

Abstract
Numerous conditions promote oxidative stress, leading to the build-up of reactive aldehydes that cause cell damage and contribute to cardiac diseases. Aldehyde dehydrogenases (ALDHs) are important enzymes that eliminate toxic aldehydes by catalysing their oxidation to non-reactive acids. The review will discuss evidence indicating a role for a specific ALDH enzyme, the mitochondrial ALDH2, in combating oxidative stress by reducing the cellular ‘aldehydic load’. Epidemiological studies in humans carrying an inactive ALDH2, genetic models in mice with altered ALDH2 levels, and small molecule activators of ALDH2 all highlight the role of ALDH2 in cardioprotection and suggest a promising new direction in cardiovascular research and the development of new treatments for cardiovascular diseases.
Keywords: ALDH2, Mitochondria, Ischaemia, Nitroglycerin, Alda-1
1. Introduction: oxidative stress and aldehyde toxicity
Ischaemia and reperfusion as well as mismatch between cardiac demand and cardiac function result in oxidative stress in the myocardium.1,2 Oxidative stress is a state in which excessive reactive oxygen species (ROS), such as 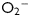 and H2O2, accumulate and result in cellular toxicity, due to imbalance between production and removal of ROS.3,4 In the myocardium, a significant ROS production occurs in the mitochondria and when it is excessive, it contributes to oxidative stress and mitochondrial dysfunction.5,6 The need to reduce oxidative stress to protect the heart led to both preclinical and clinical research. Although early research efforts focused on the deleterious effects of ROS in the myocardium,1,7 more recent work revealed that accumulation of cytotoxic and reactive aldehydes derived from ROS-induced stress8–10 or from direct insults of exogenous aldehydes11 can also severely impair cardiac functions. Although low levels of ROS and aldehydes may be regarded as second messengers that trigger stress-activated anti-oxidant mechanisms (review by Poli et al.12), it is nevertheless critical that the cells maintain a defensive capacity to prevent acute or chronic build-up of excessive oxidative stress and toxic aldehydes, as these cause irreversible injuries.
and H2O2, accumulate and result in cellular toxicity, due to imbalance between production and removal of ROS.3,4 In the myocardium, a significant ROS production occurs in the mitochondria and when it is excessive, it contributes to oxidative stress and mitochondrial dysfunction.5,6 The need to reduce oxidative stress to protect the heart led to both preclinical and clinical research. Although early research efforts focused on the deleterious effects of ROS in the myocardium,1,7 more recent work revealed that accumulation of cytotoxic and reactive aldehydes derived from ROS-induced stress8–10 or from direct insults of exogenous aldehydes11 can also severely impair cardiac functions. Although low levels of ROS and aldehydes may be regarded as second messengers that trigger stress-activated anti-oxidant mechanisms (review by Poli et al.12), it is nevertheless critical that the cells maintain a defensive capacity to prevent acute or chronic build-up of excessive oxidative stress and toxic aldehydes, as these cause irreversible injuries.
Aldehydes are generated during numerous physiological processes including catabolism of amino acids, transmitters such as GABA, serotonin, noradrenaline, adrenaline, and dopamine.13–15 Further, more than 200 different aldehydes are generated through lipid metabolism16 and metabolism of carbohydrates also generates many aldehydic intermediates.13 In addition to these endogenous aldehydes, aldehydes are ubiquitously present in the environment in smog, cigarette smoke, motor vehicle exhaust, and in a variety of industrial processes including the production of polyurethane, polyester plastics (these include formaldehyde, acetaldehyde, and acrolein). Although some dietary and aromatic aldehydes (e.g. citral, cinnamaldehyde, benzaldehyde, and retinal) are approved additives in various foods and cosmetics where they impart flavour and odour, many others are cytotoxic. A mechanism for a rapid clearance of aldehydes is essential to protect the human body, in general, and the myocardium and the brain, in particular, from the harmful effects of these aldehydes.
Aldehydes are diffusible and highly reactive agents in cells; they form adducts with lipids, proteins, and DNA, which affect the function of these macromolecules and can lead to their inactivation.16 Because of DNA damage induced by aldehyde adducts, a wide range of aldehydes have been classified as mutagenic and carcinogenic. These include even acetaldehyde, which is derived from ethanol drinking; excessive ethanol consumption has been linked to alcoholic liver diseases and various upper aerodigestive and gastrointestinal cancers.17 2,4-Dihydroxy-phenyacetaldehyde, another highly potent neurotoxin, is an intermediate of dopamine metabolism and has been implicated in neurodegenerative diseases.18,19 Methylglyoxal, an aldehyde accumulated in hyperglycaemia, is a key intermediate for the adduction formation in advanced glycation end-products found in various organs of diabetic patients.20 Many other reactive and cytotoxic aldehydes have been shown to be associated with other diseases.
Of particular interest to cardiovascular diseases are reactive aldehydes such as 4-hydroxy-2-nonenal (4-HNE) and malondialdehyde (MDA).21 In conditions like ischaemia and reperfusion, ROS-induced peroxidation of polyunsaturated fatty acids like linoleic acid and arachidonic acid present in the plasma membrane leads to 4-HNE production16,22 (Figure 1). 4-HNE is a highly reactive carbonyl compound due to the presence of α,β-unsaturated carbons.23 4-HNE readily reacts with cysteine, histidine, and lysine residues, and thus forms protein adducts via the Michaels addition.23,24 4-HNE adduct formation in the myocardium has been shown to lead to the inhibition of key metabolic enzymes such as glyceraldehyde 3-phosphate dehydrogenase24 and the 26S proteosome,25,26 thus leading to further accumulation of damaged proteins in the cell. 4-HNE also impairs ATP-generating ability in the mitochondria,27 induces opening of mitochondria permeability transition pore, and impairs mitochondrial integrity in a concentration-dependant and calcium-mediated manner28,29 (Figure 1), thus reducing the cell's ability to repair the damaged macromolecules. As a potent cardiac cytotoxin, 4-HNE has been shown to directly inhibit contractility,30 induce pro-arrhythmic effects in isolated cardiac myocytes,31 and cause tissue damage after cardiac ischaemia.32
Figure 1.

A scheme depicting aldehyde-induced mitochondrial damage and how ALDH2 reduces this aldehydic toxicity. (Left) Ischaemia and reperfusion and other oxidative stress in the heart increase ROS production, which triggers lipid peroxidation and the accumulation of reactive aldehydes, such as 4-HNE. Other xenogenic aldehydes from the environment, from ethanol metabolism, and from food additives can also increase the cellular ‘aldehydic load’ (depicted as a grey cloud in the figure). Aldehydes induce inactivation of a number of macromolecules including the proteasome, the electron transport chain (ETC) in the mitochondria, as well as inactivation of ALDH2 itself. This aldehyde-induced macromolecule inactivation contributes to mitochondrial impairment and increases in oxidative stress, thus leading to cell damage. Particularly relevant to cardiac disease, the use of nitroglycerin (GTN) can further contribute to ALDH2 inactivation, thus decreasing the cell's natural ability to reduce ROS-induced aldehydic load and cytotoxicity. Finally, a common mutation in ALDH2 (ALDH2*2) in humans further impairs the ability to reduce the aldehydic load under oxidative stress conditions. (Right) Agents that increase ALDH2 activity and protect ALDH2 from inactivation by aldehydes and by GTN will decrease the aldehydic load by enhancing the conversion of aldehydes to non-reactive acid (blue cloud in the scheme), thus leading to cytoprotection. Such one potential agent is Alda-1, an aldehyde dehydrogenase 2 activator. Alda-1 increases ALDH2 activity by about two-folds, blocks ALDH2 inactivation by both aldehydes and GTN, and thus increases the cell's natural ability to protect from oxidative stress, leading to 60% reduction from cardiac damage in an animal model of AMI. The ability of Alda 1 to increase the activity of the mutant ALDH2*2 may be of particular importance for over >0.5 billion humans who carry this mutation. (See text for details.) Reduction in aldehydic load decreases mitochondrial structural and functional damages and increases ATP generation, thus leading to cardiac protection from oxidative stress.
