his-4, argE3, lacY1, galK2, ara-14, xyl-5, mtl-1, tsx-33, rpsL31, supE44, hsdR, hsdM). JJC147 is HfrPK3-PO131 (thr1, leuB6, azi-15, tonA1, lacY1, supE44, argE86::Tn10). Strains genotypes and plasmids are described in Table S1. InvA is described in Boubakri et al. (2010), it is an MG1655 derivative, deleted for lacZ and attλ, which carries a 18 kb inversion (NT 4 025 300 to 4 043 723) encompassing the rrnA operon. Minimum medium is M9 supplied with thiamine 0.05%, CaCl2 100 µM, MgSO4 2 mM, glucose 0.4% (Miller, 1992). Casamino acids 0.2% were added for strain background other than MG1655. Most of the strains were constructed by P1 transduction (Miller, 1992). For Hfr conjugations, a mix of 10% donor cells/90% recipient cells was incubated for 15 min with low agitation at 37°C before plating on selective media. Null mutants were checked by polymerase chain reaction with external oligonucleotides that amplify a DNA fragment of different length for the wild-type and the interrupted alleles. Oligonucleotides used for checking mutations are shown in Table S4. uvrD mutants were checked for their UV-sensitive phenotypes and for their mutator phenotype (about 100-fold increase in the proportion of RifR clones in overnight cultures). The plasmids pGBts-rep (Petit and Ehrlich, 2002), pAM-rep (Lestini and Michel, 2008) and pAM-priA (Grompone et al., 2004) were described; they carry the rep or the priA gene under the control of their own promoter; they were segregated as published.
rpo mutations are rpoB A1331G (rpoBD444G), A1340G (rpoBH447R), A1552G (rpoBN518D), rpoCA338G (rpoCH113R), C1352T (rpoCP451L). Following P1 transduction or conjugation, screening for A1552G (rpoBN518D) was based the RifR phenotype it confers. Screening for A1331G (rpoBD444G) was done sequencing the rpoB region. Three mutations were screened after polymerase chain reaction amplification of the region either by sequencing or by looking for the presence of a restriction site that they create: A1340G (rpoBH447R) creates a BsiEI site, rpoCA338G (rpoCH113R) creates a BssHII site, C1352T (rpoCP451L) creates a BspMI site. Upon P1 co-transduction of rposup with thiC::Tn10 in a context where the mutation confers a phenotype, the phenotype and the presence of the mutation were tested in four to eight different TetR transductants and there was 100% correlation between the presence of the mutation and the appearance of the new phenotype. Twenty regularly spaced oligonucleotides in rpoB (10 on each strand) and 20 regularly spaced oligonucleotides in rpoC (10 on each strand) were used for sequencing of the entire genes.
Measurement of plating efficiency
Overnight cultures grown in MM (OD650 1 to 2) were diluted and plated on MM or LB plates, incubated at 37°C, 30°C or 25°C. LB plates at 37°C were counted after 24 and 48 h of incubation, MM plates at 37°C and LB plates at 30°C after 48 h and 3 days of incubation, and MM plates at 30°C and LB plates at 25°C after 3 and 4 days incubation.
Measurement of UV sensitivity and of β-galactosidase activity
Survival to UV irradiation was performed as described (Baharoglu et al., 2006). β-galactosidase assays for the measure of PrrnB activity were performed as described (Miller, 1992).
Acknowledgments
We thank O. Espeli, R.L.G. Gourse and C.J. Herbert for very helpful reading of the manuscript. We are very grateful to Annie Kolb for discussions on RNA polymerase structure, and to Audrey Lagès and Marie Le Masson for excellent technical assistance. The work was supported by ANR-05-BLAN-0204-01, ANR-08-BLAN-0230-01 and Prix ‘coup d'élan’ from the foundation Bettencourt-Schueller.
