

Abstract
Hydrogen peroxide (H2O2) produced by cell-surface NADPH Oxidase (Nox) enzymes is emerging as an important signaling molecule for growth, differentiation, and migration processes. However, how cells spatially regulate H2O2 to achieve physiological redox signaling over nonspecific oxidative stress pathways is insufficiently understood. Here we report that the water channel Aquaporin-3 (AQP3) can facilitate the uptake of H2O2 into mammalian cells and mediate downstream intracellular signaling. Molecular imaging with Peroxy Yellow 1 Methyl-Ester (PY1-ME), a new chemoselective fluorescent indicator for H2O2, directly demonstrates that aquaporin isoforms AQP3 and AQP8, but not AQP1, can promote uptake of H2O2 specifically through membranes in mammalian cells. Moreover, we show that intracellular H2O2 accumulation can be modulated up or down based on endogenous AQP3 expression, which in turn can influence downstream cell signaling cascades. Finally, we establish that AQP3 is required for Nox-derived H2O2 signaling upon growth factor stimulation. Taken together, our findings demonstrate that the downstream intracellular effects of H2O2 can be regulated across biological barriers, a discovery that has broad implications for the controlled use of this potentially toxic small molecule for beneficial physiological functions.
Keywords: growth factor signaling, redox biology, reactive oxygen species, fluorescent sensor, membrane regulation
Hydrogen peroxide (H2O2) is garnering increased attention as a molecule involved not only in immune response and oxidative stress, but also as a physiological effector in essential cellular processes (1–5). Seminal contributions have elucidated ligand stimulants (6–10) and enzymatic sources (11–13) for cellular H2O2 production as well as putative downstream targets (14–24), but principles of how this reactive oxygen species (ROS) is spatially and temporally regulated to promote redox signaling over oxidative stress pathways remain insufficiently understood. Because many of the signaling functions of H2O2 rely on its generation by nonphagocytic forms of NADPH (Nox) proteins on the extracellular side of cell membranes, understanding how cells funnel H2O2 toward beneficial pathways in these locales is of significant interest.
Despite its reactive nature, H2O2 has been long thought to be freely diffusible across biological membranes in a manner akin to the related canonical small-molecule signal nitric oxide (NO) (25). More recent studies implicate the role of AQP water channels, a class of membrane-spanning proteins that facilitate the diffusion of water and other substrates with varying specificity (26–28), in mediating H2O2 passage across the plasma membrane of reconstituted yeast (29) and plant (30) cells. However, given that the experiments described in these reports utilized nonspecific chemical reagents for determination of the redox signal that was passed into the cell, direct evidence that aquaporins influence the cellular uptake of H2O2 in a native context remains elusive, and no studies have addressed higher-order mammalian systems. More importantly, the potential physiological consequences of regulating H2O2 have not been elucidated.
We now report an H2O2-selective small-molecule fluorescent indicator, Peroxy Yellow 1 Methyl-Ester (PY1-ME), and apply this unique chemical tool to show that members of two of the three main types of aquaporins (e.g., aquaglyceroporins and unorthodox aquaporins), but not the other type (e.g., classical aquaporins), can mediate H2O2 uptake across the plasma membrane of mammalian cells. Molecular imaging with PY1-ME in flow cytometry or confocal microscopy experiments reveals that canonical examples of an aquaglyceroporin and unorthodox aquaporin (AQP3 and AQP8, respectively), which are known to be permeated by molecules in addition to water, can facilitate H2O2 uptake into mammalian cells, whereas an example of a classical aquaporin that is known only to transport water (AQP1) does not enhance membrane H2O2 permeability. Moreover, we go on to show the physiological consequences of this newly discovered form of membrane H2O2 regulation in mammalian cells. We focused these studies on the aquaglyceroporin isoform AQP3 owing to its wide expression in many tissues, including the kidney, skin, colon, spleen, stomach, small intestine, uninary bladder, and respiratory tract, with AQP3-knockout mice showing loss of kidney function, polyuric behavior, deficiencies in epidermal hydration, and propensity toward colitis (31). Along these lines, we demonstrate at the cellular level that H2O2 uptake mediated by AQP3 can amplify or diminish downstream native signaling pathways that rely on this ROS as a physiological messenger. Our findings challenge the notion that H2O2 is freely diffusible through cell membranes and define a link between its membrane regulation and consequences for downstream signal transduction.