In another study of aldehyde-induced cardiac toxicity, Wang et al.11 showed that acrolein, an exogenous reactive aldehyde that is ubiquitously present in many food sources, can exacerbate ischaemic damage to the heart. In a model of acute myocardial infarction (AMI), oral delivery of acrolein (5 mg/kg) to mice 30 min before coronary artery occlusion and 24 h reperfusion produced a significantly increased myocardial infarct size when compared with the water-treated control group.11 Interestingly, acrolein also abolished the late preconditioning cardioprotective effect offered by the pre-treatment of an NO donor, diethylenetriamine/NO. The levels of acrolein–εPKC adducts and acrolein adducts in other proteins in the mitochondrial fraction increased in an acrolein dose-dependent manner.11 High cardiovascular toxicity and statistically significant association between aldehyde-containing components of polluted air or cigarette smoke and incidents of ischaemic heart disease, arrhythmias, and heart failure have also been reported.31 These findings heightened the importance and awareness that both endogenous reactive aldehydes such as 4-HNE and aldehydes present as environmental pollutants might be contributors of these harmful cardiovascular effects and that a mechanism that protects from these aldehydes and accelerates their removal is essential to protect the myocardium as well as other organs from oxidative stress.
2. Aldehyde dehydrogenases
There is a class of detoxifying enzymes that catalyse the removal of aldehydes in the body. These are the NAD(P)+-dependent aldehyde dehydrogenase (ALDH) super gene family. Nineteen ALDH genes have been mapped in the human genome, of which 17 ALDH isozymes are expressed with different tissue distributions.15 All the ALDH genes are nuclear encoded, but at least five ALDH isozymes reside and function in the mitochondria.13 We have focused this review on the role of the mitochondrial ALDH2 in cardiovascular disease, since this enzyme has emerged as a key enzyme of cardioprotection.8,9,33–35
ALDH2 is a tetrameric enzyme and is expressed abundantly in the liver and lung, and is also present in organs that require high mitochondrial capacity for oxidative ATP generation such as heart and brain.36 ALDH2 is important in the oxidization of aldehydic substrates, such as 4-HNE, acrolein, and short chain, aromatic or polycyclic carbons.23,37 In addition to its dehydrogenase activity, ALDH2 can function as an esterase and reductase, depending on the substrates. Much current attention has also been drawn to ALDH2 for its role in the biotransformation of nitroglycerin, by its reductase activity, to 1,2-glyceryl dinitrate for the production of nitric oxide, a critical vasodilator.38,39
ALDH2 is best known for its ability as a detoxifying enzyme of acetaldehyde, an intermediate of ethanol metabolism. Importantly, more than 40% of the East Asians population carries a common ALDH2*2 mutant allele, which results in a dramatic reduction in the enzymatic activity when compared with the ALDH2*1, wild-type allele;40 carrier of this mutant ALDH2 has a characteristic acetaldehyde-induced facial flushing when drinking alcohol. The mutation is caused by a single-nucleotide substitution (G to A) in exon 12, leading to a change from glutamate to lysine at position 487.41 The biochemical characterization, molecular structure, and physiological consequences of ALDH2*2 have been extensively studied.41,42 The E487K amino acid substitution at the dimer interface of the tetrameric enzyme resulted in disruption of the co-enzyme NAD binding and reduced catalytic activity of ALDH2*2.43 Heterozygous ALDH2*1/*2 individuals retain only 10–45% of the enzymatic activity and homozygous ALDH*2/*2 individuals have 1–5% of wild-type ALDH activity, due to this single amino acid polymorphism.44 The ALDH2*2 allele has been linked to an increased risk of oesophageal and other upper aerodigestive tract cancers among alcohol drinkers.45–47 But, higher incidences of insensitivity to nitroglycerin treatment for angina,48 MI,49,50 hypertension,51,52 and other oxidative-related neurodegenerative diseases14 have also been associated with ALDH2*2 mutation in recent years (see further discussion in the following). Since the affected world population of ALDH2*2 is estimated to be at least 540 million or ∼8% of the world population,45 it is warranted that health risk for cardiac diseases be re-evaluated in ALDH2*2 carriers.
3. Mitochondrial ALDH2 and cardioprotection against ischaemia and reperfusion injury
AMI is one of the leading causes of disability and death in the USA.53 Clinical interventions such as angioplasty or thrombolytic agents have been effective in re-establishing the coronary flow. However, treatments to further reduce the injuries incurred during the ischaemic period or by reperfusion are not available. A large body of research has identified an endogenous process of cytoprotection from ischaemia/reperfusion injury.54 Cytoprotection can be induced by subjecting an organ to short bouts of ischaemia prior to the prolonged ischaemia, a process termed ischaemic preconditioning.55 Preconditioning can also be induced and/or enhanced by select hormones and neurotransmitters56 and is dependent on the activation of the diacylglycerol-dependent protein kinase C epsilon (εPKC57,58).
One pharmacological agent that mimics ischaemic-preconditioning-induced cardioprotection is ethanol.59,60 This effect may be different from the cardioprotective effect of moderate habitual consumption of alcohol;61 we found that a brief exposure to 10 mM ethanol 10–20 min prior to ischaemia can protect rat heart from prolonged ischaemic damage in both cultured cardiac myocytes and adult whole heart.60 Using an unbiased proteomic approach, we then identified the mitochondrial ALDH2 as a key enzyme downstream of ethanol-induced εPKC-dependent cardioprotection.9 ALDH2 is phosphorylated under conditions that lead to cardiac cytoprotection and its enzymatic activity is inversely correlated with the severity of the damage from cardiac ischaemia (R2 = 0.959). High-throughput screening of libraries of compounds identified N-(1,3-benzodioxol-5-ylmethyl)-2,6-dichlorobenzamide (ALDH activator-1 or Alda-1, MW = 324) and its halogen analogues as selective agonists of ALDH2 (Figure 1).
The identification of a selective activator for ALDH2 confirmed that ALDH2 activation is not only required, but sufficient to induce cardioprotection; treatment by Alda-1 increased ALDH2 activity by two-fold and reduced infarct size by 60% in an in vivo model of myocardial infarction in rats. It is likely that the benefit of ALDH2 activation is to facilitate the removal of cytotoxic aldehydes, such as 4-HNE and others that accumulate during ischaemia and reperfusion.9,32,62,63 4-HNE has been shown to be a substrate as well as a potent inhibitor of ALDH2 (due to 4-HNE adduct formation on ALDH264,65). Using a human recombinant ALDH2 enzyme, at 100 µM, 4-HNE completely inactivated ALDH2 activity in vitro. However, the 4-HNE-induced inhibition of ALDH2 activity was completely prevented in the presence of Alda-1.9 As anticipated, following ischaemia and reperfusion, the accumulation of 4-HNE–protein adducts was lower in hearts treated with Alda-1, relative to vehicle-treated controls.9 The ability of Alda-1 to reduce cardiac damage is therefore likely due to a combination of direct enzyme activation of ALDH2 and prevention of ALDH2 inactivation by its reactive substrate, 4-HNE, which is formed and accumulates under oxidative stress (Figure 1). Using an open-chest model of AMI in vivo, Churchill et al.33 further demonstrated that under cardioprotective conditions, εPKC translocates directly into the mitochondria where it interacts with ALDH2, increases its enzymatic activity, diminishes the pro-apoptotic signalling activity of JNK1/2 and ERK1/2 and reduced 4-HNE–protein adduct formation. Further, in εPKC knockout mice, direct activation of mitochondrial ALDH2 by an Alda-1 analogue, Alda-44, protects the heart against ischaemia/reperfusion injuries similar to what was achieved by ethanol preconditioning in the wild-type mice.66 These results indicate that ALDH2 is a direct downstream substrate of εPKC and suggest that the activation of ALDH2 is necessary and sufficient to confer. Hill et al.67 have shown that inhibition of 4-HNE oxidation during ischaemia may be due to a decrease in NAD/NADH ratio, which renders ALDH2 less effective. On the other hand, excessive production of 4-HNE via lipid peroxidation occurs most likely during the reperfusion period when NAD levels are rapidly restored. It is during that reperfusion period that ALDH2 may play a critical role in cardioprotection by enhancing the removal of ROS-generated 4-HNE. We found that Alda-1 increases the removal of 4-HNE to levels closer to basal levels9 by increasing the productive substrate–enzyme interaction.68 Furthermore, Beretta et al.69 have recently demonstrated that Alda-1 also increases the affinity of ALDH2 for NAD, which should result in increased ALDH2 activity even if NAD levels have not yet recovered back to basal levels, and cardioprotection by enhancing the detoxifying capability of the cells against reactive aldehydes.
