

Abstract
We present the in-vivo biosynthesis of wild-type sunflower trypsin inhibitor 1 (SFTI-1) inside E. coli cells using an intramolecular native chemical ligation in combination with a modified protein splicing unit. SFTI-1 is a small backbone cyclized polypeptide with a single disulfide bridge. A small library containing multiple Ala mutants was also biosynthesized and its activity was assayed using a trypsin-binding assay. This study clearly demonstrates the exciting possibility of generating large cyclic peptide libraries in live E. coli cells, and is a critical first step for developing in-vivo screening and directed evolution technologies using the cyclic peptide SFTI-1 as a molecular scaffold.
Keywords: Bowman-Birk inhibitor, trypsin inhibitor, backbone cyclized peptides, genetically-encoded libraries, protein splicing
INTRODUCTION
In recent years, there has been an increased interest in discovering new cyclic peptides, and also in developing these backbone-cyclized peptides to regulate targeted biological activities (Hruby and Al-Obeidi 1990; Rizo and Gierasch 1992). Much of the current efforts are centered upon exploiting cyclic peptide topology as scaffolds for libraries, which are then screened for novel biological activity. To date, several hundreds of cyclic polypeptides (ranging from 14 to 70 residues long) have been discovered in a diverse range of organisms, which includes simple prokaryotes and higher primates (Craik et al. 2003; Crovella et al. 2005; Craik et al. 2006). Backbone-cyclized peptides have the N- and C- terminal ends joined together through a natural peptide bond. By eliminating the usually flexible terminal ends, these peptides have additional protection from proteolytic enzymes, which makes them less susceptible to attack by exopeptidases, thus increasing their in-vivo lifetime (Trabi and Craik 2002). Some cyclic peptides are also noted for their exceptional stability and ability to withstand chemically harsh environments such as the gastrointestinal tract, and extreme physical environments such as boiling water. Moreover, peptide cyclization may help to stabilize structure in a manner analogous to a disulfide bond constraint; these rigid molecules then gain a thermodynamic or entropic advantage upon binding its target. Together, these characteristics suggest that cyclic peptides are ideal candidates to use as molecular scaffolds for peptide libraries, which can be screened for desired biological activity (Hilpert et al. 2000; Craik et al. 2002; Craik et al. 2006).
In a recent study, we described a method to biosynthesize cyclic peptides using intramolecular Native Chemical Ligation (NCL) in combination with a modified protein-splicing unit or intein (Kimura et al. 2006; Camarero et al. 2007). This process requires the presence, within the same polypeptide sequence, of a N-terminal Cys residue and a C-terminal α-thioester function (Camarero and Muir 1999; Camarero et al. 2001). Recombinant polypeptides containing N-terminal Cys residues can easily be produced using endogenous proteases such as methionyl aminopeptidases (MAP) (Camarero et al. 2001). Recombinant proteins containingα-thioesters can be obtained by using engineered inteins (Evans et al. 1998; Muir et al. 1998; Severinov and Muir 1998; Camarero and Muir 1999). We recently used this approach to biosynthesize several cyclotides in E. coli. (Kimura et al. 2006; Camarero et al. 2007).
Cyclotides are small globular microproteins with a unique head-to-tail cyclized backbone, which are stabilized by three disulfide bonds (Craik et al. 1999). The number and positions of cysteine residues are conserved throughout the family, forming the cyclic cystine-knot motif (CCK) (Craik et al. 1999). This CCK framework gives the cyclotides exceptional resistance to thermal and chemical denaturation and enzymatic degradation.
Based on these results we also decided to test if this technology could be also used to produce smaller cyclic peptides. For this purpose we decided to use sunflower trypsin inhibitor 1 (SFTI-1), a 14 amino acid backbone-cyclized peptide containing a single disulfide bond (Scheme 1). SFTI-1 is a naturally occurring protease inhibitor found in the seeds of sunflower (Helianthus annuus) (Luckett et al. 1999). SFTI-1 belongs to the Bowman-Birk inhibitor (BBI) family whose members, found in many plants, are potent serine protease inhibitors (Korsinczky et al. 2004). SFTI-1 is the smallest and most potent BBI, whose Ki has been reported to be in the low nanomolar range (Luckett et al. 1999). Structural analysis of the natural product reveals a well-defined double β-hairpin loop linked by two short antiparallel β-strands (Luckett et al. 1999; Korsinczky et al. 2001; Korsinczky et al. 2005) (Fig. 1). Previous studies indicate that SFTI-1 readily adopts its simple native fold in aqueous solution without need for an oxidizing redox buffer system (Marx et al. 2003). Furthermore, by virtue of SFTI-1’s ability to bind-to and inhibit trypsin, it has a built in affinity handle, which may be used to selectively pull down SFTI-1 from a complex cytoplasmic mixture. Together, these characteristics suggest that this natural product is the ideal candidate upon which to develop and graft novel bioactivities important for biosecurity, human health or agriculture.
Scheme 1.

Biosynthetic approach for the production of cyclic peptide SFTI-1 inside living E. coli cells. Backbone cyclization of the linear precursor is mediated by a modified protein splicing unit or intein. The cyclized peptide then folds spontaneously in the bacterial cytoplasm (CBD stands for chitin binding domain).
Figure 1.

Primary and tertiary structure of the cyclic peptide SFTI-1. Sequences of the linear precursors used for the in vitro and in vivo production of SFTI-1. The precursors contain an N-terminal Met residue and are fused in frame to the corresponding modified intein (Sce VMA or Mxe Gyase). The sequence corresponding to loop 1 is underlined as a reference (CBD stands for chitin binding domain).
