

Abstract
At the neuromuscular junction (NMJ), the dystrophin protein complex provides a scaffold that functions to stabilize acetylcholine receptor (AChR) clusters. Syntrophin, a key component of that scaffold, is a multidomain adapter protein that links a variety of signaling proteins and ion channels to the dystrophin protein complex. Without syntrophin, utrophin and neuronal nitric oxide synthase μ (nNOSμ) fail to localize to the NMJ and the AChRs are distributed abnormally. Here we investigate the contribution of syntrophin domains to AChR distribution and to localization of utrophin and nNOSμ at the NMJ. Transgenic mice expressing α-syntrophin lacking portions of the first pleckstrin homology (PH) domain (ΔPH1a or ΔPH1b) or the entire PDZ domain (ΔPDZ) were bred onto the α-syntrophin null background. As expected the ΔPDZ transgene did not restore the NMJ localization of nNOS. The ΔPH1a transgene did restore postsynaptic nNOS but surprisingly did not restore sarcolemmal nNOS (although sarcolemmal aquaporin-4 was restored). Mice lacking the α-syntrophin PDZ domain or either half of the PH1 domain were able to restore utrophin to the NMJ but did not correct the aberrant AChR distribution of the α-syntrophin knock-out mice. However, mice expressing both the transgenic ΔPDZ and the transgenic ΔPH1a constructs did restore normal AChR distribution, demonstrating that both domains are required but need not be confined within the same protein to function. We conclude that the PH1 and PDZ domains of α-syntrophin work in concert to facilitate the localization of AChRs and nNOS at the NMJ.
Introduction
Dystrophin is concentrated at the postsynaptic neuromuscular junction (NMJ) along with its orthologs, utrophin and dystrobrevin (Bewick et al., 1996). Mouse models lacking any one of these proteins exhibit reduced AChR expression and/or aberrant distribution (Lyons and Slater, 1991; Grady et al., 1997, 2000). The dystrophin (or utrophin) complex consists of a myriad of proteins that function to link the extracellular matrix to the actin cytoskeleton and to provide scaffolding for precise localization of signaling proteins. This scaffolding function relies primarily on the ability of each of the dystrophin orthologs to bind two syntrophin molecules (Newey et al., 2000).
Syntrophins are a family of adapter proteins that link signaling proteins to the dystrophin protein complex. Of the five syntrophin isoforms (α, β1, β2, γ1, γ2), four are present at the mammalian NMJ (Froehner et al., 1987; Peters et al., 1994, 1997; Alessi et al., 2006). However, only α-syntrophin is at the crests of the postjunctional folds colocalizing with the AChRs and utrophin (Kramarcy and Sealock, 2000).
Each of the syntrophins has four core domains, a PDZ domain, two PH (pleckstrin homology) domains, and a C-terminal SU (syntrophin unique) domain. The second PH domain (PH2) combined with the SU domain is critical for binding dystrophin proteins (Ahn and Kunkel, 1995; Kachinsky et al., 1999). This leaves the first PH domain (PH1) and the PDZ domain free to interact with other molecules. The syntrophin PDZ domain binds ion channels (Gee et al., 1998; Connors et al., 2004; Leonoudakis et al., 2004), G-protein-coupled receptors (Chen et al., 2006), water channels (Neely et al., 2001), kinases and associated proteins (Hasegawa et al., 1999; Lumeng et al., 1999; Hogan et al., 2001; Luo et al., 2005), and nNOS (Brenman et al., 1996). The syntrophin PH1 domain binds phosphoinositides but may interact with proteins as well (Chockalingam et al., 1999; Yan et al., 2005).
The syntrophin scaffold is involved in a variety of diseases, including multiple forms of muscular dystrophy (Compton et al., 2005, 2008; Wakayama et al., 2006), brain edema (Amiry-Moghaddam et al., 2003, 2004), and long QT syndrome (Ueda et al., 2008). Syntrophin is also associated with key physiological processes including insulin secretion (Ort et al., 2001) and blood pressure regulation (Lyssand et al., 2008). At the NMJ the absence of α-syntrophin results in structurally aberrant NMJs with reduced levels of AChRs (Adams et al., 2000). Instead of localizing only opposite the nerve terminal, the AChRs radiate away from the nerve in small spikes.
Utrophin is absent from the postsynaptic membrane of the α-syntrophin knock-out mouse NMJ (Adams et al., 2000). Mice lacking α-syntrophin do not have nNOS on the sarcolemma or at the NMJ, although it is still present in the cytosol (Adams et al., 2000; Thomas et al., 2003). In this work, we use an in vivo approach to determine the contribution of syntrophin domains to stabilizing AChR clusters and localizing utrophin and nNOS at the postsynaptic NMJ.
Materials and Methods
Transgenic mice.
Generation of the α-syntrophin knock-out mice and mice expressing the full-length and ΔPDZ α-syntrophin transgenes has been described previously (Adams et al., 2000, 2001). Here we generated two additional transgenic mice expressing mouse α-syntrophin lacking either the first half of the PH domain (ΔPH1a) or the second half of the first PH domain (ΔPH1b). For the ΔPH1a construct, we replaced the DNA encoding the first 71 aa of α-syntrophin with DNA encoding the FLAG epitope tag (MDYKDDDDK). For the ΔPH1b construct, we replaced DNA encoding amino acids 174-271 with a hemagglutinin epitope tag (YPYDVPDYASL). Both constructs were cloned downstream of the MCK promoter for muscle-specific expression (Adams et al., 2001). Constructs were injected into oocytes at the Transgenic Resources Program, University of Washington. Resulting founder mice (3 for ΔPH1a, 4 for ΔPH1b) were bred onto the α-syntrophin knock-out mouse background. Similar results were found in all founder lines for each construct. Experiments were performed using muscles from both male and female mice with no gender-specific differences observed. The experimental procedures performed on mice were approved by the Institutional Animal Care and Use Committee of the University of Washington.
Antibodies.