Recent epidemiological studies indicated that ALDH2 is critical in cardiovascular diseases. A study conducted in Korea showed an association of higher risk of MI with the E487K mutation carried by both ALDH*1/*2 heterozygous and ALDH2*2/*2 homozygous individuals in older Korean men.49 An independent study conducted in Japan also identified ALDH2*2/*2 genotype as a risk factor for MI incidents in Japanese me.50 An important feature of Alda-1 is its ability to increase the activity of the inactive ALDH2*2 mutant.9 Alda-1 increased the enzymatic activity of ALDH2*2 homotetramers and ALDH2*2 heterotetramers by 11- and two-fold, respectively. The ability of Alda-1 to partially complement or restore the activity of the mutant, ALDH2*2, is striking, as it is rare to find a small molecule that can specifically rescue a mutation in humans. This enhancement of catalytic activity translates to a possibility of rescuing some enzymatic activity for the ALDH2*2/*2 individuals and restoring the activity of ALDH2*1/*2 individuals to almost the basal level in the wild-type ALDH2*1/*1 individual.9 Co-crystal structures of Alda-1 with both ALDH2*1 wild-type and ALDH2*2 mutant enzymes have recently been resolved and offered detailed mechanistic explanations on how Alda-1 can enhance the enzymatic activity, protect ALDH2 against 4-HNE inactivation, and restore the function of ALDH2*2 mutation.68 Structural comparison revealed that Alda-1 interacts with ALDH2 at the substrate-binding tunnel and kinetically increased the rate of catalysis by reducing the probability of non-productive substrate hydrolysis. The unique binding site of Alda-1 also positions this compound as a shield to the key sulfhydryl amino acid, Cys302, at the catalytic centre and likely protects the ALDH2 molecule against enzyme inactivation by its reactive aldehyde, 4-HNE. Finally, even though Alda-1 made no direct contact with the mutated E487K residue, it nevertheless functioned as a molecular chaperone and restored the electron density and structural abnormality of the co-enzyme-binding site within ALDH2*2. Alda-1 therefore represents as a new pharmacological agonist and opens the possibility that modulation of ALDH2 by small molecule enzyme activators should have therapeutic value for cardiovascular and other ALDH2-related diseases.
4. Angina, nitroglycerin bioactivation, tolerance, and ALDH2
Since its discovery in 1860s, nitroglycerin has become one of the most widely used drugs, treating patients with stable and unstable angina and with AMI and heart failure. About 9.8 million Americans experience angina annually and many of them are given nitroglycerin for both acute and chronic symptom relief.70 The beneficial effect of nitroglycerin is achieved due to its ability to increase blood flow to the heart by dilating coronary arteries and decreasing cardiac preload due to venodilation.71 Although acute treatment including sublingual regimen is immediate and effective, the benefit of nitroglycerin has been limited because of in vivo tolerance that rapidly develops on continuous treatment.72 (Nitroglycerin tolerance is manifested as a reduced vasodilatation effect and requirement of high doses of the drug after continuous treatment.)
Previous experimental and clinical investigations have uncovered several critical mechanisms of nitroglycerin tolerance, including oxidative stress, endothelial dysfunction, and increased sensitivity to vasocontrictors.72,73 Recently, Chen et al. reported a pathway involving the mitochondrial enzyme, ALDH2, that provides a novel mechanistic insight into the development of nitroglycerin tolerance.38,39,74 In their study measuring conversion of nitroglycerin to nitric oxide, ex vivo, they found that bioactivation of nitroglycerin into 1,2-glyceral dinitrate, the physiologically relevant metabolite of nitroglycerin, was significantly diminished in aortas from ALDH2 knockout mice. Similar results were observed using ALDH2-selective inhibitors in animals as well as in humans.75,76 These studies have defined an unequivocal dependence of nitroglycerin bioactivation on ALDH2. Further studies in murine and human tissues have demonstrated a more targeted role for ALDH2 that explains nitroglycerin tolerance77,78 (Figure 1). In rabbit aorta made tolerant by large doses of nitroglycerin, ALDH2 dehydrogenase activity was inhibited by ∼50%.74 Similar inhibition was observed in studies using rat hearts, ex vivo.9 Although it remains to be determined how prolonged treatment with nitroglycerin leads to inactivation of ALDH2, these studies have opened a new avenue for examining the role of ALDH2 in the myocardium.
As described above, activation of ALDH2 reduced cardiac damage caused by ischaemia insult, indicating a cardioprotective role for ALDH2.9 That study also demonstrated that inactivation of ALDH2 associated with nitroglycerin tolerance resulted in an increase in infarct size.9 These data suggest a potential risk to patients who experience an AMI while on continuous nitroglycerin treatment (Figure 1). Whereas clinical treatments such as the intermittent use of organic nitrates have proven effective in reducing tolerance, other consequences of nitroglycerin tolerance have been poorly understood. Correlation studies analysing existing patients have yielded useful but incomplete data. In a 5-year follow-up study examining the long-term efficacy of nitrate therapy for the treatment of AMI in Japan, Yamauchi et al. found higher mortality rate in nitrate-treated group (18.9%), compared with the control patients (11.0%). However, the author acknowledged the limitation of the study, as the difference may be attributed to, among other uncontrolled factors, an increased frequency of nitrate use in patients with more severe conditions.79 Information on the consequence of nitroglycerin tolerance will be valuable for a better clinical use of nitroglycerin and other nitrates in the treatment of patients with angina-related cardiovascular diseases.
In the light of the ALDH2-dependent bioactivation of nitroglycerin in Asians carrying the E487K mutation, it is predicted that the substantially diminished ALDH2 activity would lead to a decreased response to nitroglycerin treatment. Indeed, in vitro experiments showed that the E487K mutant enzyme was ∼10-fold slower in catalysing nitroglycerin conversion to 1,2-glyceral dinitrate.69 Moreover, previous clinical studies confirmed a marked decrease in nitroglycerin efficacy in patients carrying the mutant ALDH2*2 relatively to carriers of the wild-type enzyme.48,75 Not surprisingly, larger doses of nitroglycerin were required to achieve sufficient vasodilatation in subjects with the ALDH2*2 form. Because the Asian ALDH2*2 mutation may be associated with a higher risk of various diseases including ischaemic damage9,13 due to a significant loss of ALDH2 activity, the consequence of nitroglycerin tolerance in the background of E487K polymorphism needs to be further investigated.