MATERIALS AND METHODS
General materials and methods
Analytical HPLC was performed on a HP1100 series instrument with 220 nm and 280 nm detection using a Vydac C18 column (5 μm, 4.6 × 150 mm) at a flow rate of 1 mL/min. Semipreparative HPLC was performed on a Waters Delta Prep system fitted with a Waters 2487 Ultraviolet-Visible (UV-vis) detector using a Vydac C18 column (15–20 μm, 10 × 250 mm) at a flow rate of 5 mL/min. All runs used linear gradients of 0.1% aqueous trifluoroacetic acid (TFA, solvent A) vs. 0.1% TFA, 90% acetonitrile in H2O (solvent B). UV-vis spectroscopy was carried out on an Agilent 8453 diode array spectrophotometer. Electrospray mass spectrometry (ES-MS) analysis was routinely applied to all cyclized peptides. ES-MS was performed on a Sciex API-150EX single quadrupole electrospray mass spectrometer, MS/MS was performed on an Applied Biosystems API 3000 triple quadrupole mass spectrometer. Calculated masses were obtained by using ProMac v1.5.3. Protein samples were analyzed by SDS-PAGE. Samples were run on Invitrogen (Carlsbad, CA) 4–20% Tris-Glycine Gels. The gels were then stained with Pierce (Rockford, IL) Gelcode Blue, photographed/digitized using a Kodak (Rochester, NY) EDAS 290, and quantified using NIH Image-J software (http://rsb.info.nih.gov/ij/). Amino acid analysis was performed by the Molecular Structure Facility at University of California Davis on a Hitachi L-8800 amino acid analyzer. DNA sequencing was performed by Davis Sequencing (Davis, CA) using an ABI 3730 DNA sequencer, and the sequence data was analyzed with DNAStar (Madison, WI) Lasergene v5.5.2. All chemicals were obtained from Sigma-Aldrich (Milwaukee, WI) unless otherwise indicated.
Construction of expression plasmids
Plasmids expressing the linear sunflower trypsin Inhibitor-1 (SFTI-1) precursors were constructed using either the pTYB1 or pTXB1 expression plasmids (New England Biolabs), which contain an engineered Sce VMA or Mxe Gyrase intein, respectively, and a chitin-binding domain (CBD). Oligonucleotides coding for the SFTI-1 wild type and mutant sequences (Table 1) were synthesized, phosphorylated and PAGE purified by IDT DNA (Coralville, IA). Complementary strands were annealed in 0.3 M NaCl and the resulting double stranded DNA (dsDNA) was purified using Qiagen’s (Valencia, CA) miniprep column and buffer PN. pTXB1 or pTYB1 plasmids was double digested with NdeI and SapI (NEB). The linearized vectors and the SFTI-1 dsDNA fragments were ligated at 15°C overnight using T4 DNA Ligase (New England Biolabs). The ligated plasmids were transformed into DH5α cells (Invitrogen) and plated on Luria Broth (LB)- agar containing ampicillin. Positive colonies were grown in 5 mL LB containing ampicillin at 37°C overnight and the corresponding plasmids purified using a Miniprep Kit (Qiagen). Plasmids were initially screened by EcoRI digestion, as this restriction site is removed during cloning. Preliminary positives were expressed (see below) and fully characterized by ES-MS.
Table 1.
Forward (p5) and reverse (p3) 5’-phosphorylated oligonucleotides used to clone the different SFTI-based peptides.
| SFTI-C1 | p5 TATGTGCACCAAATCTATCCCGCCGATCTGCTTCCCGGACGGTCGT |
| SFTI-C1 | p3 GCAACGACCGTCCGGGAAGCAGATCGGCGGGATAGATTTGGTGCACA |
| T2A | p5 TATGTGCGCTAAATCTATCCCGCCGATCTGCTTCCCGGACGGTCGT |
| T2A | p3 GCAACGACCGTCCGGGAAGCAGATCGGCGGGATAGATTTAGCGCACA |
| K3A | p5 TATGTGCACCGCTTCTATCCCGCCGATCTGCTTCCCGGACGGTCGT |
| K3A | p3 GCAACGACCGTCCGGGAAGCAGATCGGCGGGATAGAAGCGGTGCACA |
| S4A | p5 TATGTGCACCAAAGCTATCCCGCCGATCTGCTTCCCGGACGGTCGT |
| S4A | p3 GCAACGACCGTCCGGGAAGCAGATCGGCGGGATAGCTTTGGTGCACA |
| I5A | p5 TATGTGCACCAAATCTGCTCCGCCGATCTGCTTCCCGGACGGTCGT |
| I5A | p3 GCAACGACCGTCCGGGAAGCAGATCGGCGGAGCAGATTTGGTGCACA |
| P6A | p5 TATGTGCACCAAATCTATCGCTCCGATCTGCTTCCCGGACGGTCGT |
| P6A | p3 GCAACGACCGTCCGGGAAGCAGATCGGAGCGATAGATTTGGTGCACA |
| P7A | p5 TATGTGCACCAAATCTATCCCGGCTATCTGCTTCCCGGACGGTCGT |
| P7A | p3 GCAACGACCGTCCGGGAAGCAGATAGCCGGGATAGATTTGGTGCACA |
| I8A | p5 TATGTGCACCAAATCTATCCCGCCGGCTTGCTTCCCGGACGGTCGT |
| I8A | p3 GCAACGACCGTCCGGGAAGCAAGCCGGCGGGATAGATTTGGTGCACA |
| C9A | p5 TATGTGCACCAAATCTATCCCGCCGATCGCTTTCCCGGACGGTCGT |
| C9A | p3 GCAACGACCGTCCGGGAAAGCGATCGGCGGGATAGATTTGGTGCACA |
| F10A | p5 TATGTGCACCAAATCTATCCCGCCGATCTGCGCTCCGGACGGTCGT |
| F10A | p3 GCAACGACCGTCCGGAGCGCAGATCGGCGGGATAGATTTGGTGCACA |
| I11A | p5 TATGTGCACCAAATCTATCCCGCCGATCTGCTTCGCTGACGGTCGT |
| I11A | p3 GCAACGACCGTCAGCGAAGCAGATCGGCGGGATAGATTTGGTGCACA |
| D12A | p5 TATGTGCACCAAATCTATCCCGCCGATCTGCTTCCCGGCTGGTCGT |
| D12A | p3 GCAACGACCAGCCGGGAAGCAGATCGGCGGGATAGATTTGGTGCACA |