Rabbit polyclonal antibodies to utrophin and α-syntrophin have been described previously (Kramarcy et al., 1994; Peters et al., 1997). Commercial antibodies were purchased as follows: anti -HA from Zymed/Invitrogen, anti-Flag from Sigma Aldrich, anti-aquaporin-4 from Millipore Bioscience Research Reagents, and nNOS from Immunostar. Fluorescently tagged secondary antibodies and bungarotoxin were purchased from Invitrogen.
Immunoblotting.
Muscle protein extracts were prepared as previously described (Peters et al., 1997) and subjected to electrophoresis on a 4–15% gradient polyacrylamide gels (Bio-Rad), transferred to Immobilon-P membrane (Millipore), and incubated with α-syntrophin-specific antibodies as previously described (Peters et al., 1997).
Fluorescence microscopy.
Immunofluorescence labeling of cross sections was performed on unfixed quadriceps muscle using 8 μm cryosections as described previously (Peters et al., 1997). Images were obtained using a Leica TCS-NT confocal microscope. For en face views, teased fibers of the sternomastoid muscle were incubated with Alexa 488 α-bungarotoxin to visualize AChRs. Images were obtained using a Zeiss Meta 510 confocal microscope. All microscopy was performed at the W. M. Keck Center for Advanced Studies in Neural Signaling at the University of Washington.
Real-time PCR.
RNA was purified from the quadriceps of 8–12-week-old mice using the fibrous tissue kit from Qiagen. Real-time quantitative RT-PCR was performed using TaqMan chemistry and the ABI 7000 sequence detection system (Applied Biosystems) with 50 ng of RNA, the One-Step RT-PCR Master Mix reagents (Applied Biosystems), and primers specific for the A isoform of utrophin (primer 1—GGACTCTTGGCATCCCTCACT, primer 2—AGGCTTTCCCAGATCCTCTTCT). The utrophin-A probe spanned an intron splicing site and had the sequence CAACTGACAAGATTTAGCCTT. Samples were normalized using an internal GAPDH control primer/probe set (Applied Biosystems). Samples were obtained from 3 mice of each genotype and each sample was run in triplicate.
Results
We have previously shown that the α-syntrophin null mice have structurally aberrant neuromuscular junctions (Adams et al., 2000). Without α-syntrophin, the levels of AChRs are reduced and the remaining receptors are disorganized and have a shallow neural groove with radiating spikes of receptor. Furthermore, the proteins nNOS and utrophin both fail to localize to the NMJ in the absence of α-syntrophin. In this work, we address the mechanism of these defects by determining the functional regions of syntrophin required for proper formation of the NMJ.
Generation and characterization of transgenic mice
Since NMJ formation cannot be fully reproduced in cell culture, we took the in vivo approach of using transgenic mice to restore full-length α-syntrophin or altered α-syntrophin to the α-syntrophin knock-out mouse. We have previously generated transgenic mice expressing full-length α-syntrophin and α-syntrophin lacking the PDZ domain (Adams et al., 2001). In addition to these mice, we generated transgenic mice lacking portions of the first PH domain (Fig. 1). In one construct, we replaced the first half of the α-syntrophin PH1 domain (PH1a) with a Flag epitope tag. In the second construct, we replaced the PH1b domain with a hemagglutinin epitope (HA) epitope tag. All of the transgenes were under control of the same promoter (MCK) that is active only in striated muscle. Each of the transgenic mice was bred onto the α-syntrophin null background so that the only α-syntrophin expressed is that produced by the transgene. All of the experiments below were performed using mice bred onto the α-syntrophin null background.
Figure 1.
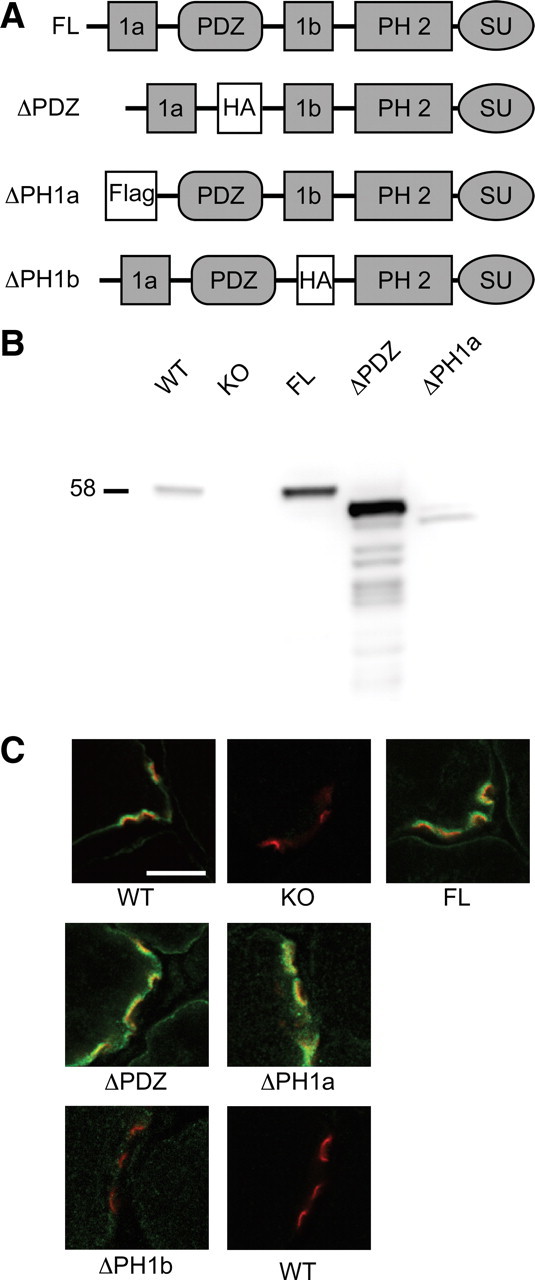
Generation of syntrophin transgenic mice. A, The protein domains of syntrophin are depicted in the four constructs used to generate transgenic mice. FL is the full-length α-syntrophin transgene. The first pleckstrin homology (PH) domain of syntrophin is naturally split by the PDZ domain into PH1a and PH1b. To generate the ΔPDZ transgene the PDZ domain was replaced by a hemagglutinin epitope tag (HA). In the ΔPH1a and ΔPH1b constructs, the 1a and 1b portions of the first PH domain were replaced by Flag and HA epitope tags, respectively. SU refers to the syntrophin unique region. B, Western blot analysis shows the relative expression levels of α-syntrophin in muscle isolated from the transgenic mice. Note the reduced size of syntrophins missing domains. WT, Wild type; KO, α-syntrophin knock-out. C, Immunofluorescence of NMJs in transverse sections of quadriceps muscle. Fluorescent α-bungarotoxin (red) was used to label the postsynaptic acetylcholine receptors. α-Syntrophin is labeled in green. The ΔPH1b sample (and wild-type control) was labeled using the HA antibody since the α-syntrophin-specific antibody epitope is absent in this construct. Scale bar, 10 μm.