Since Alda-1 activates both the dehydrogenase activity and esterase activity of ALDH2,9,68 Beretta et al. evaluated the effect of Alda-1 on bioactivation of nitroglyercin.69 Surprisingly, Alda-1 failed to increase GTN denitration and bioactivation in these assays using either the ALDH2 wild-type and ALDH2*2 recombinant enzyme in vitro. A drug that can increase the potency of nitroglycerin either by enhancing its bioconversion to NO and/or by preventing the inhibitory effect of nitroglycerin on ALDH2 will clearly be beneficial, especially for the ALDH2*2 human subjects.
5. ALDH2 in transgenic mice, ethanol/acetaldehyde metabolism, and cardiovascular disease
Several independent ALDH2 transgenic mice have been established and studied extensively, especially with regard to the role of ALDH2 in ethanol metabolism. In one model, ALDH2 knockout mice were produced by gene interruption at the ALDH2 locus.80 As expected, these ALDH2-null mice lacked any detectable ALDH2 enzyme activity, accumulated a high level of acetaldehyde when exposed to ethanol, and were significantly more sensitive to alcohol and acetaldehyde toxicity and damage.81–83 Surprisingly, in these ALDH2-null mice, both acute and chronic administration of ethanol seem to produce a smaller extent of oxidative stress in the liver as measured by the decreased levels of MDA, alanine aminotransferase, TNF-α in the serum and increased level of the anti-oxidant, glutathione, when compared with the wild-type ALDH mice.84,85 The molecular mechanism for the reduction of these oxidative stress biomarkers is not clear, but may be associated with the metabolism of ethanol itself through the microsomal CYP2E1 pathway in the liver. It appears unlikely, though, that such ethanol-induced protective effect exists in hearts of the ALDH2*2 carriers. In fact, using the same ALDH2-null mice, Wenzel et al.86 demonstrated that the loss of ALDH2 enzyme activity led to increased mitochondrial oxidative stress in aortic endothelia by three pro-oxidant stimuli, nitroglycerin, doxorubicin, and acetaldehyde (Figure 1).
Overexpression of ALDH2 wild-type enzymes appeared to confer multiple beneficial effects to the heart tissue and cardiac functions in another transgenic mice model. Ma et al.87 explored the effect of ALDH2 overexpression on acute ethanol-induced myocardium damage. Acute ethanol challenge (3 g/kg) severely impaired myocardial and myocyte functions in the wild-type FVB mice as a result of acetaldehyde toxicity. This is evidenced by the reduction in maximal velocity of pressure development and decline (±dP/dt), left ventricular developed pressure, cell shortening, prolonged relengthening duration, and an increase in cardiac protein carbonyl level within 24 h after ethanol administration. These negative outcomes were much reduced in mice expressing four-fold higher levels of ALDH2. In this study, overexpression of ALDH2 was associated with a suppression of phosphatase activity which leads to enhanced Akt and AMP-activated protein kinase activities and regulation of their downstream targets Foxo3 transcription factor and caspase-3.87,88 Thus, ALDH2 appears critical in protecting the heart from aldehyde toxicity and is able to elicit signalling events that can protect against myocardial damages caused by acute ethanol toxicity.
Acetaldehyde toxicity also results in an exacerbated cardiac hypertrophy and contractile defect.89,90 In models of chronic alcohol ingestion, mimicking human alcoholism, overexpression of the ALDH2 enzyme dramatically reduced the complications of the cardiovascular system. Control mice placed on a 4% alcohol liquid diet for 14-week developed cardiac hypertrophy and contractile defects, whereas mice overexpressing ALDH2 exhibited a significant attenuation of cardiac hypertrophy and contractile dysfunction.34 These improvements were attributed to better calcium handling, reduction in myocardial fibrosis, reduction in protein carbonyl formation, and reduction in apoptosis, likely through a molecular mechanism associated with phosphorylation of apoptosis-stimulated kinase, glycogen synthase kinase-3β, GATA4, and cAMP-response element-binding protein. Related to alcohol-induced cardiomyopathy, overexpression of ALDH2 can also overcome insulin resistance in the heart of chronically alcohol-fed mice via an improvement of insulin signalling.91 This ALDH2-mediated protection mechanism also extends to the brain as apoptosis and cerebral damages were ablated in ALDH2 overexpressing mice after 12 weeks of continuous ethanol ingestion.92
Another approach to studying the potential consequences of E487K mutation represented in the ALDH2*2 carrying East Asians is the use of transgenic mice overexpressing ALDH2*2. Endo et al.93 created these loss-of-function ALDH2 transgenic mice by overexpressing the E487K human form under a strong promoter. In these mice, expression of the dominant-negative ALDH2*2 resulted in impaired ALDH activity when using a variety of aldehydic substrates. These mice also showed increased mitochondrial oxidative stress demonstrated by an elevated level of 4-HNE–protein adducts, a decline in mitochondria respiratory function, smaller body size, reduced muscle mass, diminished fat content, osteopenia, kyphosis, and smaller heart morphology.93 Surprisingly, despite the presence of substantial mitochondrial oxidative damage, the hearts of these ALDH2*2 transgenic mice demonstrated a greater tolerance to oxidative stress. This metabolic remodelling was attributed to increased glutathione biosynthesis to compensate for the diminished ALDH activity and the elevated oxidative stress. It should be noted that since ALDHs are tetramers, it is possible that overexpression of ALDH2*2 monomer inactivates not only the wild-type ALDH2, but may also form heterotetramers and inactive other ALDH isozymes (e.g. heterotetramer formation between ALDH2 and another highly homologous ALDH1B1).81,94 Since ALDH2*2 levels were eight-fold higher than that of the endogenous ALDH2 wild-type protein in the mitochondria, ALDH2*2 may therefore have pleiotropic effects. This may explain why the ALDH2-null mice and mice that express ALDH2*2 only one- to three-fold over the endogenous ALDH2 protein do not show these major pathologies.95 Whether metabolic remodelling and cardiac adaptation exist in ALDH2*2 Asians subjects therefore remains to be determined. A better transgenic animal model for the study of human ALDH2*2 deficiency would be the introduction, by gene targeting, of a single true genomic E487K point mutation to replace the wild-type ALDH2*1 allele on mouse chromosome 5 via homologous recombination.
6. Conclusion
Aldehydes are commonly formed inside cells as metabolic products (e.g. products of lipid peroxidation) or can gain access to the body from the environment (e.g. industry pollutants and food additives). Because aldehydes are chemically reactive moieties, forming adducts on a variety of macromolecules, which result in impairment or inactivation of these macromolecules, aldehydes catabolism is critical for cytoprotection, in general, and for cardioprotection, in particular. A natural cardioprotective mechanism involves a family of ALDHs and in particular the intra-mitochondrial enzyme, ALDH2. Although ALDH2 is constitutively active, it can be further activated by ischaemic preconditioning (due to εPKC-mediated phosphorylation) as well as by small molecule activators of this enzyme, Aldas. In addition for its role in removal of toxic aldehydes, the role of ALDH2 in nitroglycerin conversion to the bioactive NO highlights the importance of this enzyme in a variety of cardiovascular diseases. Epidemiological studies in humans carrying an inactivating mutation in ALDH2, combined with genetic and pharmacological studies in animal models, suggest ALDH2 as an important target for generating new treatments for heart diseases.
Funding
The work in DM-R laboratory was supported by an NIG grant, AA11147. Funding to pay the Open Access publication charges for this article was provided by the National Institute on Alcohol Abuse and Alcoholism (NIAAA), grant number AA011147.
Acknowledgements
The authors thank Daniel Cheng for the design of the figure.