| G13A | p5 TATGTGCACCAAATCTATCCCGCCGATCTGCTTCCCGGACGCTCGT |
| G13A | p3 GCAACGAGCGTCCGGGAAGCAGATCGGCGGGATAGATTTGGTGCACA |
| R14A | p5 TATGTGCACCAAATCTATCCCGCCGATCTGCTTCCCGGACGGTGCT |
| R14A | p3 GCAAGCACCGTCCGGGAAGCAGATCGGCGGGATAGATTTGGTGCACA |
| SFTI-C9 | p5 TATGTGCACCAAATCTATCCCGCCGATCGCTTTCCCGGACGGTCGT |
| SFTI-C9 | p3 GCAACGACCGTCCGGGAAAGCGATCGGCGGGATAGATTTGGTGCACA |
Expression and purification of recombinant proteins
E.coli BL21(DE3) or Origami(DE3) cells (Novagen, San Diego, CA) were transformed with the SFTI-1 plasmids (see above). Expression was carried out in LB medium (1–2 L) containing ampicillin at room temperature or 30°C for 2 h to overnight. Briefly, 5 mL of an overnight starter culture derived from either a single clone or single plate (Ala-scan library) were used to inoculate 1 L of LB media. Cells were grown to an OD at 600 nm of 0.5 at 37°C, and expression was induced by the addition of isopropyl-β-D-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM at the temperatures and times mentioned above and in the manuscript. The cells were then harvested by centrifugation. For fusion protein purification, the cells were resuspended in 30 mL of lysis buffer (0.1 mM EDTA, 1 mM PMSF, 50 mM sodium phosphate, 250 mM NaCl buffer at pH 7.2 containing 5% glycerol) and lysed by sonication. The lysate was clarified by centrifugation at 15,000 rpm in a Sorval SS-34 rotor for 30 min. The clarified supernatant was incubated with chitin-beads (2 mL beads/L cells, New England Biolabs), previously equilibrated with column buffer (0.1 mM EDTA, 50 mM sodium phosphate, 250 mM NaCl buffer at pH 7.2) at 4°C for 1 h with gentle rocking. The beads were extensively washed with 100 bead-volumes of column buffer containing 0.1% Triton X100 and then rinsed and equilibrated with 100 bead-volumes of column buffer. In-vivo cleavage was quantified by SDS-PAGE analysis of the purified fusion proteins using the NIH Image-J software package.
Concomitant cleavage, cyclization and folding of SFTI-1 with EtSH, MESNA and GSH
Chitin beads containing the different purified SFTI-1–Intein-CBD fusion proteins were cleaved with 1% EtSH, 50 mM MESNA or 50 mM GSH in degassed column buffer. The cyclization/folding reactions were kept for up to 4 days at 25°C with gentle rocking. For small scale reactions, aliquots were taken each day (when necessary) and analyzed by HPLC. The reduced and oxidized circular SFTI-1 product was analyzed by ES-MS (Table 2). Larger-scale (2 L culture) purification of the SFTI-1 wild type, T2A, K3A linear precursors were also performed. The supernatant of the cyclization reaction was separated by filtration and the beads were washed with additional column buffer (1 column volume per each mL of beads). The supernatant and washes were pooled, and the oxidized-circular peptide was purified by semipreparative HPLC using a linear gradient of 15–45% solvent B over 30 min. The isolated yield for purified the SFTI-1 peptides was around 300 μg/L.
Table 2.
Sequences and molecular weights found for the different SFTI-based peptides used in this work.
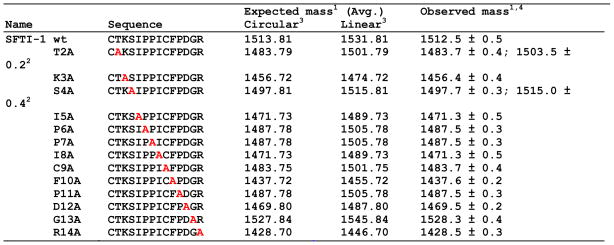 |
Mass in Daltons;
Mass corresponds to hydrolyzed peptide;
Mass corresponds to the oxidized peptide except for mutant C9A;
Mass found for in vitro cyclized peptides. Identical values, within error, were obtained for in vivo cyclized peptides.
Purification of SFTI-1 using trypsin-sepharose beads
Preparation of trypsin-sepharose beads: NHS-activated Sepharose was washed with 15 volumes of ice-cold 1 mM HCl. Each volume of beads was incubated with an equal volume of coupling buffer (250 mM NaCl, 200 mM sodium phosphate buffer at pH 6.0) containing 2 mg of Porcine Pancreas Trypsin type IX-S (14,000 units/mg) for 3 h with gentle rocking at room temperature. The beads were then rinsed with 10 volumes of coupling buffer, and incubated with excess coupling buffer containing 100 mM ethanolamine (Eastman Kodak) for 3 h with gentle rocking at room temperature. Finally, the beads were washed with 50 volumes of wash buffer (200 mM sodium acetate buffer at pH 3, 250 mM NaCl) and stored in one volume of wash buffer.
30 mL of clarified lysate was incubated with 0.2 mL of trypsin-sepharose for one hour at room temperature with gentle rocking, and centrifuged at 3000 rpm for 1 min. The beads were washed with 50 volumes of PBS containing 0.1% Triton X100, then rinsed with 50 volumes of PBS, and drained of excess PBS. After the addition of 0.5 mL of 8 M Gdm HCl, the reaction was incubated in a 90°C heat block for 5 minutes to elute the bound peptide. The peptide was analyzed by RP-HPLC and ES-MS/MS.