Immunoblotting of skeletal muscle homogenates using our α-syntrophin-specific antibody shows the relative expression levels of the transgene product for 3 of the 4 mouse lines compared to wild-type muscle (Fig. 1B). The epitope for our α-syntrophin-specific antibody is located in the PH1b region and is therefore missing in the ΔPH1b construct. We performed similar blots using an anti-HA antibody but could not detect the ΔPH1b protein in muscle homogenates (data not shown). However, we were able to detect small amounts of the ΔPH1b protein at the NMJ using the anti-HA antibody by immunofluorescence (Fig. 1C). The ΔPH1b construct was expressed at very low levels in all four founder lines, suggesting that the PH1b region is required for stable expression of a-syntrophin. The other 3 transgenes express well and are localized properly to the sarcolemma and NMJ (Fig. 1).
NMJ structure
In the absence of α-syntrophin, the postsynaptic NMJ is disorganized with a shallow neural groove and AChRs radiating away from beneath the nerve in small spikes [Adams et al. (2000) and Fig. 2]. We tested whether the transgenic syntrophins lacking the PH1a, PH1b, or PDZ domains could restore normal structure to the NMJs of the α-syntrophin knock-out mouse. We collected single myofibers from the sternomastoid muscle of each transgenic and control strain. The muscle fibers were labeled with α-bungarotoxin and examined en face by confocal microscopy.
Figure 2.
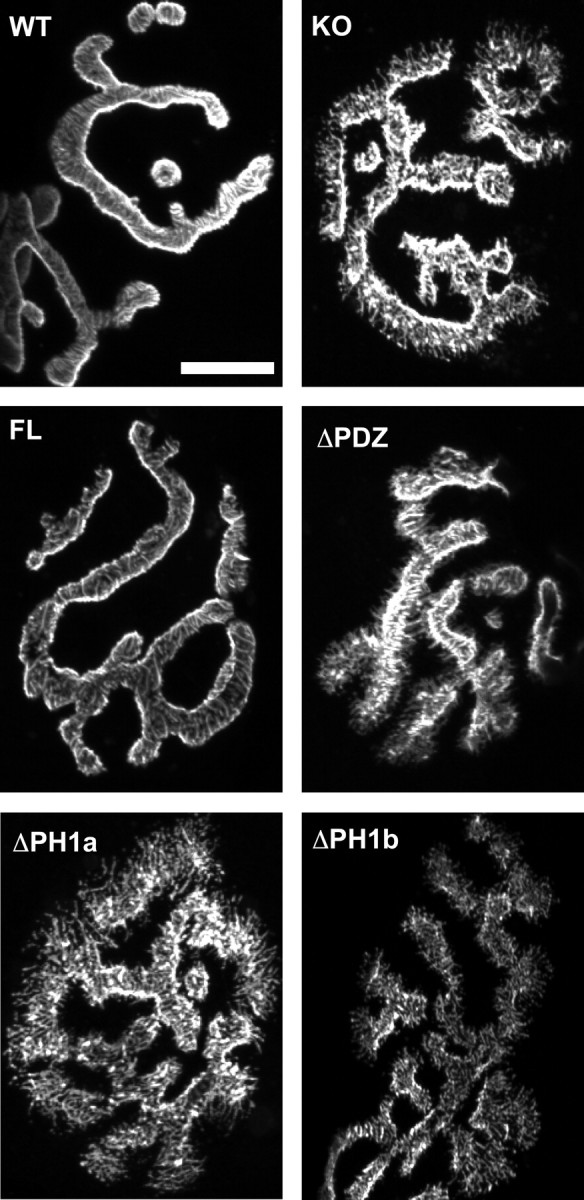
Transgenic rescue of aberrant NMJ structure in the α-syntrophin KO muscle. En face views of NMJs from the sternomastoid muscle visualized using fluorescent α-bungarotoxin. The spikes observed in the α-syntrophin KO mice are not present in the FL transgenic, which has continuous smooth edges similar to those of the WT mouse. NMJs of mice lacking the PDZ or either portion of the PH1 domain resemble the KO with spicules and fragmented edges. Scale bar, 20 μm.
The full-length syntrophin transgene restored the AChR distribution similar to that of wild-type mice (Fig. 2). Both the wild-type and full-length transgenic mice have AChR distributions with smooth edges and well defined neural groove borders. Muscle fibers from the other three transgenic mouse strains (ΔPDZ, ΔPH1a, and ΔPH1b) had AChR distributions indistinguishable from that of the α-syntrophin knock-out mouse (Fig. 2). Therefore both the PH1 and PDZ domains of syntrophin are required for proper distribution of AChRs at the NMJ.