Conflict of interest: D.M.-R. is the founder of KAI Pharmaceuticals, Inc. However, none of the research in her laboratory is supported by or is in collaboration with the company.
References
- 1.Bolli R, Jeroudi MO, Patel BS, DuBose CM, Lai EK, Roberts R, et al. Direct evidence that oxygen-derived free radicals contribute to postischemic myocardial dysfunction in the intact dog. Proc Natl Acad Sci USA. 1989;86:4695–4699. doi: 10.1073/pnas.86.12.4695. doi:10.1073/pnas.86.12.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang M, Shah AM. Role of reactive oxygen species in myocardial remodeling. Curr Heart Fail Rep. 2007;4:26–30. doi: 10.1007/s11897-007-0022-5. doi:10.1007/s11897-007-0022-5. [DOI] [PubMed] [Google Scholar]
- 3.Poyton RO, Ball KA, Castello PR. Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol Metab. 2009;20:332–340. doi: 10.1016/j.tem.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 4.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. doi:10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Downey JM. Free radicals and their involvement during long-term myocardial ischemia and reperfusion. Annu Rev Physiol. 1990;52:487–504. doi: 10.1146/annurev.ph.52.030190.002415. doi:10.1146/annurev.ph.52.030190.002415. [DOI] [PubMed] [Google Scholar]
- 6.Hori M, Nishida K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc Res. 2009;81:457–464. doi: 10.1093/cvr/cvn335. doi:10.1093/cvr/cvn335. [DOI] [PubMed] [Google Scholar]
- 7.Misra MK, Sarwat M, Bhakuni P, Tuteja R, Tuteja N. Oxidative stress and ischemic myocardial syndromes. Med Sci Monit. 2009;15:RA209–219. [PubMed] [Google Scholar]
- 8.Budas GR, Disatnik MH, Mochly-Rosen D. Aldehyde dehydrogenase 2 in cardiac protection: a new therapeutic target? Trends Cardiovasc Med. 2009;19:158–164. doi: 10.1016/j.tcm.2009.09.003. doi:10.1016/j.tcm.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–1495. doi: 10.1126/science.1158554. doi:10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srivastava S, Chandra A, Wang LF, Seifert WE, Jr, DaGue BB, Ansari NH, et al. Metabolism of the lipid peroxidation product, 4-hydroxy-trans-2-nonenal, in isolated perfused rat heart. J Biol Chem. 1998;273:10893–10900. doi: 10.1074/jbc.273.18.10893. doi:10.1074/jbc.273.18.10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang GW, Guo Y, Vondriska TM, Zhang J, Zhang S, Tsai LL, et al. Acrolein consumption exacerbates myocardial ischemic injury and blocks nitric oxide-induced PKCepsilon signaling and cardioprotection. J Mol Cell Cardiol. 2008;44:1016–1022. doi: 10.1016/j.yjmcc.2008.03.020. doi:10.1016/j.yjmcc.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 12.Poli G, Schaur RJ, Siems WG, Leonarduzzi G. 4-Hydroxynonenal: a membrane lipid oxidation product of medicinal interest. Med Res Rev. 2008;28:569–631. doi: 10.1002/med.20117. doi:10.1002/med.20117. [DOI] [PubMed] [Google Scholar]
- 13.Marchitti SA, Brocker C, Stagos D, Vasiliou V. Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert Opin Drug Metab Toxicol. 2008;4:697–720. doi: 10.1517/17425250802102627. doi:10.1517/17425255.4.6.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marchitti SA, Deitrich RA, Vasiliou V. Neurotoxicity and metabolism of the catecholamine-derived 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenylglycolaldehyde: the role of aldehyde dehydrogenase. Pharmacol Rev. 2007;59:125–150. doi: 10.1124/pr.59.2.1. doi:10.1124/pr.59.2.1. [DOI] [PubMed] [Google Scholar]
- 15.Vasiliou V, Nebert DW. Analysis and update of the human aldehyde dehydrogenase (ALDH) gene family. Hum Genomics. 2005;2:138–143. doi: 10.1186/1479-7364-2-2-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. doi:10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 17.Yokoyama A, Omori T. Genetic polymorphisms of alcohol and aldehyde dehydrogenases and risk for esophageal and head and neck cancers. Jpn J Clin Oncol. 2003;33:111–121. doi: 10.1093/jjco/hyg026. doi:10.1093/jjco/hyg026. [DOI] [PubMed] [Google Scholar]
- 18.Burke WJ. 3,4-Dihydroxyphenylacetaldehyde: a potential target for neuroprotective therapy in Parkinson's disease. Curr Drug Targets CNS Neurol Disord. 2003;2:143–148. doi: 10.2174/1568007033482913. doi:10.2174/1568007033482913. [DOI] [PubMed] [Google Scholar]
- 19.Burke WJ, Li SW, Williams EA, Nonneman R, Zahm DS. 3,4-Dihydroxyphenylacetaldehyde is the toxic dopamine metabolite in vivo: implications for Parkinson's disease pathogenesis. Brain Res. 2003;989:205–213. doi: 10.1016/s0006-8993(03)03354-7. doi:10.1016/S0006-8993(03)03354-7. [DOI] [PubMed] [Google Scholar]
- 20.Cantero AV, Portero-Otin M, Ayala V, Auge N, Sanson M, Elbaz M, et al. Methylglyoxal induces advanced glycation end product (AGEs) formation and dysfunction of PDGF receptor-beta: implications for diabetic atherosclerosis. FASEB J. 2007;21:3096–3106. doi: 10.1096/fj.06-7536com. doi:10.1096/fj.06-7536com. [DOI] [PubMed] [Google Scholar]
- 21.Lee SH, Oe T, Blair IA. Vitamin C-induced decomposition of lipid hydroperoxides to endogenous genotoxins. Science. 2001;292:2083–2086. doi: 10.1126/science.1059501. doi:10.1126/science.1059501. [DOI] [PubMed] [Google Scholar]
- 22.Sayre LM, Lin D, Yuan Q, Zhu X, Tang X. Protein adducts generated from products of lipid oxidation: focus on HNE and one. Drug Metab Rev. 2006;38:651–675. doi: 10.1080/03602530600959508. doi:10.1080/03602530600959508. [DOI] [PubMed] [Google Scholar]
- 23.Petersen DR, Doorn JA. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free Radic Biol Med. 2004;37:937–945. doi: 10.1016/j.freeradbiomed.2004.06.012. doi:10.1016/j.freeradbiomed.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 24.Uchida K, Stadtman ER. Covalent attachment of 4-hydroxynonenal to glyceraldehyde-3-phosphate dehydrogenase. A possible involvement of intra- and intermolecular cross-linking reaction. J Biol Chem. 1993;268:6388–6393. [PubMed] [Google Scholar]