Supporting information
Additional supporting information may be found in the online version of this article.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Aguilera A, Gomez-Gonzalez B. Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet. 2008;9:204–217. doi: 10.1038/nrg2268. [DOI] [PubMed] [Google Scholar]
- Azvolinsky A, Giresi PG, Lieb JD, Zakian VA. Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol Cell. 2009;34:722–734. doi: 10.1016/j.molcel.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baharoglu Z, Petranovic M, Flores MJ, Michel B. RuvAB is essential for replication forks reversal in certain replication mutants. EMBO J. 2006;25:596–604. doi: 10.1038/sj.emboj.7600941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker MM, Gaal T, Gourse RL. Mechanism of regulation of transcription initiation by ppGpp. II. Models for positive control based on properties of RNAP mutants and competition for RNAP. J Mol Biol. 2001a;305:689–702. doi: 10.1006/jmbi.2000.4328. [DOI] [PubMed] [Google Scholar]
- Barker MM, Gaal T, Josaitis CA, Gourse RL. Mechanism of regulation of transcription initiation by ppGpp. I. Effects of ppGpp on transcription initiation in vivo and in vitro. J Mol Biol. 2001b;305:673–688. doi: 10.1006/jmbi.2000.4327. [DOI] [PubMed] [Google Scholar]
- Bartlett MS, Gaal T, Ross W, Gourse RL. RNA polymerase mutants that destabilize RNA polymerase-promoter complexes alter NTP-sensing by rrn P1 promoters. J Mol Biol. 1998;279:331–345. doi: 10.1006/jmbi.1998.1779. [DOI] [PubMed] [Google Scholar]
- Bartlett MS, Gaal T, Ross W, Gourse RL. Regulation of rRNA transcription is remarkably robust: FIS compensates for altered nucleoside triphosphate sensing by mutant RNA polymerases at Escherichia coli rrn P1 promoters. J Bacteriol. 2000;182:1969–1977. doi: 10.1128/jb.182.7.1969-1977.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borukhov S, Lee J, Laptenko O. Bacterial transcription elongation factors: new insights into molecular mechanism of action. Mol Microbiol. 2005;55:1315–1324. doi: 10.1111/j.1365-2958.2004.04481.x. [DOI] [PubMed] [Google Scholar]
- Boubakri H, de Septenville AL, Viguera E, Michel B. The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J. 2010;29:145–157. doi: 10.1038/emboj.2009.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broccoli S, Rallu F, Sanscartier P, Cerritelli SM, Crouch RJ, Drolet M. Effects of RNA polymerase modifications on transcription-induced negative supercoiling and associated R-loop formation. Mol Microbiol. 2004;52:1769–1779. doi: 10.1111/j.1365-2958.2004.04092.x. [DOI] [PubMed] [Google Scholar]
- Garibyan L, Huang T, Kim M, Wolff E, Nguyen A, Nguyen T, et al. Use of the rpoB gene to determine the specificity of base substitution mutations on the Escherichia coli chromosome. DNA Repair (Amst) 2003;2:593–608. doi: 10.1016/s1568-7864(03)00024-7. [DOI] [PubMed] [Google Scholar]
- Grompone G, Bidnenko V, Ehrlich SD, Michel B. PriA is essential for viability of the Escherichia coli topoisomerase IV parE10(Ts) mutant. J Bacteriol. 2004;186:1197–1199. doi: 10.1128/JB.186.4.1197-1199.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy CP, Atkinson J, Gupta MK, Mahdi AA, Gwynn EJ, Rudolph CJ, et al. Rep provides a second motor at the replisome to promote duplication of protein-bound DNA. Mol Cell. 2009;36:654–666. doi: 10.1016/j.molcel.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzheva N, Mustaev A, Kozlov M, Malhotra A, Nikiforov V, Goldfarb A, Darst SA. A structural model of transcription elongation. Science. 2000;289:619–625. doi: 10.1126/science.289.5479.619. [DOI] [PubMed] [Google Scholar]
- Kuzminov A. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol Mol Biol Rev. 1999;63:751–813. doi: 10.1128/mmbr.63.4.751-813.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane HE, Denhardt DT. The rep mutation. IV. Slower movement of replication forks in Escherichia coli rep strains. J Mol Biol. 1975;97:99–112. doi: 10.1016/s0022-2836(75)80025-8. [DOI] [PubMed] [Google Scholar]
- Lestini R, Michel B. UvrD controls the access of recombination proteins to blocked replication forks. EMBO J. 2007;26:3804–3814. doi: 10.1038/sj.emboj.7601804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lestini R, Michel B. UvrD and UvrD252 counteract RecQ, RecJ, and RecFOR in a rep mutant of Escherichia coli. J Bacteriol. 2008;190:5995–6001. doi: 10.1128/JB.00620-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel B, Boubakri H, Baharoglu Z, Lemasson M, Lestini R. Recombination proteins and rescue of arrested replication forks. DNA Repair (Amst) 2007;6:967–980. doi: 10.1016/j.dnarep.2007.02.016. [DOI] [PubMed] [Google Scholar]