Utrophin
In our previous characterization of the α-syntrophin null mouse, we observed that in the absence of α-syntrophin, the protein utrophin failed to localize properly at the NMJ. We hypothesized that the reason that the syntrophin domain deleted transgenes are unable to restore normal NMJ structures may be that they are unable to restore utrophin to the NMJ. We tested this idea by performing immunofluorescence studies on transverse sections of mouse quadriceps muscle (Fig. 3). For these experiments, we used an antibody that recognizes both the A (NMJ) and B (nerve and blood vessel) forms of utrophin (Weir et al., 2002). This allowed us to visualize the presynaptic utrophin, which serves as an internal antibody control when no postsynaptic utrophin is present (Fig. 3). We found that all four transgenes were able to restore utrophin to the postsynaptic NMJ. This result indicates that it is not the lack of utrophin that results in the inability of the ΔPDZ and ΔPH1 transgenes to rescue the NMJ structural defects.
Figure 3.
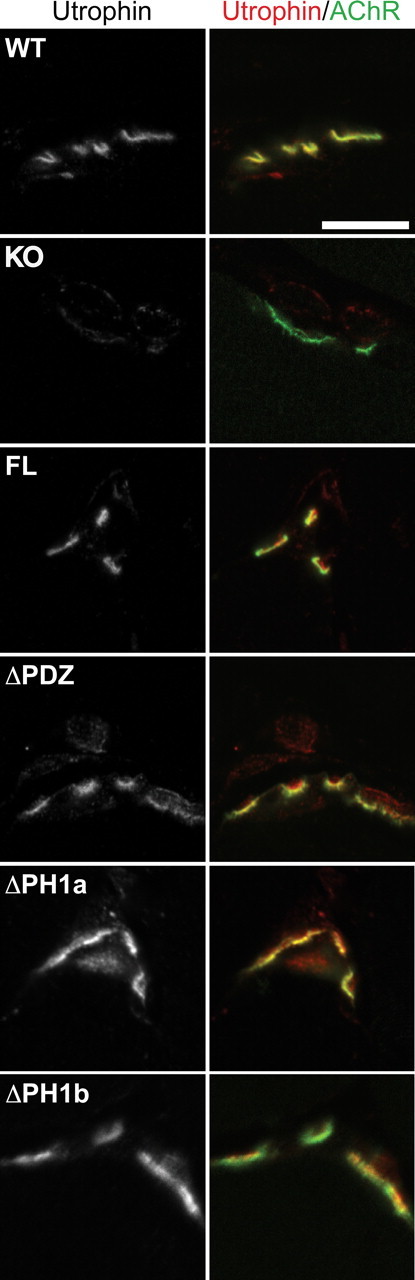
Postsynaptic utrophin is rescued by all transgenic mice. Immunofluorescence of quadriceps transverse sections shows that postsynaptic utrophin (left panel and red right panel) is present in all transgenic mice whereas it is absent in the α-syntrophin KO. α-Bungarotoxin labeling of acetylcholine receptors (AChR) is shown in green. Scale bar, 10 μm.
It is particularly surprising that the ΔPH1b mouse, which expresses very low levels of the transgene at the NMJ, (see Fig. 1C) was still able to restore utrophin to the NMJ. We currently do not know the basis for the lack of postsynaptic utrophin in the absence of α-syntrophin. The most likely possibilities are that α-syntrophin affects the stability, transport, or transcription of utrophin. We investigated the latter by determining the relative levels of utrophin-A mRNA expression in wild-type, α-syntrophin knock-out, and the full-length α-syntrophin transgenic mouse muscle. Real-time PCR using primers and probe specific for the utrophin-A isoform showed no significant difference in mRNA expression in the wild-type, α-syntrophin knock-out, and FL transgenic mice (Fig. 4). We conclude that utrophin-A transcription is independent of α-syntrophin expression.
Figure 4.

Utrophin-A mRNA expression in mouse muscle. Real-time PCR of mRNA derived from quadriceps muscle of wild type (WT), α-syntrophin knock-out (KO), and the full-length syntrophin transgenic (FL) shows similar levels of expression in each of these mice. Error bars are SDs with n = 3 mice.
nNOSμ
In addition to utrophin, the protein nNOSμ also depends on α-syntrophin for proper localization in muscle (Kameya et al., 1999; Adams et al., 2000). In the absence of α-syntrophin, nNOSμ fails to localize to either the sarcolemma or postsynaptic NMJ. We therefore investigated whether the transgenes could restore nNOSμ to its proper locations in skeletal muscle (Fig. 5). The full-length α-syntrophin transgene restored nNOSμ to both the NMJ and the sarcolemma (Fig. 5). As expected, the ΔPDZ transgene was unable to restore nNOSμ at either location. However, it was curious that the ΔPH1a transgene was able to restore nNOSμ at the NMJ but not on the sarcolemma. Following this surprising observation, we were concerned that by deleting the PH1a domain, we may have inadvertently altered the function of the PDZ domain. Therefore we investigated the distribution of another protein that is known to depend on the α-syntrophin PDZ domain for sarcolemmal expression, aquaporin-4. Aquaporin-4 is normally present on the sarcolemma of fast fibers but is absent from all fibers in the α-syntrophin knock-out muscle and the ΔPDZ transgenic muscle (Frigeri et al., 1998; Adams et al., 2000, 2001) (Fig. 5). Aquaporin-4 is restored to the sarcolemma by either the full-length α-syntrophin or the ΔPH1a α-syntrophin, indicating that the ΔPH1a transgene and its PDZ domain can function properly.
Figure 5.

The ΔPH1a transgenic mouse rescues nNOS expression at the NMJ but not on the sarcolemma. Immunofluorescence of quadriceps transverse sections shows that nNOS is absent from the NMJs of the KO and ΔPDZ mice. NMJ nNOS expression is rescued by both the full-length and ΔPH1a transgenes. However, sarcolemmal nNOS is rescued only by the full-length transgene, not by the ΔPH1a transgene. Another sarcolemmal protein, aquaporin-4 (AQP4) is rescued by the ΔPH1a transgene. Red, nNOS; AChR (green), α-Bungarotoxin-labeled acetylcholine receptors. Scale bar, 10 μm.