- 25.Farout L, Mary J, Vinh J, Szweda LI, Friguet B. Inactivation of the proteasome by 4-hydroxy-2-nonenal is site specific and dependant on 20S proteasome subtypes. Arch Biochem Biophys. 2006;453:135–142. doi: 10.1016/j.abb.2006.02.003. doi:10.1016/j.abb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Ferrington DA, Kapphahn RJ. Catalytic site-specific inhibition of the 20S proteasome by 4-hydroxynonenal. FEBS Lett. 2004;578:217–223. doi: 10.1016/j.febslet.2004.11.003. doi:10.1016/j.febslet.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 27.Yan LJ, Sohal RS. Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proc Natl Acad Sci USA. 1998;95:12896–12901. doi: 10.1073/pnas.95.22.12896. doi:10.1073/pnas.95.22.12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Echtay KS, Brand MD. 4-Hydroxy-2-nonenal and uncoupling proteins: an approach for regulation of mitochondrial ROS production. Redox Rep. 2007;12:26–29. doi: 10.1179/135100007X162158. doi:10.1179/135100007X162158. [DOI] [PubMed] [Google Scholar]
- 29.Kristal BS, Park BK, Yu BP. 4-Hydroxyhexenal is a potent inducer of the mitochondrial permeability transition. J Biol Chem. 1996;271:6033–6038. doi: 10.1074/jbc.271.11.6033. [DOI] [PubMed] [Google Scholar]
- 30.Aberle NS, 2nd, Picklo MJ, Sr, Amarnath V, Ren J. Inhibition of cardiac myocyte contraction by 4-hydroxy-trans-2-nonenal. Cardiovasc Toxicol. 2004;4:21–28. doi: 10.1385/ct:4:1:21. doi:10.1385/CT:4:1:21. [DOI] [PubMed] [Google Scholar]
- 31.Bhatnagar A. Environmental cardiology: studying mechanistic links between pollution and heart disease. Circ Res. 2006;99:692–705. doi: 10.1161/01.RES.0000243586.99701.cf. doi:10.1161/01.RES.0000243586.99701.cf. [DOI] [PubMed] [Google Scholar]
- 32.Lucas DT, Szweda LI. Cardiac reperfusion injury: aging, lipid peroxidation, and mitochondrial dysfunction. Proc Natl Acad Sci USA. 1998;95:510–514. doi: 10.1073/pnas.95.2.510. doi:10.1073/pnas.95.2.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Churchill EN, Disatnik MH, Mochly-Rosen D. Time-dependent and ethanol-induced cardiac protection from ischemia mediated by mitochondrial translocation of varepsilonPKC and activation of aldehyde dehydrogenase 2. J Mol Cell Cardiol. 2009;46:278–284. doi: 10.1016/j.yjmcc.2008.09.713. doi:10.1016/j.yjmcc.2008.09.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doser TA, Turdi S, Thomas DP, Epstein PN, Li SY, Ren J. Transgenic overexpression of aldehyde dehydrogenase-2 rescues chronic alcohol intake-induced myocardial hypertrophy and contractile dysfunction. Circulation. 2009;11:1941–1949. doi: 10.1161/CIRCULATIONAHA.108.823799. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Perlman DH, Bauer SM, Ashrafian H, Bryan NS, Garcia-Saura MF, Lim CC, et al. Mechanistic insights into nitrite-induced cardioprotection using an integrated metabolomic/proteomic approach. Circ Res. 2009;104:796–804. doi: 10.1161/CIRCRESAHA.108.187005. doi:10.1161/CIRCRESAHA.108.187005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stewart MJ, Malek K, Crabb DW. Distribution of messenger RNAs for aldehyde dehydrogenase 1, aldehyde dehydrogenase 2, and aldehyde dehydrogenase 5 in human tissues. J Investig Med. 1996;44:42–46. [PubMed] [Google Scholar]
- 37.Klyosov AA, Rashkovetsky LG, Tahir MK, Keung WM. Possible role of liver cytosolic and mitochondrial aldehyde dehydrogenases in acetaldehyde metabolism. Biochemistry. 1996;35:4445–4456. doi: 10.1021/bi9521093. doi:10.1021/bi9521093. [DOI] [PubMed] [Google Scholar]
- 38.Chen Z, Foster MW, Zhang J, Mao L, Rockman HA, Kawamoto T, et al. An essential role for mitochondrial aldehyde dehydrogenase in nitroglycerin bioactivation. Proc Natl Acad Sci USA. 2005;102:12159–12164. doi: 10.1073/pnas.0503723102. doi:10.1073/pnas.0503723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Z, Stamler JS. Bioactivation of nitroglycerin by the mitochondrial aldehyde dehydrogenase. Trends Cardiovasc Med. 2006;16:259–265. doi: 10.1016/j.tcm.2006.05.001. doi:10.1016/j.tcm.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 40.Yoshida A, Huang IY, Ikawa M. Molecular abnormality of an inactive aldehyde dehydrogenase variant commonly found in Orientals. Proc Natl Acad Sci USA. 1984;81:258–261. doi: 10.1073/pnas.81.1.258. doi:10.1073/pnas.81.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goedde HW, Agarwal DP, Harada S, Meier-Tackmann D, Ruofu D, Bienzle U, et al. Population genetic studies on aldehyde dehydrogenase isozyme deficiency and alcohol sensitivity. Am J Hum Genet. 1983;35:769–772. [PMC free article] [PubMed] [Google Scholar]
- 42.Eng MY, Luczak SE, Wall TL. ALDH2, ADH1B, and ADH1C genotypes in Asians: a literature review. Alcohol Res Health. 2007;30:22–27. [PMC free article] [PubMed] [Google Scholar]
- 43.Larson HN, Weiner H, Hurley TD. Disruption of the coenzyme binding site and dimer interface revealed in the crystal structure of mitochondrial aldehyde dehydrogenase ‘Asian’ variant. J Biol Chem. 2005;280:30550–30556. doi: 10.1074/jbc.M502345200. doi:10.1074/jbc.M502345200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seitz HK, Matsuzaki S, Yokoyama A, Homann N, Vakevainen S, Wang XD. Alcohol and cancer. Alcohol Clin Exp Res. 2001;25:137S–143S. doi: 10.1097/00000374-200105051-00024. [DOI] [PubMed] [Google Scholar]
- 45.Brooks PJ, Enoch MA, Goldman D, Li TK, Yokoyama A. The alcohol flushing response: an unrecognized risk factor for esophageal cancer from alcohol consumption. PLoS Med. 2009;6:e50. doi: 10.1371/journal.pmed.1000050. doi:10.1371/journal.pmed.1000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen YJ, Chen C, Wu DC, Lee CH, Wu CI, Lee JM, et al. Interactive effects of lifetime alcohol consumption and alcohol and aldehyde dehydrogenase polymorphisms on esophageal cancer risks. Int J Cancer. 2006;119:2827–2831. doi: 10.1002/ijc.22199. doi:10.1002/ijc.22199. [DOI] [PubMed] [Google Scholar]
- 47.Seitz HK, Stickel F. Acetaldehyde as an underestimated risk factor for cancer development: role of genetics in ethanol metabolism. Genes Nutr. 2010;5:121–128. doi: 10.1007/s12263-009-0154-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Y, Zhang D, Jin W, Shao C, Yan P, Xu C, et al. Mitochondrial aldehyde dehydrogenase-2 (ALDH2) Glu504Lys polymorphism contributes to the variation in efficacy of sublingual nitroglycerin. J Clin Invest. 2006;116:506–511. doi: 10.1172/JCI26564. doi:10.1172/JCI26564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jo SA, Kim EK, Park MH, Han C, Park HY, Jang Y, et al. A Glu487Lys polymorphism in the gene for mitochondrial aldehyde dehydrogenase 2 is associated with myocardial infarction in elderly Korean men. Clin Chim Acta. 2007;382:43–47. doi: 10.1016/j.cca.2007.03.016. doi:10.1016/j.cca.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 50.Takagi S, Iwai N, Yamauchi R, Kojima S, Yasuno S, Baba T, et al. Aldehyde dehydrogenase 2 gene is a risk factor for myocardial infarction in Japanese men. Hypertens Res. 2002;25:677–681. doi: 10.1291/hypres.25.677. doi:10.1291/hypres.25.677. [DOI] [PubMed] [Google Scholar]