Results and Discussion
Design, Synthesis, and Properties of the New Chemoselective H2O2 Indicator PY1-ME.
Traditional fluorescent dyes used to probe H2O2 typically rely on nonspecific general oxidations that can be modulated by a variety of ROS, which can complicate data interpretation (32). We therefore sought to ensure that we could directly discriminate between H2O2 uptake and transfer of the redox signal by other means. Accordingly, we developed a unique H2O2-specific fluorescent indicator, PY1-ME (Fig. S1A), which combines a boronate-masked phenol for the selective detection of H2O2 (10, 33–39) and an appended methyl-ester group designed to increase cellular uptake and retention upon cleavage by intracellular esterases; this latter feature makes the dye suitable for both fluorescence imaging and flow cytometry experiments. Because this probe relies on a specific molecular reaction between H2O2 and the boronate to generate a fluorescence signal, rather than thiol-based chemistry that can occur by nonspecific ROS oxidation or disulfide exchange, our chemical method affords a direct measure of H2O2 in cellular specimens. Spectroscopic data show that PY1-ME features high selectivity for H2O2 over other ROS, visible excitation and emission profiles, and a 12-fold turn-on dynamic range with sensitivity to micromolar levels of H2O2 (Fig. S1 B and C).
Application of PY1-ME to Directly Establish that Select Types of AQP Channels Mediate H2O2 Uptake in Mammalian Cells.
Initial experiments tested whether overexpression of various types of AQPs would lead to increased intracellular H2O2 uptake in a reconstituted mammalian system. Previous work established that the unorthodox aquaporin AQP8 but not the classical aquaporin AQP1 can facilitate the uptake of H2O2 in yeast models (29). We sought to test whether this same pattern would hold in mammalian cells, as well as explore whether aquaglyceroporins, the third major type of these channels, could affect membrane H2O2 permeability. To this end, we transfected HEK 293 cells with either AQP expression or control vectors. Overexpression of each AQP was confirmed by Western blot analysis on cell lysates (Fig. S2 A and B). Aquaporin expressing or control cells were then loaded with PY1-ME, stimulated with H2O2 or vehicle control, and analyzed by flow cytometry. As shown in Fig. 1A, the addition of H2O2 to both control and AQP1 expressing HEK 293 cells results in enhanced PY1-ME fluorescent signal, indicating that this unique fluorescent probe is capable of measuring intracellular uptake of H2O2. AQP1 expression does not cause an increased signal from PY1-ME upon H2O2 stimulation as compared to control cells also stimulated with H2O2. However, as shown in Fig. 1B, addition of 50 μM H2O2 to AQP8-overexpressing HEK 293 cells loaded with PY1-ME results in a greater relative increase in intracellular fluorescence compared to control cells. These results confirm the yeast studies and demonstrate that some members of the AQP family of proteins are capable of allowing enhanced uptake of H2O2 into mammalian cells, whereas others appear to lack this ability.
Fig. 1.

Application of PY1-ME to show some aquaporins can increase the uptake of H2O2 in mammalian cells. HEK 293 cells transfected with either a control vector or AQP1 (A), AQP8 (B), or AQP3 (C) expression vectors, loaded with PY1-ME, treated with 0 or 50 μM H2O2 for 30 min, and analyzed by flow cytometry (n = 3). (D) HEK 293 cells transfected with either a control vector or AQP3 and mRFP as a transfection marker and measured by confocal microscopy (n = 8). (E) Representative images of experiment represented in (D). Scale bar = 20 μm. For all panels data were normalized to controls and statistical analyses were performed with a one-tailed Student’s t-test. Error bars are ± s.e.m.