The ΔPDZ and the ΔPH1a transgenes both retain the C-terminal domains known to associate with dystrophin (and related proteins) and both were able to localize to the sarcolemma and NMJ (Figs. 1, 6). Since the ΔPDZ transgene retains the PH1a and the ΔPH1a constructs retains the PDZ domain, we postulated that if expressed together these two syntrophin mutants may be able to act in concert to restore syntrophin function. We therefore crossed the two transgenic lines to produce mice expressing both transgenes (ΔPDZ/ΔPH1a). Immunofluorescence using antibodies to the HA (ΔPDZ) and Flag (ΔPH1a) showed that both proteins were expressed on the sarcolemma and enriched at the NMJ as expected (Fig. 6). However, the coexpression of both transgenes was unable to restore nNOSμ to the sarcolemma. Instead the distribution of nNOSμ looked indistinguishable from that of the ΔPH1a alone with nNOSμ only detectable at the NMJ. However, when we looked at the NMJ structure in these mice, we found that the combination of transgenes was able to restore normal AChR distribution. The NMJs of these mice have smooth, well defined edges and lack the AChR spikes observed in the α-syntrophin knock-out mice. This is in direct contrast to the NMJs of either parent strain, which remain structurally aberrant. We conclude that at the NMJ the PH1a and PDZ domains of syntrophin can function without being covalently attached.
Figure 6.
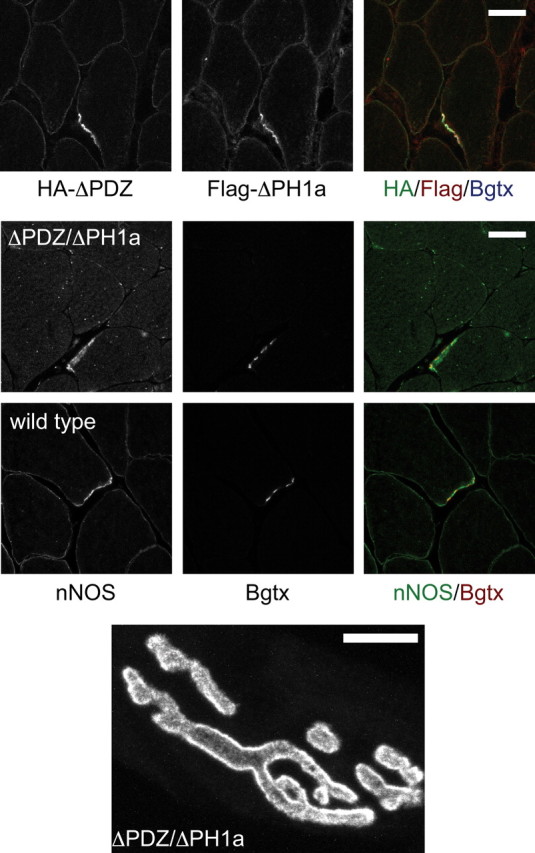
NMJ structure is rescued when both the ΔPH1a and ΔPDZ transgenes are present. We generated mice expressing both the ΔPH1a and ΔPDZ transgene as demonstrated by immunofluorescent labeling using antibodies to the epitope tags present in each construct (see Fig. 1). Like the muscles of mice expressing only the ΔPH1a transgene, the ΔPDZ/ΔPH1a muscles express nNOS only at the NMJ and not on the sarcolemma. An en face view of the NMJ (labeled using fluorescent α-bungarotoxin) shows that the NMJs of these mice have smooth continuous edges and lack the radiating spikes seen in the α-syntrophin knock-out mouse and in mice expressing only one of the parent transgenes (see Fig. 2). Scale bar, 20 μm.
Discussion
At the mammalian NMJ, AChRs are tightly packed into an array at the crest of the postjunctional folds directly opposed to the nerve terminal. Maintenance of this array is crucial for proper neuromuscular signal transmission. The submembrane protein assembly that provides stability to the AChR array includes several members of the dystrophin protein complex, specifically utrophin, dystrobrevin, and syntrophin. The absence of any of these proteins leads to reduced levels of AChRs (Lyons and Slater, 1991; Grady et al., 1997, 2000; Adams et al., 2000). Additionally, the NMJs of the α-syntrophin knock-out mouse have an abnormal distribution of AChRs and do not properly localize the proteins nNOS and utrophin (Adams et al., 2000). Experiments described in this paper address the specific role of the protein domains of α-syntrophin in determining the distribution of AChRs, nNOSμ, and utrophin at the NMJ. The key findings are summarized in Table 1.
Table 1.
Summary of mouse phenotypes
| Mouse | NMJ structure | NMJ utrophin | NMJ nNOS | Sarcolemma nNOS |
|---|---|---|---|---|
| Wild type | Normal | P | P | P |
| α-KO | Aberrant | A | A | A |
| FLA | Normal | P | P | P |
| ΔPDZ | Aberrant | P | A | A |
| ΔPH1a | Aberrant | P | P | A |
| ΔPH1b | Aberrant | P | ND | ND |
| ΔPDZ/ΔPH1a | Normal | P | P | A |
P, Present; A, absent; ND, not determined.
Utrophin is highly homologous with dystrophin but has a very different distribution in skeletal muscle. Dystrophin is present on the sarcolemma and concentrated at the NMJ where it is only in the depths of the postjunctional folds (Bewick et al., 1996). In normal, mature skeletal muscle utrophin is confined to the NMJ, where it is localized at the crests of the postjunctional folds with the AChRs. Both dystrophin and utrophin directly bind α-syntrophin, which is localized on the sarcolemma and throughout the postjunctional folds (Kramarcy and Sealock, 2000). In the complete absence of α-syntrophin, utrophin is no longer present at the NMJ, but the distribution of dystrophin is unaffected (Adams et al., 2000). The loss of utrophin in α-syntrophin null mice suggests that α-syntrophin is important for utrophin stabilization, trafficking, or transcription. Our real-time PCR results indicate that utrophin mRNA levels are not significantly affected by the loss of syntrophin (Fig. 4). Therefore, we conclude that α-syntrophin most likely modulates utrophin stabilization or trafficking to the NMJ.