- Miller JH. A Short Course in Bacterial Genetic: Cold Spring Harbor. New York: Cold Spring Harbor Press; 1992. [Google Scholar]
- Mirkin EV, Mirkin SM. Replication fork stalling at natural impediments. Microbiol Mol Biol Rev. 2007;71:13–35. doi: 10.1128/MMBR.00030-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nudler E. RNA polymerase active center: the molecular engine of transcription. Annu Rev Biochem. 2009;78:335–361. doi: 10.1146/annurev.biochem.76.052705.164655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BJ, Berkmen MB, Gourse RL. DksA potentiates direct activation of amino acid promoters by ppGpp. Proc Natl Acad Sci USA. 2005;102:7823–7828. doi: 10.1073/pnas.0501170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit MA, Ehrlich D. Essential bacterial helicases that counteract the toxicity of recombination proteins. EMBO J. 2002;21:3137–3147. doi: 10.1093/emboj/cdf317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz RT, O'Donnell M. Direct restart of a replication fork stalled by a head-on RNA polymerase. Science. 2010;327:590–592. doi: 10.1126/science.1179595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potrykus K, Cashel M. (p)ppGpp: still magical? Annu Rev Microbiol. 2008;62:35–51. doi: 10.1146/annurev.micro.62.081307.162903. [DOI] [PubMed] [Google Scholar]
- Rudolph CJ, Dhillon P, Moore T, Lloyd RG. Avoiding and resolving conflicts between DNA replication and transcription. DNA Repair (Amst) 2007;6:981–993. doi: 10.1016/j.dnarep.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Rutherford ST, Villers CL, Lee JH, Ross W, Gourse RL. Allosteric control of Escherichia coli rRNA promoter complexes by DksA. Genes Dev. 2009;23:236–248. doi: 10.1101/gad.1745409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigneur M, Bidnenko V, Ehrlich SD, Michel B. RuvAB acts at arrested replication forks. Cell. 1998;95:419–430. doi: 10.1016/s0092-8674(00)81772-9. [DOI] [PubMed] [Google Scholar]
- Szalewska-Palasz A, Johansson LU, Bernardo LM, Skarfstad E, Stec E, Brannstrom K, Shingler V. Properties of RNA polymerase bypass mutants: implications for the role of ppGpp and its co-factor DksA in controlling transcription dependent on sigma54. J Biol Chem. 2007;282:18046–18056. doi: 10.1074/jbc.M610181200. [DOI] [PubMed] [Google Scholar]
- Taucher-Scholtz G, Abdel-Monem M, Hoffmann-Berling H. Functions of helicases in E. coli. In: Cozzarelli NR, editor. Mechanisms of DNA Replication and Recombination. New York: Alan Liss Inc; 1983. pp. 65–76. [Google Scholar]
- Trautinger BW, Lloyd RG. Modulation of DNA repair by mutations flanking the DNA channel through RNA polymerase. EMBO J. 2002;21:6944–6953. doi: 10.1093/emboj/cdf654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautinger BW, Jaktaji RP, Rusakova E, Lloyd RG. RNA polymerase modulators and DNA repair activities resolve conflicts between DNA replication and transcription. Mol Cell. 2005;19:247–258. doi: 10.1016/j.molcel.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Vassylyev DG, Sekine S, Laptenko O, Lee J, Vassylyeva MN, Borukhov S, Yokoyama S. Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 A resolution. Nature. 2002;417:712–719. doi: 10.1038/nature752. [DOI] [PubMed] [Google Scholar]
- Vassylyev DG, Vassylyeva MN, Perederina A, Tahirov TH, Artsimovitch I. Structural basis for transcription elongation by bacterial RNA polymerase. Nature. 2007a;448:157–162. doi: 10.1038/nature05932. [DOI] [PubMed] [Google Scholar]
- Vassylyev DG, Vassylyeva MN, Zhang J, Palangat M, Artsimovitch I, Landick R. Structural basis for substrate loading in bacterial RNA polymerase. Nature. 2007b;448:163–168. doi: 10.1038/nature05931. [DOI] [PubMed] [Google Scholar]
- Veaute X, Delmas S, Selva M, Jeusset J, Le Cam E, Matic I, et al. UvrD helicase, unlike Rep helicase, dismantles RecA nucleoprotein filaments in Escherichia coli. EMBO J. 2005;24:180–189. doi: 10.1038/sj.emboj.7600485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancey-Wrona JE, Matson SW. Bound Lac repressor protein differentially inhibits the unwinding reactions catalyzed by DNA helicases. Nucleic Acids Res. 1992;20:6713–6721. doi: 10.1093/nar/20.24.6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Campbell EA, Minakhin L, Richter C, Severinov K, Darst SA. Crystal structure of Thermus aquaticus core RNA polymerase at 3.3 A resolution. Cell. 1999;98:811–824. doi: 10.1016/s0092-8674(00)81515-9. [DOI] [PubMed] [Google Scholar]
- Zhou YN, Jin DJ. The rpoB mutants destabilizing initiation complexes at stringently controlled promoters behave like ‘stringent’ RNA polymerases in Escherichia coli. Proc Natl Acad Sci USA. 1998;95:2908–2913. doi: 10.1073/pnas.95.6.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.