- 51.Amamoto K, Okamura T, Tamaki S, Kita Y, Tsujita Y, Kadowaki T, et al. Epidemiologic study of the association of low-Km mitochondrial acetaldehyde dehydrogenase genotypes with blood pressure level and the prevalence of hypertension in a general population. Hypertens Res. 2002;25:857–864. doi: 10.1291/hypres.25.857. doi:10.1291/hypres.25.857. [DOI] [PubMed] [Google Scholar]
- 52.Ohsawa I, Kamino K, Nagasaka K, Ando F, Niino N, Shimokata H, et al. Genetic deficiency of a mitochondrial aldehyde dehydrogenase increases serum lipid peroxides in community-dwelling females. J Hum Genet. 2003;48:404–409. doi: 10.1007/s10038-003-0046-y. doi:10.1007/s10038-003-0046-y. [DOI] [PubMed] [Google Scholar]
- 53.Heart disease and stroke statistics. Am Heart Assoc. 2003:1–42. Update. [Google Scholar]
- 54.Parratt JR. Protection of the heart by ischaemic preconditioning: mechanisms and possibilities for pharmacological exploitation. Trends Pharmacol Sci. 1994;15:19–25. doi: 10.1016/0165-6147(94)90129-5. doi:10.1016/0165-6147(94)90129-5. [DOI] [PubMed] [Google Scholar]
- 55.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 56.Nakano A, Cohen MV, Downey JM. Ischemic preconditioning: from basic mechanisms to clinical applications. Pharmacol Ther. 2000;86:263–275. doi: 10.1016/s0163-7258(00)00058-9. doi:10.1016/S0163-7258(00)00058-9. [DOI] [PubMed] [Google Scholar]
- 57.Dawn B, Bolli R. Role of nitric oxide in myocardial preconditioning. Ann N Y Acad Sci. 2002;962:18–41. doi: 10.1111/j.1749-6632.2002.tb04053.x. doi:10.1111/j.1749-6632.2002.tb04053.x. [DOI] [PubMed] [Google Scholar]
- 58.Yellon DM, Downey JM. Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev. 2003;83:1113–1151. doi: 10.1152/physrev.00009.2003. [DOI] [PubMed] [Google Scholar]
- 59.Chen C, Mochly-Rosen D. Opposing effects of delta and xi PKC in ethanol-induced cardioprotection. J Mol Cell Cardiol. 2001;33:581–585. doi: 10.1006/jmcc.2000.1330. doi:10.1006/jmcc.2000.1330. [DOI] [PubMed] [Google Scholar]
- 60.Chen CH, Gray MO, Mochly-Rosen D. Cardioprotection from ischemia by a brief exposure to physiological levels of ethanol: role of epsilon protein kinase C. Proc Natl Acad Sci USA. 1999;96:12784–12789. doi: 10.1073/pnas.96.22.12784. doi:10.1073/pnas.96.22.12784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Collins MA, Neafsey EJ, Mukamal KJ, Gray MO, Parks DA, Das DK, et al. Alcohol in moderation, cardioprotection, and neuroprotection: epidemiological considerations and mechanistic studies. Alcohol Clin Exp Res. 2009;33:206–219. doi: 10.1111/j.1530-0277.2008.00828.x. doi:10.1111/j.1530-0277.2008.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen J, Henderson GI, Freeman GL. Role of 4-hydroxynonenal in modification of cytochrome c oxidase in ischemia/reperfused rat heart. J Mol Cell Cardiol. 2001;33:1919–1927. doi: 10.1006/jmcc.2001.1454. doi:10.1006/jmcc.2001.1454. [DOI] [PubMed] [Google Scholar]
- 63.Eaton P, Li JM, Hearse DJ, Shattock MJ. Formation of 4-hydroxy-2-nonenal-modified proteins in ischemic rat heart. Am J Physiol. 1999;276:H935–H943. doi: 10.1152/ajpheart.1999.276.3.H935. [DOI] [PubMed] [Google Scholar]
- 64.Doorn JA, Hurley TD, Petersen DR. Inhibition of human mitochondrial aldehyde dehydrogenase by 4-hydroxynon-2-enal and 4-oxonon-2-enal. Chem Res Toxicol. 2006;19:102–110. doi: 10.1021/tx0501839. doi:10.1021/tx0501839. [DOI] [PubMed] [Google Scholar]
- 65.Mitchell DY, Petersen DR. Inhibition of rat hepatic mitochondrial aldehyde dehydrogenase-mediated acetaldehyde oxidation by trans-4-hydroxy-2-nonenal. Hepatology. 1991;13:728–734. doi: 10.1016/0270-9139(91)92572-p. [DOI] [PubMed] [Google Scholar]
- 66.Budas GR, Disatnik MH, Chen CH, Mochly-Rosen D. Activation of aldehyde dehydrogenase 2 (ALDH2) confers cardioprotection in protein kinase C epsilon (PKCvarepsilon) knockout mice. J Mol Cell Cardiol. 2010;48:757–764. doi: 10.1016/j.yjmcc.2009.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hill BG, Awe SO, Vladykovskaya E, Ahmed Y, Liu SQ, Bhatnagar A, et al. Myocardial ischaemia inhibits mitochondrial metabolism of 4-hydroxy-trans-2-nonenal. Biochem J. 2009;417:513–524. doi: 10.1042/BJ20081615. doi:10.1042/BJ20081615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perez-Miller S, Younus H, Vanam R, Chen CH, Mochly-Rosen D, Hurley TD. Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nat Struct Mol Biol. 2010;17:159–164. doi: 10.1038/nsmb.1737. doi:10.1038/nsmb.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Beretta M, Gorren AC, Wenzl MV, Weis R, Russwurm M, Koesling D, et al. Characterization of the East Asian variant of aldehyde dehydrogenase-2: bioactivation of nitroglycerin and effects of Alda-1. J Biol Chem. 2010;285:943–952. doi: 10.1074/jbc.M109.014548. doi:10.1074/jbc.M109.014548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Parker JD, Parker JO. Nitrate therapy for stable angina pectoris. N Engl J Med. 1998;338:520–531. doi: 10.1056/NEJM199802193380807. doi:10.1056/NEJM199802193380807. [DOI] [PubMed] [Google Scholar]
- 71.Abrams J. Hemodynamic effects of nitroglycerin and long-acting nitrates. Am Heart J. 1985;110:216–224. [PubMed] [Google Scholar]
- 72.Munzel T, Daiber A, Mulsch A. Explaining the phenomenon of nitrate tolerance. Circ Res. 2005;97:618–628. doi: 10.1161/01.RES.0000184694.03262.6d. doi:10.1161/01.RES.0000184694.03262.6d. [DOI] [PubMed] [Google Scholar]
- 73.Klemenska E, Beresewicz A. Bioactivation of organic nitrates and the mechanism of nitrate tolerance. Cardiol J. 2009;16:11–19. [PubMed] [Google Scholar]
- 74.Chen Z, Zhang J, Stamler JS. Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proc Natl Acad Sci USA. 2002;99:8306–8311. doi: 10.1073/pnas.122225199. doi:10.1073/pnas.122225199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mackenzie IS, Maki-Petaja KM, McEniery CM, Bao YP, Wallace SM, Cheriyan J, et al. Aldehyde dehydrogenase 2 plays a role in the bioactivation of nitroglycerin in humans. Arterioscler Thromb Vasc Biol. 2005;25:1891–1895. doi: 10.1161/01.ATV.0000179599.71086.89. doi:10.1161/01.ATV.0000179599.71086.89. [DOI] [PubMed] [Google Scholar]