It is interesting that even very low levels of α-syntrophin expression are able to restore utrophin to the NMJ. We detected very little product of the ΔPH1b transgene (Fig. 1) but the low levels were sufficient to restore utrophin to the NMJ (Fig. 3). This is consistent with results from the Takeda laboratory (Hosaka et al., 2002). This group generated an α-syntrophin exon 2-knock-out mouse, which eliminated nearly all the α-syntrophin, but due to low levels of exon 1 to exon 3 splicing, small amounts of α-syntrophin are still present at the NMJ. These mice retain postsynaptic utrophin (Hosaka et al., 2002).
We have previously shown that the PDZ domain of α-syntrophin is necessary for the sarcolemmal localization of nNOSμ (Adams et al., 2001). The present work shows that the postsynaptic NMJ localization of nNOSμ also requires the α-syntrophin PDZ domain (Fig. 5). The transgene expressing the ΔPH1a construct restores nNOSμ to the NMJ (Fig. 5). This was the anticipated result since this construct contains a functional PDZ domain. The surprising result is that the ΔPH1a transgene does not restore sarcolemmal nNOSμ. This is despite evidence that the ΔPH1a transgene product is localized to the sarcolemma and its PDZ domain is functional, as demonstrated by its ability to restore sarcolemmal localization of aquaporin-4 (Fig. 5). It is possible nNOSμ is present on the sarcolemma of the ΔPH1a mice at levels below the detection limit of our immunofluorescence assay, but such a reduction would still be in sharp contrast to the nNOSμ levels at the NMJ, which are similar to those in wild-type mice.
The presence of an α-syntrophin PDZ domain is necessary but not sufficient for sarcolemmal localization of nNOSμ. Patients with Becker muscular dystrophy affecting dystrophin exons 45–48 lack sarcolemmal nNOSμ despite expression of sarcolemmal α-syntrophin (Wells et al., 2003). Similarly, mdx mice expressing α-syntrophin artificially directed to the sarcolemma fail to localize nNOSμ to the sarcolemma (Adams et al., 2008). Furthermore, mdx mice expressing shortened forms of dystrophin only recruit nNOSμ to the sarcolemma if the region encoded by exons 45–48 (spectrin repeats 16 and 17) is included in the construct (Lai et al., 2009). A likely possibility is that the PH1 domain of syntrophin is linked to the exon 45–48 region of dystrophin either directly or indirectly to facilitate nNOSμ association with the dystrophin complex. This could explain why nNOSμ fails to localize to the sarcolemma in the absence of the α-syntrophin PH1a domain.
Results presented here show that proper AChR distribution at the NMJ requires the presence of both the α-syntrophin PDZ domain and the α-syntrophin PH1 domain. However, these domains do not necessarily need to be on the same syntrophin molecule to function (Fig. 6). Transgenic expression of both the ΔPDZ and ΔPH1a syntrophin mutants in the same mouse restored normal AChR distribution to the α-syntrophin knock-out mouse. One interpretation of this result is that the two domains are involved in separate signaling pathways and both are required to facilitate AChR distribution. An alternate interpretation is that these two domains work together in a common signaling pathway. In this case, it is likely that both the PH1 and PDZ domains need to be in close proximity (as they are in the intact molecule) to function. Such close proximity could be facilitated in the transgenic mice by the tandem syntrophin binding sites present on dystrophin protein family members (Newey et al., 2000). Alternatively, it is also possible that the two α-syntrophin transgenic proteins form an intermolecular interaction that brings the domains in close proximity. Since the PH1 domain is naturally split (by the PDZ domain) the PH1a of the ΔPDZ transgene could potential combine with the PH1b of the ΔPH1a transgene to restore a functional unit where the PH1 and PDZ domains are together in a manner similar to that of intact syntrophin. Such intermolecular PH domain formations occur in other molecules with split PH domains (van Rossum et al., 2005). Investigating the α-syntrophin-based signaling mechanisms leading to AChR stability at the mammalian NMJ will be the focus of future studies.
Footnotes
This work supported by National Institutes of Health Grant NS33145. We thank Drs. Jeffrey Chamberlain and Stephen Hauschka (University of Washington) for providing the MCK promoter construct, Dr. Bernard Jasmin for initial determination that utrophin-A transcription was unaltered in α-syntrophin null muscle, and our laboratory colleagues for critical reading of the manuscript.