Determination of Ki
SFTI-1 concentrations were quantified by amino acid analysis (UC Davis Molecular Structure facility). Trypsin inhibitory assay was performed in a solution containing 20 mM CaCl2, 50 mM Tris HCl buffer at pH 8.0. Inhibitor solutions of SFTI-1 ranging from 10 nM to 100 nM were preincubated with a freshly prepared solution of 2 nM trypsin in a final reaction volume of 3.0 mL. After incubation for 20 minutes, residual trypsin activity was measured by adding the fluorogenic substrate Nα-Cbz-L-Arg-AMC (AMC: 7-amido-4-methyl-coumarin, Cbz: benzyloxycarbonyl) to a final concentration of 2 μM and then following the release of AMC using a Fluorolog-3 spectrofluorometer (Jobin Yvon Inc., Edison, NJ). Measurements were taken every 60 seconds using an excitation wavelength of 366 nm and an emission wavelength of 465 nm. The initial velocities for the hydrolysis of substrate Nα-Cbz-L-Arg-AMC by trypsin in the presence of different concentrations of SFTI-1 were fitted to a one-site competitive binding equation using the software package Prism (GraphPad Software). Ki was calculated using the equation of Cheung and Prusoff (Cheng and Prusoff 1973) and a Km value of 12 μM (Baird et al. 2000) (Fig. S2).
RESULTS
Biosynthesis of SFTI-1 via Cys1 and Cys9
SFTI-1 is a relatively simple cyclic peptide that contains two cysteine residues that may be used for NCL. Two different SFTI-1 linear precursors were cloned in frame with a modified Sce VMA intein. The N-terminal methionine residue is efficiently removed by the endogenous MAP, which yields the required N-terminal Cys residue required for intramolecular NCL. At the same time, the modified intein generates the α-thioester at the junction between C-terminal end of the SFTI-1 and the N-terminal Cys of the intein (Fig. 1). Linear versions of SFTI-1 have shown to adopt a native-like fold, so we anticipated that this conformational predisposition would bring the N-terminal Cys in close proximity to the α-thioester thus facilitating the intramolecular cyclization through NCL.
SFTI-1 intein fusion proteins (SFTI-C1 and SFTI-C9) were expressed in E. coli BL21(DE3) cells, purified by affinity chromatography using a chitin binding domain and analyzed by SDS PAGE. As shown in Fig. 2A, both intein precursors had comparable levels of expression in E. coli cells, however they showed different rates and propensities for in-vivo cleavage. SFTI-C1 shows 80% in-vivo cleavage, and SFTI-C9 shows 70% in-vivo cleavage (Fig. 2A). This difference could be attributed to the close proximity of two consecutive Pro residues to the extein-intein junction in the precursor SFTI-C9, which may partially affect the equilibrium between the thioester and amide bonds at the junction.
Figure 2.

Biosynthesis of SFTI-1 using a modified Sce VMA intein. A. SDS-PAGE analysis of the expression levels and in vivo cleavage of precursors SFTI-C1 and SFT-C9. B. Analytical reversed-phase HPLC trace of the in vitro cyclization/folding of SFTI-C1 with 50 mM GSH after 24 h. C. Analytical reversed-phase HPLC trace of the soluble cellular fraction of E. coli BL21 (DE3) cells expressing precursor SFTI-C1 after it was purified on trypsin-immobilized sepharose beads. The arrow denotes the natively folded SFTI-1 peptide. The large peak eluting at 16–17 min and small peaks around SFTI-1 are not related to SFTI-1 or SFTI-intein precursor. D. ES-MS spectrum of affinity purified SFTI-1 peptide. HPLC gradients are 0–70% B over 20 min (see materials and methods section for buffer composition).
Next, we investigated in vitro cyclization of intein-containing precursor proteins. Affinity purified fusion precursors SFTI-C1 and SFTI-C9 were incubated in phosphate buffer at pH 7.2 containing either 1% ethanethiol (EtSH), 50 mM sodium mercapto-ethanesulfonate (MESNA) or 50 mM reduced glutathione (GSH) to facilitate intramolecular cyclization. After 12 h of reaction, SFTI-C1 was completely cleaved in-vitro by any of the thiols tested, whereas SFTI-C9 showed no additional cleavage even after 48 h of reaction.
RP-HPLC and ES-MS analysis confirmed the presence of both reduced-circular SFTI-1 and native SFTI-1 when using SFTI-C1, but neither reduced nor cyclic peptide using the SFTI-C9 precursor. This initial in-vitro analysis indicated that the SFTI-C1 linear precursor is more efficient for intramolecular cyclization. Interestingly, when the cyclization reaction was performed in the presence of GSH, the only product obtained was the native SFTI-1 as indicated by ES-MS (Fig. 2B and 2D). This could be attributed to the partial air oxidation of GSH during the reaction to give a GSSG/GSH redox buffer that facilitated the formation of the native disulfide between Cys1 and Cys9. This effect has been also observed in the cyclization of cyclotides using GSH-containing buffers (Kimura et al. 2006; Camarero et al. 2007). The obtained SFTI-1 was purified and its biological activity tested using a trypsin inhibitory assay (Jaulent and Leatherbarrow 2004; Camarero et al. 2007). In vitro biosynthesized SFTI-I was able to inhibit pancreatic trypsin with a Ki of 26 ± 2 nM (Fig. S2). This Ki constant closely parallels previously reported values, thus confirming the native fold of recombinant SFTI-1.