We next turned our attention to evaluating whether an aquaglyceroporin-type AQP would regulate membrane H2O2 uptake in mammalian cells. We focused these studies on AQP3, owing to the wide tissue distribution of this aquaglyceroporin and phenotypes shown by AQP3 knockout mice (31), including kidney duct malfunctions and polyuric behavior (40), epidermal hydration deficiencies (41), and propensity toward colitis (42). At the cellular level, this aquaglyceroporin is known to facilitate diffusion of larger, alternative substrates to water (26) and is involved in cell migration and wound healing processes (43). Because some of these cell migration and wound healing processes are redox dependent (43, 44), we hypothesized that AQP3 may be involved in second-messenger signaling cascades utilizing H2O2. Indeed, many cell types, particularly colon cells (45, 46) and keratinocytes (47, 48), possess the endogenous machinery to produce H2O2 at the membrane through NADPH oxidase (Nox) proteins as well as high levels of AQP channels.
Overexpression of AQP3 was confirmed by Western blot analysis on cell lysates as well as immunostaining (Fig. S2 C and D and Figs. S3, S4, and S5). As shown in Fig. 1C, addition of 50 μM H2O2 to AQP3-overexpressing HEK 293 cells loaded with PY1-ME results in a greater relative increase in intracellular fluorescence compared to control cells. These results were further verified by live cell imaging (Fig. 1 D and E). AQP3-expressing cells exhibit a 130 ± 9% increase in fluorescence upon treatment with H2O2, whereas control cells show only a 80 ± 18% increase upon H2O2 addition (Fig. 2E). These experiments directly demonstrate that AQP3 is also able to increase the permeability of mammalian cells to exogenous H2O2.
Fig. 2.

AQP3 mediated exogenous H2O2 uptake can effect intracellular signaling. (A) Live-cell imaging of changes in HyPer fluorescence upon treatment of HEK 293 cells with 10 μM H2O2. “pNice,” HEK 293 cells expressing HyPer and transfected with pNice as a control vector before addition of H2O2. “+AQP3,” HEK 293 cells expressing HyPer and AQP3 before addition of H2O2. “+H2O2,” cells 2 min after addition of 10 μM H2O2. 20 μm scale bar. (B) Live-cell imaging of changes in HyPer fluorescence upon treatment of HeLa cells with 10 μM H2O2. “pNice,” HeLa cells expressing HyPer and transfected with pNice as a control vector before addition of H2O2. “+AQP3,” HeLa cells expressing HyPer and AQP3 before addition of H2O2. “+H2O2,” cells 2 min after addition of 10 μM H2O2. 20 μm scale bar. (C) Time-course and quantification of (B). HeLa cells expressing HyPer and transfected with pNice (black line) or AQP3 (blue line) were stimulated with 10 μM H2O2 at t = 0 and the changes in HyPer fluorescence monitored over time. Error bars are ± s.e.m. (n = 3). (D) Western blot showing changes in pAKT1/2/3 levels in HeLa cells treated with H2O2. HeLa cells expressing AQP3 or control vector were serum-starved and then treated with 200 μM H2O2 for 20 min at 37 °C and then lysed. Phospho-AKT and AQP3 levels were measured by Western blot analysis of whole cell extracts, followed by stripping and reprobing for total AKT or Actin, respectively.
Application of HyPer to Further Validate that AQP3 Facilitates H2O2 Uptake in Mammalian Cells.