References
- Adams ME, Kramarcy N, Krall SP, Rossi SG, Rotundo RL, Sealock R, Froehner SC. Absence of alpha-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J Cell Biol. 2000;150:1385–1398. doi: 10.1083/jcb.150.6.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams ME, Mueller HA, Froehner SC. In vivo requirement of the alpha-syntrophin PDZ domain for the sarcolemmal localization of nNOS and aquaporin-4. J Cell Biol. 2001;155:113–122. doi: 10.1083/jcb.200106158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams ME, Tesch Y, Percival JM, Albrecht DE, Conhaim JI, Anderson K, Froehner SC. Differential targeting of nNOS and AQP4 to dystrophin-deficient sarcolemma by membrane-directed alpha-dystrobrevin. J Cell Sci. 2008;121:48–54. doi: 10.1242/jcs.020701. [DOI] [PubMed] [Google Scholar]
- Ahn AH, Kunkel LM. Syntrophin binds to an alternatively spliced exon of dystrophin. J Cell Biol. 1995;128:363–371. doi: 10.1083/jcb.128.3.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi A, Bragg AD, Percival JM, Yoo J, Albrecht DE, Froehner SC, Adams ME. gamma-Syntrophin scaffolding is spatially and functionally distinct from that of the alpha/beta syntrophins. Exp Cell Res. 2006;312:3084–3095. doi: 10.1016/j.yexcr.2006.06.019. [DOI] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Williamson A, Palomba M, Eid T, de Lanerolle NC, Nagelhus EA, Adams ME, Froehner SC, Agre P, Ottersen OP. Delayed K+ clearance associated with aquaporin-4 mislocalization: phenotypic defects in brains of alpha-syntrophin-null mice. Proc Natl Acad Sci U S A. 2003;100:13615–13620. doi: 10.1073/pnas.2336064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Xue R, Haug FM, Neely JD, Bhardwaj A, Agre P, Adams ME, Froehner SC, Mori S, Ottersen OP. Alpha-syntrophin deletion removes the perivascular but not endothelial pool of aquaporin-4 at the blood–brain barrier and delays the development of brain edema in an experimental model of acute hyponatremia. FASEB J. 2004;18:542–544. doi: 10.1096/fj.03-0869fje. [DOI] [PubMed] [Google Scholar]
- Bewick GS, Young C, Slater CR. Spatial relationships of utrophin, dystrophin, beta-dystroglycan and beta-spectrin to acetylcholine receptor clusters during postnatal maturation of the rat neuromuscular junction. J Neurocytol. 1996;25:367–379. [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC, Bredt DS. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- Chen Z, Hague C, Hall RA, Minneman KP. Syntrophins regulate alpha1D-adrenergic receptors through a PDZ domain-mediated interaction. J Biol Chem. 2006;281:12414–12420. doi: 10.1074/jbc.M508651200. [DOI] [PubMed] [Google Scholar]
- Chockalingam PS, Gee SH, Jarrett HW. Pleckstrin homology domain 1 of mouse alpha1-syntrophin binds phosphatidylinositol 4,5-bisphosphate. Biochemistry. 1999;38:5596–5602. doi: 10.1021/bi982564+. [DOI] [PubMed] [Google Scholar]
- Compton AG, Cooper ST, Hill PM, Yang N, Froehner SC, North KN. The syntrophin-dystrobrevin subcomplex in human neuromuscular disorders. J Neuropathol Exp Neurol. 2005;64:350–361. doi: 10.1093/jnen/64.4.350. [DOI] [PubMed] [Google Scholar]
- Compton AG, Albrecht DE, Seto JT, Cooper ST, Ilkovski B, Jones KJ, Challis D, Mowat D, Ranscht B, Bahlo M, Froehner SC, North KN. Mutations in contactin-1, a neural adhesion and neuromuscular junction protein, cause a familial form of lethal congenital myopathy. Am J Hum Genet. 2008;83:714–724. doi: 10.1016/j.ajhg.2008.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors NC, Adams ME, Froehner SC, Kofuji P. The potassium channel Kir4.1 associates with the dystrophin glycoprotein complex via alpha-syntrophin in glia. J Biol Chem. 2004;279:28387–28392. doi: 10.1074/jbc.M402604200. [DOI] [PubMed] [Google Scholar]
- Frigeri A, Nicchia GP, Verbavatz JM, Valenti G, Svelto M. Expression of aquaporin-4 in fast twitch fibers of mammalian skeletal muscle. J Clin Invest. 1998;102:695–703. doi: 10.1172/JCI2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froehner SC, Murnane AA, Tobler M, Peng HB, Sealock R. A postsynaptic Mr 58,000 (58K) protein concentrated at acetylcholine receptor-rich sites in Torpedo electroplaques and skeletal muscle. J Cell Biol. 1987;104:1633–1646. doi: 10.1083/jcb.104.6.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee SH, Madhavan R, Levinson SR, Caldwell JH, Sealock R, Froehner SC. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J Neurosci. 1998;18:128–137. doi: 10.1523/JNEUROSCI.18-01-00128.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady RM, Merlie JP, Sanes JR. Subtle neuromuscular defects in utrophin-deficient mice. J Cell Biol. 1997;136:871–882. doi: 10.1083/jcb.136.4.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady RM, Zhou H, Cunningham JM, Henry MD, Campbell KP, Sanes JR. Maturation and maintenance of the neuromuscular synapse: genetic evidence for roles of the dystrophin–glycoprotein complex. Neuron. 2000;25:279–293. doi: 10.1016/s0896-6273(00)80894-6. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Cuenda A, Spillantini MG, Thomas GM, Buée-Scherrer V, Cohen P, Goedert M. Stress-activated protein kinase-3 interacts with the PDZ domain of alpha1-syntrophin. A mechanism for specific substrate recognition. J Biol Chem. 1999;274:12626–12631. doi: 10.1074/jbc.274.18.12626. [DOI] [PubMed] [Google Scholar]
- Hogan A, Shepherd L, Chabot J, Quenneville S, Prescott SM, Topham MK, Gee SH. Interaction of {gamma}1-syntrophin with diacylglycerol kinase-{zeta}: regulation of nuclear localization by PDZ interactions. J Biol Chem. 2001;276:26526–26533. doi: 10.1074/jbc.M104156200. [DOI] [PubMed] [Google Scholar]