In-vivo biosynthesis of SFTI-1
Due to the highly efficient cleavage of precursor SFTI-C1 and the high propensity to cyclize and fold in the presence of GSH-containing buffers, we explored whether any folded SFTI-1 was formed in vivo. We used trypsin-sepharose to capture folded SFTI-1 formed inside living cells expressing the SFTI-C1 precursor. After extensive washings, the bound fraction was eluted with 8 M GdmCl solution. HPLC analysis of the bound fraction revealed the presence of a major peak with the same retention time and mass as that the folded SFTI-1, thus confirming the in-vivo biosynthesis of SFTI-1 inside living E. coli cells (Fig. 2C). Approximately 250 μg of folded SFTI-1 were produced per gram of wet E. coli cells. This corresponds to an intracellular concentration of 100 nM (Camarero et al. 2007). Interestingly, although SFTI-C9 is cleaved efficiently in vivo ( 70%), no cyclic folded SFTI-1 was found in the corresponding lysate (data not shown).
Encouraged by these results, we tested in vivo cyclization of SFTI-C1 using the modified Mxe Gyrase intein. This mini-intein is a bacterial intein that can be expressed very efficiently in E. coli cells at 30°C. The Sce VMA intein starts to accumulate to high levels in the insoluble cellular fraction when expressed at 30°C, thus reducing the yield of folded intein. The expression of the Mxe Gyrase intein, however, can be induced at this temperature for 6 h without any significant aggregation. On average, the Gyrase intein is able to produce between 2–3 times more precursor protein than the corresponding VMA intein precursor.
Using the Mxe Gyrase intein system, close to 70% of the SFTI-C1 precursor was cleaved in vivo as indicated by SDS-PAGE analysis (Fig. S1). This value is similar to that found for the Sce VMA intein. Analysis of the cell lysate using trypsin-sepharose affinity chromatography showed the presence of folded SFTI-1 in E. coli cytoplasm (Fig. S1). The intracellular concentration of folded SFTI-1 was estimated to be approximately 1 μM. A similar value has was observed for intracellular biosynthesis of the cyclotide MCoTI-II in E. coli cells using the Mxe Gyrase intein (Camarero et al. 2007).
Biosynthesis of an SFTI-based library
In order to evaluate whether this biosynthetic approach may produce genetically- encoded libraries of SFTI mutants, we performed alanine scanning mutagenesis to determine whether this stable double β-hairpin scaffold could accommodate amino acid substitutions throughout its primary structure, retain its bioactivity and importantly, whether these mutants can be biosynthesized in-vivo.
A small library of 14 Ala mutants was designed, such that every amino acid, with the exception of Cys1, was sequentially substituted by Ala (Table 2). Since Cys1 was found to be the better of the two cysteines for native chemical ligation, the wild type member as well as each Ala-mutant member incorporates this cysteine at the N-terminal end of the corresponding intein precursors. Synthetic dsDNA fragments encoding the different SFTI-1 mutants were ligated into plasmid pTXB1 in frame with Mxe Gyrase intein. The resulting plasmid library was transformed into competent DH5α E. coli cells obtaining approximately 104 colonies (data not shown). All colonies were pooled and the corresponding plasmid library was transformed into different E. coli expression cell lines. Expression of the library in E. coli BL21(DE3) cells produced the corresponding Gyrase intein precursors with similar yields to that of the wild-type SFTI-1. The level of in vivo cleavage was estimated to be 70% following induction for 5 h at 30°C. Interestingly, when SFTI-Gyrase encoded plasmid library was expressed in E. coli Origami(DE3) cells under the same conditions (5 h at 30°C), only 20% in-vivo cleavage was observed (data not shown). Origami cells have a more oxidizing cytoplasm, which may favor the formation disulfide bonds over the competing intramolecular NCL reaction. We have found, however, that in both cellular backgrounds (BL21 and Origami cells), the in-vivo cleavage ratio can be increased by inducing the culture at lower temperatures for longer times (Camarero et al. 2007). This allows for experimental to accommodate different screening methods. For in vitro screening, cyclization can be accomplished in vitro under controlled conditions, and therefore short induction times at relatively higher temperatures will yield more uncleaved linear precursor. Alternatively, for high throughput in vivo screening, cyclization yields must be maximized. Levels of in vivo cleavage may be increased by longer induction times and lower induction temperatures (18–20 h at 20°C for example).
In order to characterize the SFTI-based library, uncleaved SFTI-Gyrase fusion proteins were obtained from E. coli BL21(DE3) cells which were induced at 30°C for 5 h. The uncleaved fusion precursors ( 30% of the total induced protein) were cyclized with phosphate buffer at pH 7.2 containing 50 mM GSH for 24 h. This treatment resulted in nearly 100% cleavage of the intein precursors. The soluble fraction was analyzed by HPLC and ES-MS/MS to determine the presence of relative representation of library members (Fig. 3A). All library members were observed with the exception of D12A. This mutant was found in the cell lysate (see results below). Mutants with the same or similar molecular weight were distinguished by MS/MS and/or differences in their HPLC retention times.
Figure 3.

Analysis of the SFTI-based library expressed in E. coli BL21(DE3) cells. A. ES-MS spectrum of the SFTI-based library generated in vitro by cyclization/folding using 50 mM GSH. B. ES-MS spectrum of the of the soluble cellular fraction of E. coli BL21 (DE3) cells expressing precursor SFTI-C1 using a modified Mxe Gyrase after it was purified on trypsin-immobilized sepharose beads. Peptides hydrolyzed by trypsin are indicated with an asterisk.