After establishing that AQP3 and AQP8 but not AQP1 mediate H2O2 uptake utilizing a chemoselective small-molecule, we sought to utilize a secondary method of H2O2 measurement. For these experiments, we utilized the genetically encoded fluorescent H2O2 sensor HyPer (49), which has reasonable selectivity for and high sensitivity to H2O2, and thus should permit the detection of very low levels of H2O2 uptake and assure intracellular localization of the fluorescent reporter. Control HEK 293 cells exposed to 10 μM H2O2 show no statistically significant fluorescence enhancements as determined by HyPer fluorescence (Fig. 2A and Fig. S6). In contrast, AQP3-overexpressing HEK 293 cells exhibit a fluorescence increase within 2 min of H2O2 addition (Fig. 2A and Fig. S6). These data further show that AQP3 increases the permeability of mammalian cells to exogenous H2O2, and validates the use of HyPer as an alternative methodology for the detection of H2O2 uptake.
AQP3-Mediated H2O2 Uptake Influences Intracellular Signaling Cascades.
After establishing that AQP3 can facilitate the uptake of H2O2 by two independent imaging methods, we sought to determine whether AQP3-mediated H2O2 accumulation could affect downstream intracellular processes. For these experiments we utilized HeLa cells, owing to their high number of signaling proteins and lack of endogenous AQP3 expression, to provide a clean reconstituted mammalian cell model. First we confirmed that AQP3 could also mediate H2O2 uptake in this cell line. Control HeLa cells exposed to 10 μM H2O2 show no statistically significant fluorescence enhancements as determined by HyPer fluorescence (Fig. 2 B and C). In contrast, AQP3-overexpressing HeLa cells exhibit a 50 ± 4% fluorescence increase within 2 min of H2O2 addition (Fig. 2 B and C). We then sought to probe whether these differences in H2O2 uptake obtained by the overexpression of AQP3 could affect downstream cell signaling pathways that rely on this ROS as a physiological effector molecule. In this context, a compelling relationship has emerged between H2O2 signaling and the phosphorylation status of the serine/threonine kinase AKT/protein kinase B, a major node in cellular signal transduction (50). ROS signals can amplify AKT signaling cascades in the forward direction through transient oxidative inactivation of the phosphatase and tensin homolog (PTEN) and other phosphatases that oppose AKT phosphorylation (21). Accordingly, we utilized the phosphorylation status of AKT as a central marker for signal transduction that can be modulated by H2O2. Initial experiments tested the direct effects of H2O2 on AKT phosphorylation. Serum-starved control HeLa cells do not exhibit increases in pAKT/AKT ratios upon addition of H2O2 (Fig. 2D). However, AQP3-overexpressing, serum-starved HeLa cells treated with H2O2 for 20 min triggers a marked enhancement in AKT phosphorylation (Fig. 2D). When correlated with the imaging data this result shows that AQP3 can enhance permeability of cells to H2O2 and amplify downstream signaling that relies on this ROS as an intracellular messenger.
Endogenous AQP3 Levels Regulate H2O2 Uptake.
We next tested whether endogenously expressed AQP3 could also facilitate H2O2 uptake and downstream intracellular signaling. For these experiments we turned our attention to HT29 human colon cancer adenocarcinoma cells, as this model naturally expresses both AQP3 (45) and Nox proteins (46) and possesses the requisite AKT signaling hub. We utilized an shRNA construct targeted against AQP3 and validated in HEK 293 cells (Fig. S2D) to knock down the expression of endogenous AQP3 in HT29 cells (Fig. 3B). When control HT29 cells transfected with an shRNA construct targeting AQP4, an AQP isoform not found in this cell line, were treated with H2O2, we observed a ca. 20% increase in fluorescence intensity measured by HyPer (Fig. 3 A, C, and D). In contrast, AQP3-knockdown cells showed no significant fluorescence increase when exposed to an equivalent amount of H2O2 (Fig. 3 A, C, and D), establishing that natural levels of AQP3 can affect H2O2 uptake.
Fig. 3.