- Hosaka Y, Yokota T, Miyagoe-Suzuki Y, Yuasa K, Imamura M, Matsuda R, Ikemoto T, Kameya S, Takeda S. Alpha1-syntrophin-deficient skeletal muscle exhibits hypertrophy and aberrant formation of neuromuscular junctions during regeneration. J Cell Biol. 2002;158:1097–1107. doi: 10.1083/jcb.200204076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachinsky AM, Froehner SC, Milgram SL. A PDZ-containing scaffold related to the dystrophin complex at the basolateral membrane of epithelial cells. J Cell Biol. 1999;145:391–402. doi: 10.1083/jcb.145.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameya S, Miyagoe Y, Nonaka I, Ikemoto T, Endo M, Hanaoka K, Nabeshima Y, Takeda S. Alpha-syntrophin gene disruption results in the absence of neuronal-type nitric oxide synthase at the sarcolemma but does not induce muscle degeneration. J Biol Chem. 1999;274:2193–2200. doi: 10.1074/jbc.274.4.2193. [DOI] [PubMed] [Google Scholar]
- Kramarcy NR, Sealock R. Syntrophin isoforms at the neuromuscular junction: developmental time course and differential localization. Mol Cell Neurosci. 2000;15:262–274. doi: 10.1006/mcne.1999.0823. [DOI] [PubMed] [Google Scholar]
- Kramarcy NR, Vidal A, Froehner SC, Sealock R. Association of utrophin and multiple dystrophin short forms with the mammalian M(r) 58,000 dystrophin-associated protein (syntrophin) J Biol Chem. 1994;269:2870–2876. [PubMed] [Google Scholar]
- Lai Y, Thomas GD, Yue Y, Yang HT, Li D, Long C, Judge L, Bostick B, Chamberlain JS, Terjung RL, Duan D. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest. 2009;119:624–635. doi: 10.1172/JCI36612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonoudakis D, Conti LR, Anderson S, Radeke CM, McGuire LM, Adams ME, Froehner SC, Yates JR, 3rd, Vandenberg CA. Protein trafficking and anchoring complexes revealed by proteomic analysis of inward rectifier potassium channel (Kir2.x)-associated proteins. J Biol Chem. 2004;279:22331–22346. doi: 10.1074/jbc.M400285200. [DOI] [PubMed] [Google Scholar]
- Lumeng C, Phelps S, Crawford GE, Walden PD, Barald K, Chamberlain JS. Interactions between beta2-syntrophin and a family of microtubule-associated serine/threonine kinases. Nat Neurosci. 1999;2:611–617. doi: 10.1038/10165. [DOI] [PubMed] [Google Scholar]
- Luo S, Chen Y, Lai KO, Arévalo JC, Froehner SC, Adams ME, Chao MV, Ip NY. {alpha}-Syntrophin regulates ARMS localization at the neuromuscular junction and enhances EphA4 signaling in an ARMS-dependent manner. J Cell Biol. 2005;169:813–824. doi: 10.1083/jcb.200412008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons PR, Slater CR. Structure and function of the neuromuscular junction in young adult mdx mice. J Neurocytol. 1991;20:969–981. doi: 10.1007/BF01187915. [DOI] [PubMed] [Google Scholar]
- Lyssand JS, DeFino MC, Tang XB, Hertz AL, Feller DB, Wacker JL, Adams ME, Hague C. Blood pressure is regulated by an alpha1D-adrenergic receptor/dystrophin signalosome. J Biol Chem. 2008;283:18792–18800. doi: 10.1074/jbc.M801860200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely JD, Amiry-Moghaddam M, Ottersen OP, Froehner SC, Agre P, Adams ME. Syntrophin-dependent expression and localization of Aquaporin-4 water channel protein. Proc Natl Acad Sci U S A. 2001;98:14108–14113. doi: 10.1073/pnas.241508198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newey SE, Benson MA, Ponting CP, Davies KE, Blake DJ. Alternative splicing of dystrobrevin regulates the stoichiometry of syntrophin binding to the dystrophin protein complex. Curr Biol. 2000;10:1295–1298. doi: 10.1016/s0960-9822(00)00760-0. [DOI] [PubMed] [Google Scholar]
- Ort T, Voronov S, Guo J, Zawalich K, Froehner SC, Zawalich W, Solimena M. Dephosphorylation of beta2-syntrophin and Ca2+/mu-calpain-mediated cleavage of ICA512 upon stimulation of insulin secretion. EMBO J. 2001;20:4013–4023. doi: 10.1093/emboj/20.15.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters MF, Kramarcy NR, Sealock R, Froehner SC. β2-Syntrophin: localization at the neuromuscular junction in skeletal muscle. Neuroreport. 1994;5:1577–1580. [PubMed] [Google Scholar]
- Peters MF, Adams ME, Froehner SC. Differential association of syntrophin pairs with the dystrophin complex. J Cell Biol. 1997;138:81–93. doi: 10.1083/jcb.138.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GD, Shaul PW, Yuhanna IS, Froehner SC, Adams ME. Vasomodulation by skeletal muscle-derived nitric oxide requires alpha-syntrophin-mediated sarcolemmal localization of neuronal nitric oxide synthase. Circ Res. 2003;92:554–560. doi: 10.1161/01.RES.0000061570.83105.52. [DOI] [PubMed] [Google Scholar]
- Ueda K, Valdivia C, Medeiros-Domingo A, Tester DJ, Vatta M, Farrugia G, Ackerman MJ, Makielski JC. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci U S A. 2008;105:9355–9360. doi: 10.1073/pnas.0801294105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rossum DB, Patterson RL, Sharma S, Barrow RK, Kornberg M, Gill DL, Snyder SH. Phospholipase Cgamma1 controls surface expression of TRPC3 through an intermolecular PH domain. Nature. 2005;434:99–104. doi: 10.1038/nature03340. [DOI] [PubMed] [Google Scholar]
- Wakayama Y, Inoue M, Kojima H, Jimi T, Yamashita S, Kumagai T, Shibuya S, Hara H, Oniki H. Altered alpha1-syntrophin expression in myofibers with Duchenne and Fukuyama muscular dystrophies. Histol Histopathol. 2006;21:23–34. doi: 10.14670/HH-21.23. [DOI] [PubMed] [Google Scholar]
- Weir AP, Burton EA, Harrod G, Davies KE. A- and B-utrophin have different expression patterns and are differentially up-regulated in mdx muscle. J Biol Chem. 2002;277:45285–45290. doi: 10.1074/jbc.M205177200. [DOI] [PubMed] [Google Scholar]
- Wells KE, Torelli S, Lu Q, Brown SC, Partridge T, Muntoni F, Wells DJ. Relocalization of neuronal nitric oxide synthase (nNOS) as a marker for complete restoration of the dystrophin associated protein complex in skeletal muscle. Neuromuscul Disord. 2003;13:21–31. doi: 10.1016/s0960-8966(02)00191-8. [DOI] [PubMed] [Google Scholar]
- Yan J, Wen W, Xu W, Long JF, Adams ME, Froehner SC, Zhang M. Structure of the split PH domain and distinct lipid-binding properties of the PH-PDZ supramodule of alpha-syntrophin. EMBO J. 2005;24:3985–3995. doi: 10.1038/sj.emboj.7600858. [DOI] [PMC free article] [PubMed] [Google Scholar]