In-vivo biosynthesis and screening of SFTI-based library
In vivo biosynthesis of the SFTI-libray was performed in E. coli BL21(DE3) cells by inducing the expression of the SFTI-intein fusion library at 20°C for 18 h. Under these conditions, in vivo cleavage was close to 95%. The soluble cellular fraction was incubated with trypsin-sepharose, and the bound fraction was analyzed by HPLC and ES-MS. As shown in Fig. 3B, the MS analysis was able to detect the SFTI-1 wt, and the mutants G13A, R14A, S4A, I5A, I8A, P7A, P11A, F10A and D12A. Visual inspection of the structural complex formed between SFTI-1 and trypsin (Luckett et al. 1999) indicates that the side-chains of these residues are solvent exposed. Thus, these residues may be more tolerable to amino acid substitution with relatively minor consequences to biological activity (Luckett et al. 1999). For example, a study by (Descours et al. 2002) showed that the Ki values of mutants P8A and P13A are only slightly affected, and I7A and I9A are only moderately affected with Ki values ranging from pM to low μM. These findings are also corroborated by the results reported by (Hilpert et al. 2000) on the complete substitutional analysis of SFTI-1 with different serine proteases and more recently by (Daly et al. 2006), where a complete suite of Ala mutants of SFTI-1 was chemically synthesized and characterized structurally and functionally.
As expected, the SFTI-1 K3A was not found in the trypsin-bound fraction. This residue determines binding affinity and specificity, and can only be replaced by Arg to maintain biological activity (Hilpert et al. 2000). SFTI-1 mutants P6A and C9A were also not found in the trypsin-bound fraction. In agreement with Hilpert et al. 2000, bioactivity of Pro6 mutants is maintained only by Asp or Glu substitutions. A more recent study by (Daly et al. 2006) showed the importance of this residue as well. Mutation of Cys9 to Ala is also known to disrupt the biological activity due to the necessary disulfide bond (Korsinczky et al. 2005).
Interestingly, two masses corresponding to linear Ala mutants were also found in the in-vivo trypsin-bound fraction. Linear oxidized S4A binds trypsin beads demonstrating not only tight binding of this library member, but suggests an equilibrium between circular and linear (hydrolyzed) forms. In addition, a mass corresponding to the linear-oxidized T2A was also detected. Previously, it has been determined that an active acyclic permutant of SFTI-1 which opens at its scissile bond (i.e. Lys3-Ser4) can be enzymatically backbone-cyclized by trypsin. The resulting ratio of cyclic SFTI-1 to hydrolyzed SFTI-1 is 10:1 regardless of whether trypsin is incubated with the acyclic or cyclic form of SFTI-1 (Marx et al. 2003). Therefore, the results of mutants T2A and S4A may be attributed to the proximity of these mutations with respect to the scissile bond, which may stabilize the hydrolyzed form.
DISCUSSION
A substantial number of natural products with wide range of pharmacological activities are cyclic polypeptides. Peptide cyclization is widely adopted in medicinal chemistry to improve the biochemical and biophysical properties of peptide-based drug candidates (Hruby and Al-Obeidi 1990; Rizo and Gierasch 1992). Among the different approaches used to cyclize polypeptides, backbone or head-to-tail cyclization remains one of the most extensively used to introduce structural constraints into biologically active peptides. Cyclization generally increases stability and rigidifies structure, thereby minimizing the entropic cost of receptor binding.
Chemical synthesis of cyclic peptides has been well explored and a number different approaches involving solid-phase or liquid-phase have been reported (Camarero and Muir 1997; Zhang and Tam 1997; Camarero et al. 1998; Camarero et al. 1998; Shao et al. 1998). Recent developments in the fields of molecular biology and protein engineering also allow biosynthesis of cyclic peptides.
The biosynthesis of cyclic polypeptides offers many advantages over purely synthetic methods. Using the tools of molecular biology, large combinatorial libraries of cyclic peptides may be generated and screened in vivo. A typical chemical synthesis may generate 104 different molecules. It is not uncommon for a recombinant library to contain as many as 109 members. The molecular diversity generated by this approach is analogous to phage-display technology. Moreover, this approach takes advantage of the enhanced pharmacological properties of backbone-cyclized peptides as opposed to linear peptides or disulfide-stabilized polypeptides. However, unlike phage-display, backbone-cyclized polypeptides are not fused to or displayed on the surface, but remain on the inside of the living cell where they can be further screened for biological activity. The complex cellular cytoplasm provides the appropriate environment to address the physiological relevance of potential leads.
Protein trans-splicing has been successfully used by Benkovic and co-workers to generate backbone cyclized or polypeptides in vivo (Scott et al. 1999). In this approach, the peptide to be cyclized is nested between the two split intein fragments of the naturally occurring Ssp DnaE split intein (Wu et al. 1998) (usually referred as N- and C-inteins) in such way that the N-terminus of the peptide template is fused to C-intein fragment and vice versa. Protein splicing of this chimeric protein leads to the formation of the desired cyclic peptide inside E. coli cells. A potential limitation of this approach, however, is the requirement for specific N- and C-extein residues at the intein junction sites (Evans et al. 2000). These amino acids are necessary for efficient protein splicing to occur, which restricts the sequence diversity within the sequence of the cyclic peptide library members.
An attractive alternative approach is the use of modified protein splicing units to perform an intramolecular version of NCL reaction in vivo (Camarero and Muir 1999; Iwai and Pluckthum 1999; Evans et al. 2000; Camarero et al. 2001). Here we have shown the biosynthesis inside living E. coli cells of the cyclic peptide SFTI-1 and a small SFTI-based peptide library containing different Ala mutants using a modified protein-splicing unit. SFTI-1 is a bicyclic 14-amino acid peptide isolated from sunflower seeds that is related to the Bowman-Birk inhibitors and is one of the most potent inhibitors of trypsin of any naturally occurring peptide (Luckett et al. 1999). SFTI-1 forms a tightly folded scaffold, either when complexed with trypsin or free in solution (Luckett et al. 1999; Korsinczky et al. 2001). Its compact structure (Fig. 1) and high potency have led to suggestions that it may serve as a scaffold for the design of novel peptide-based drug leads (Korsinczky et al. 2004).