- 76.Zhang J, Chen Z, Cobb FR, Stamler JS. Role of mitochondrial aldehyde dehydrogenase in nitroglycerin-induced vasodilation of coronary and systemic vessels: an intact canine model. Circulation. 2004;110:750–755. doi: 10.1161/01.CIR.0000138105.17864.6B. doi:10.1161/01.CIR.0000138105.17864.6B. [DOI] [PubMed] [Google Scholar]
- 77.DiFabio J, Ji Y, Vasiliou V, Thatcher GR, Bennett BM. Role of mitochondrial aldehyde dehydrogenase in nitrate tolerance. Mol Pharmacol. 2003;64:1109–1116. doi: 10.1124/mol.64.5.1109. doi:10.1124/mol.64.5.1109. [DOI] [PubMed] [Google Scholar]
- 78.Hink U, Daiber A, Kayhan N, Trischler J, Kraatz C, Oelze M, et al. Oxidative inhibition of the mitochondrial aldehyde dehydrogenase promotes nitroglycerin tolerance in human blood vessels. J Am Coll Cardiol. 2007;50:2226–2232. doi: 10.1016/j.jacc.2007.08.031. doi:10.1016/j.jacc.2007.08.031. [DOI] [PubMed] [Google Scholar]
- 79.Yamauchi T, Hagiwara N, Kasanuki H, Koyanagi R, Ogawa H. Long-term nitrate use in acute myocardial infarction (the Heart Institute of Japan, Department of Cardiology nitrate evaluation program) Cardiovasc Drugs Ther. 2008;22:177–184. doi: 10.1007/s10557-008-6089-8. doi:10.1007/s10557-008-6089-8. [DOI] [PubMed] [Google Scholar]
- 80.Kitagawa K, Kawamoto T, Kunugita N, Tsukiyama T, Okamoto K, Yoshida A, et al. Aldehyde dehydrogenase (ALDH) 2 associates with oxidation of methoxyacetaldehyde; in vitro analysis with liver subcellular fraction derived from human and Aldh2 gene targeting mouse. FEBS Lett. 2000;476:306–311. doi: 10.1016/s0014-5793(00)01710-5. doi:10.1016/S0014-5793(00)01710-5. [DOI] [PubMed] [Google Scholar]
- 81.Isse T, Matsuno K, Oyama T, Kitagawa K, Kawamoto T. Aldehyde dehydrogenase 2 gene targeting mouse lacking enzyme activity shows high acetaldehyde level in blood, brain, and liver after ethanol gavages. Alcohol Clin Exp Res. 2005;29:1959–1964. doi: 10.1097/01.alc.0000187161.07820.21. doi:10.1097/01.alc.0000187161.07820.21. [DOI] [PubMed] [Google Scholar]
- 82.Kiyoshi A, Weihuan W, Mostofa J, Mitsuru K, Toyoshi I, Toshihiro K, et al. Ethanol metabolism in ALDH2 knockout mice–blood acetate levels. Leg Med (Tokyo) 2009;11(Suppl. 1):S413–S415. doi: 10.1016/j.legalmed.2009.02.043. [DOI] [PubMed] [Google Scholar]
- 83.Oyama T, Kim YD, Isse T, Phuong PT, Ogawa M, Yamaguchi T, et al. A pilot study on subacute ethanol treatment of ALDH2 KO mice. J Toxicol Sci. 2007;32:421–428. doi: 10.2131/jts.32.421. doi:10.2131/jts.32.421. [DOI] [PubMed] [Google Scholar]
- 84.Matsumoto A, Ichiba M, Horita M, Yamashita Z, Takahashi T, Isse T, et al. Lack of aldehyde dehydrogenase ameliorates oxidative stress induced by single-dose ethanol administration in mouse liver. Alcohol. 2007;41:57–59. doi: 10.1016/j.alcohol.2007.01.004. doi:10.1016/j.alcohol.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 85.Matsumoto A, Kawamoto T, Mutoh F, Isse T, Oyama T, Kitagawa K, et al. Effects of 5-week ethanol feeding on the liver of aldehyde dehydrogenase 2 knockout mice. Pharmacogenet Genomics. 2008;18:847–852. doi: 10.1097/FPC.0b013e328307a0a9. doi:10.1097/FPC.0b013e328307a0a9. [DOI] [PubMed] [Google Scholar]
- 86.Wenzel P, Muller J, Zurmeyer S, Schuhmacher S, Schulz E, Oelze M, et al. ALDH-2 deficiency increases cardiovascular oxidative stress–evidence for indirect antioxidative properties. Biochem Biophys Res Commun. 2008;367:137–143. doi: 10.1016/j.bbrc.2007.12.089. doi:10.1016/j.bbrc.2007.12.089. [DOI] [PubMed] [Google Scholar]
- 87.Ma H, Li J, Gao F, Ren J. Aldehyde dehydrogenase 2 ameliorates acute cardiac toxicity of ethanol: role of protein phosphatase and forkhead transcription factor. J Am Coll Cardiol. 2009;54:2187–2196. doi: 10.1016/j.jacc.2009.04.100. doi:10.1016/j.jacc.2009.04.100. [DOI] [PubMed] [Google Scholar]
- 88.Karliner JS. Lessons from the besotted heart. J Am Coll Cardiol. 2009;54:2197–2198. doi: 10.1016/j.jacc.2009.06.052. doi:10.1016/j.jacc.2009.06.052. [DOI] [PubMed] [Google Scholar]
- 89.Duan J, McFadden GE, Borgerding AJ, Norby FL, Ren BH, Ye G, et al. Overexpression of alcohol dehydrogenase exacerbates ethanol-induced contractile defect in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2002;282:H1216–H1222. doi: 10.1152/ajpheart.00780.2001. [DOI] [PubMed] [Google Scholar]
- 90.Hintz KK, Relling DP, Saari JT, Borgerding AJ, Duan J, Ren BH, et al. Cardiac overexpression of alcohol dehydrogenase exacerbates cardiac contractile dysfunction, lipid peroxidation, and protein damage after chronic ethanol ingestion. Alcohol Clin Exp Res. 2003;27:1090–1098. doi: 10.1097/01.ALC.0000075823.73536.DD. doi:10.1097/01.ALC.0000075823.73536.DD. [DOI] [PubMed] [Google Scholar]
- 91.Li SY, Gilbert SA, Li Q, Ren J. Aldehyde dehydrogenase-2 (ALDH2) ameliorates chronic alcohol ingestion-induced myocardial insulin resistance and endoplasmic reticulum stress. J Mol Cell Cardiol. 2009;47:247–255. doi: 10.1016/j.yjmcc.2009.03.017. doi:10.1016/j.yjmcc.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ren J, Babcock SA, Li Q, Huff AF, Li SY, Doser TA. Aldehyde dehydrogenase-2 transgene ameliorates chronic alcohol ingestion-induced apoptosis in cerebral cortex. Toxicol Lett. 2009;187:149–156. doi: 10.1016/j.toxlet.2009.02.019. doi:10.1016/j.toxlet.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Endo J, Sano M, Katayama T, Hishiki T, Shinmura K, Morizane S, et al. Metabolic remodeling induced by mitochondrial aldehyde stress stimulates tolerance to oxidative stress in the heart. Circ Res. 2009;105:1118–1127. doi: 10.1161/CIRCRESAHA.109.206607. doi:10.1161/CIRCRESAHA.109.206607. [DOI] [PubMed] [Google Scholar]
- 94.Steinmetz CG, Xie P, Weiner H, Hurley TD. Structure of mitochondrial aldehyde dehydrogenase: the genetic component of ethanol aversion. Structure. 1997;5:701–711. doi: 10.1016/s0969-2126(97)00224-4. doi:10.1016/S0969-2126(97)00224-4. [DOI] [PubMed] [Google Scholar]
- 95.Ohsawa I, Nishimaki K, Murakami Y, Suzuki Y, Ishikawa M, Ohta S. Age-dependent neurodegeneration accompanying memory loss in transgenic mice defective in mitochondrial aldehyde dehydrogenase 2 activity. J Neurosci. 2008;28:6239–6249. doi: 10.1523/JNEUROSCI.4956-07.2008. doi:10.1523/JNEUROSCI.4956-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]