In-vivo biosynthesis of small backbone cyclic peptides like SFTI-1 containing a disulfide bond may have tremendous potential for drug discovery. This study shows that SFTI-1 may provide an ideal scaffold for the biosynthesis of large combinatorial libraries inside of living E. coli cells. Coupled to an appropriate in-vivo reporter system, this library may rapidly be screened using high throughput technologies such as fluorescence activated cell sorting (Kimura et al. 2007).
Supplementary Material
Acknowledgments
Work was supported by funding from the School of Pharmacy at the University of Southern California and Lawrence Livermore National Laboratory.
Abbreviations
- AMC
7-amido-4-methyl-coumarin
- CBD
chitin binding domain
- Cbz
benzyloxycarbonyl
- CCK
cyclic cystine-knot motif
- BBI
Bowman-Birk inhibitor
- EDTA
ethylenediaminetetraacetic acid
- EtSH
ethanethiol
- GdmCl
guanidinium hydrochloride
- GSH
glutathione
- HPLC
high performance liquid chromatography
- LB
Luria-Bertani
- MAP
methionyl aminopeptidase
- MESNA
mercaptoethanesulfonic acid
- NHS
N-hydroxysuccinimide ester
- PAGE
poly-acrylamide gel electrophoresis
- PMSF
phenylmethylsulphonyl fluoride
- SDS
sodium dodecyl sulfate
- SFTI-1
sunflower trypsin inhibitor 1
- TFA
trifluoroacetic acid
- UV-Vis
Ultraviolet visible
References
- Baird T, Wang B, Lodder M, Hecht S, Craik CS. Generation of active trypsin by chemcial cleavage. Tetrahedron. 2000;56:9477–9485. [Google Scholar]
- Camarero JA, Cotton GJ, Adeva A, Muir TW. Chemical ligation of unprotected peptides directly form a solid support. J Pept Res. 1998;51:303–316. doi: 10.1111/j.1399-3011.1998.tb00428.x. [DOI] [PubMed] [Google Scholar]
- Camarero JA, Fushman D, Cowburn D, Muir TW. Peptide chemical ligation inside living cells: in vivo generation of a circular protein domain. Bioorg Med Chem. 2001;9:2479–2484. doi: 10.1016/s0968-0896(01)00217-6. [DOI] [PubMed] [Google Scholar]
- Camarero JA, Kimura RH, Woo YH, Shekhtman A, Cantor J. Biosynthesis of a fully functional cyclotide inside living bacterial cells. Chembiochem. 2007;8:1363–1366. doi: 10.1002/cbic.200700183. [DOI] [PubMed] [Google Scholar]
- Camarero JA, Muir TW. Chemoselective backbone cyclization of unprotected peptides. J Chem Soc, Chem Comm. 1997:1369–1370. [Google Scholar]
- Camarero JA, Muir TW. Biosynthesis of a Head-to-Tail Cyclized Protein with Improved Biological Activity. J Am Chem Soc. 1999;121:5597–5598. [Google Scholar]
- Camarero JA, Muir TW. Native Chemical Ligation of Polypeptides. Current Protocols in Protein Science. 1999:1–21. doi: 10.1002/0471140864.ps1804s15. [DOI] [PubMed] [Google Scholar]
- Camarero JA, Pavel J, Muir TW. Chemical Synthesis of a Circular Protein Domain: Evidence for Folding-Assisted Cyclization. Angew Chem Int Ed. 1998;37:347–349. doi: 10.1002/(SICI)1521-3773(19980216)37:3<347::AID-ANIE347>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Craik DJ, Cemazar M, Daly NL. The cyclotides and related macrocyclic peptides as scaffolds in drug design. Curr Opin Drug Discov Devel. 2006;9:251–260. [PubMed] [Google Scholar]
- Craik DJ, Cemazar M, Wang CK, Daly NL. The cyclotide family of circular miniproteins: nature's combinatorial peptide template. Biopolymers. 2006;84:250–266. doi: 10.1002/bip.20451. [DOI] [PubMed] [Google Scholar]
- Craik DJ, Daly NL, Bond T, Waine C. Plant cyclotides: A unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif. J Mol Biol. 1999;294:1327–1336. doi: 10.1006/jmbi.1999.3383. [DOI] [PubMed] [Google Scholar]
- Craik DJ, Daly NL, Saska I, Trabi M, Rosengren KJ. Structures of naturally occurring circular proteins from bacteria. J Bacteriol. 2003;185:4011–4021. doi: 10.1128/JB.185.14.4011-4021.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craik DJ, Simonsen S, Daly NL. The cyclotides: novel macrocyclic peptides as scaffolds in drug design. Curr Opin Drug Discov Devel. 2002;5:251–260. [PubMed] [Google Scholar]
- Crovella S, Antcheva N, Zelezetsky I, Boniotto M, Pacor S, Verga Falzacappa MV, Tossi A. Primate beta-defensins--structure, function and evolution. Curr Protein Pept Sci. 2005;6:7–21. doi: 10.2174/1389203053027593. [DOI] [PubMed] [Google Scholar]
- Daly NL, Chen YK, Foley FM, Bansal PS, Bharathi R, Clark RJ, Sommerhoff CP, Craik DJ. The absolute structural requirement for a proline in the P3'-position of Bowman-Birk protease inhibitors is surmounted in the minimized SFTI-1 scaffold. J Biol Chem. 2006;281:23668–23675. doi: 10.1074/jbc.M601426200. [DOI] [PubMed] [Google Scholar]
- Descours A, Moehle K, Renard A, Robinson JA. A new family of beta-hairpin mimetics based on a trypsin inhibitor from sunflower seeds. Chembiochem. 2002;3:318–323. doi: 10.1002/1439-7633(20020402)3:4<318::AID-CBIC318>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Evans TC, Benner J, Xu M-Q. Semisynthesis of cytotoxic proteins using a modified protein splicing element. Protein Sci. 1998;7:2256–2264. doi: 10.1002/pro.5560071103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans TC, Jr, Martin D, Kolly R, Panne D, Sun L, Ghosh I, Chen L, Benner J, Liu XQ, Xu MQ. Protein trans-splicing and cyclization by a naturally split intein from the dnaE gene of Synechocystis species PCC6803. J Biol Chem. 2000;275:9091–9094. doi: 10.1074/jbc.275.13.9091. [DOI] [PubMed] [Google Scholar]
- Hilpert K, Hansen G, Wessner H, Schneider-Mergener J, Hohne W. Characterizing and optimizing protease/peptide inhibitor interactions, a new application for spot synthesis. J Biochem. 2000;128:1051–1057. doi: 10.1093/oxfordjournals.jbchem.a022833. [DOI] [PubMed] [Google Scholar]
- Hruby VJ, Al-Obeidi F. Emerging approaches in the molecular design of receptor-selective peptide ligands: conformational, topographical and dynamic considerations. J Biochem. 1990;268:249–262. doi: 10.1042/bj2680249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai H, Pluckthum A. Circular b-lactamase: stability enhancement by cyclizing the backbone. FEBS Lett. 1999:166–172. doi: 10.1016/s0014-5793(99)01220-x. [DOI] [PubMed] [Google Scholar]
- Jaulent AM, Leatherbarrow RJ. Design, synthesis and analysis of novel bicyclic and bifunctional protease inhibitors. Protein Eng Des Sel. 2004;17:681–687. doi: 10.1093/protein/gzh077. [DOI] [PubMed] [Google Scholar]
- Kimura RH, Steenblock ER, Camarero JA. Development of a cell-based fluorescence resonance energy transfer reporter for Bacillus anthracis lethal factor protease. Anal Biochem. 2007;369:60–70. doi: 10.1016/j.ab.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Kimura RH, Tran AT, Camarero JA. Biosynthesis of the cyclotide kalata B1 by using protein splicing. Angew Chem Int Ed Engl. 2006;45:973–976. doi: 10.1002/anie.200503882. [DOI] [PubMed] [Google Scholar]
- Korsinczky ML, Clark RJ, Craik DJ. Disulfide bond mutagenesis and the structure and function of the head-to-tail macrocyclic trypsin inhibitor SFTI-1. Biochemistry. 2005;44:1145–1153. doi: 10.1021/bi048297r. [DOI] [PubMed] [Google Scholar]
- Korsinczky ML, Schirra HJ, Craik DJ. Sunflower trypsin inhibitor-1. Curr Protein Pept Sci. 2004;5:351–364. doi: 10.2174/1389203043379594. [DOI] [PubMed] [Google Scholar]
- Korsinczky ML, Schirra HJ, Rosengren KJ, West J, Condie BA, Otvos L, Anderson MA, Craik DJ. Solution structures by 1H NMR of the novel cyclic trypsin inhibitor SFTI-1 from sunflower seeds and an acyclic permutant. J Mol Biol. 2001;311:579–591. doi: 10.1006/jmbi.2001.4887. [DOI] [PubMed] [Google Scholar]
- Luckett S, Garcia RS, Barker JJ, Konarev AV, Shewry PR, Clarke AR, Brady RL. High-resolution structure of a potent, cyclic proteinase inhibitor from sunflower seeds. J Mol Biol. 1999;290:525–533. doi: 10.1006/jmbi.1999.2891. [DOI] [PubMed] [Google Scholar]
- Marx UC, Korsinczky ML, Schirra HJ, Jones A, Condie B, Otvos L, Jr, Craik DJ. Enzymatic cyclization of a potent bowman-birk protease inhibitor, sunflower trypsin inhibitor-1, and solution structure of an acyclic precursor peptide. J Biol Chem. 2003;278:21782–21789. doi: 10.1074/jbc.M212996200. [DOI] [PubMed] [Google Scholar]
- Muir TW, Sondhi D, Cole PA. Expressed protein ligation: a general method for protein engineering. Proc Natl Acad Sci U S A. 1998;95:6705–6710. doi: 10.1073/pnas.95.12.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J, Gierasch LM. Constrained peptides: models of bioactive peptides and protein substructures. Ann Rev Biochem. 1992;61:387–418. doi: 10.1146/annurev.bi.61.070192.002131. [DOI] [PubMed] [Google Scholar]
- Scott CP, Abel-Santos E, Wall M, Wahnon D, Benkovic SJ. Production of cyclic peptides and proteins in vivo. Proc Natl Acad Sci USA. 1999;96:13638–13643. doi: 10.1073/pnas.96.24.13638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severinov K, Muir TW. Expressed protein ligation, a novel method for studying protein-protein interactions in transcription. J Biol Chem. 1998;273:16205–16209. doi: 10.1074/jbc.273.26.16205. [DOI] [PubMed] [Google Scholar]
- Shao Y, Lu WY, Kent SBH. A novel method to synthesize cyclic peptides. Tetrahedron Lett. 1998;39:3911–3914. [Google Scholar]
- Trabi M, Craik DJ. Circular proteins--no end in sight. Trends Biochem Sci. 2002;27:132–138. doi: 10.1016/s0968-0004(02)02057-1. [DOI] [PubMed] [Google Scholar]
- Wu H, Hu Z, Liu XQ. Protein trans-splicing by a split intein encoded in a split DnaE gene of Synechocystis sp. PCC6803. Proc Natl Acad Sci USA. 1998;95:9226–9231. doi: 10.1073/pnas.95.16.9226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Tam JP. Synthesis and application of unprotected cyclic peptides as building blocks for peptide dendrimers. J Am Chem Soc. 1997;119:2363–2370. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.