Natural levels of AQP3 can mediate exogenous H2O2 uptake. (A) HT29s transfected with either HyPer and shRNA targeted against AQP3(241) or AQP4(801) as a control with 100 μM H2O2 at t = 0 and the changes in HyPer fluorescence monitored over time. Images show cells at t = 0 and t = 2 min (“+H2O2”). 10 μm scale bar. (B) Knockdown of endogenous AQP3 in HT29 cells. HT29 cells were transfected with either shRNA targeted against AQP3(241), or AQP4(801) as a control, then lysed and probed for AQP3 levels by Western blot analysis of whole cell extracts, followed by stripping and reprobing for Actin. (C) Endogenous AQP3 can mediate H2O2 uptake. HT29 cells expressing HyPer and transfected with AQP4 shRNA as a control (black line) or AQP3 shRNA (blue line) were stimulated with 100 μM H2O2 at t = 0 and the changes in HyPer fluorescence monitored over time. Error bars are ± s.e.m. (n = 3). (D) Quantification of C at t = 2. (n = 3). Data were normalized to controls and statistical analyses were performed with a one-tailed Student’s t test. Error bars are ± s.e.m.
AQP3 Regulates Endogenous H2O2 Uptake and Downstream Cell Signaling Upon Growth Factor Stimulation.
We then sought to determine whether AQP3-mediated uptake of naturally generated H2O2 could also mediate intracellular redox signaling. In this context, previous work has shown that EGF stimulation causes the activation of membrane-bound Nox proteins to produce H2O2, which in turn regulates intracellular signaling (51). In agreement with experiments in HEK 293 and HeLa cells, addition of 100 μM H2O2 causes a marked increase intracellular fluorescence in both natural and AQP3-overexpressing HT29 cells, with a greater relative increase in the AQP3-elevated samples (Figs. S7 and S8). Moreover, HT29 cells overexpressing AQP3 and stimulated with EGF exhibit a ca. 10% increase in intracellular fluorescence (Fig. 4 A and B). We were unable to directly image Nox-generated H2O2 upon EGF stimulation of basal HT29 cells under similar conditions, which is presumably due to the low levels of H2O2 produced for signaling purposes that are under the detection limit of HyPer. These experiments do, however, establish that AQP3 can also enhance the uptake of endogenous H2O2.
Fig. 4.

Natural levels of AQP3 can mediate uptake of Nox-generated H2O2 and effect intracellular signaling. (A) Live-cell imaging of changes in HyPer fluorescence upon treatment of HT29 cells with 100 ng/mL EGF. HT29 cells expressing HyPer and transfected with pNice as a control (“pNice” ) or AQP3 (“+AQP3”) before addition of EGF. Cells images at t = 0 and then again at t = 20 min (“+EGF”). 20 μm scale bar. (B) Time-course and quantification of A. HT29 cells expressing HyPer and transfected with pNice (black line) or AQP3 (blue line) stimulated with 100 ng/mL EGF at t = 0 and the changes in HyPer fluorescence monitored over time. Error bars are ± s.e.m. (n = 3). (C) Schematic illustrating where the H2O2 redox signal can by intercepted. EGF stimulation can activate a membrane-bound NADPH oxidase (Nox), which will then produce extracellular H2O2, which can then pass into the cell and modulate intracellular redox signaling. Diphenylene iodonium (DPI) will block the Nox production of H2O2 and extracellular catalase (Cat) will destroy any H2O2 produced outside of the cell. The genetic manipulation of AQP3 using shRNA will block any potential factilitated uptake of extracellular H2O2 by this protein. (D) Extracellular catalase and DPI abrogate EGF signaling in HT29 cells. Serum-starved HT29 cells were either preincubated with 5 μM DPI or DMSO as a carrier control for 30 min. 5 mg/mL catalase or water as a carrier control was then added while the cells were simulataneously stimulated with 100 ng/mL EGF. Phospho-AKT was measured by Western blot analysis of whole cell extracts, followed by stripping and reprobing for total AKT. (E) HT29 cells were transfected with either AQP3 shRNA or AQP4 shRNA as a control, serum starved, stimulated with 100 ng/mL EGF, and lysed at the various time points. Phospho-AKT was measured by Western blot analysis of whole cell extracts, followed by stripping and reprobing for total AKT. (F) Quantification of experiment as represented in (E) by analyzing 4 separate trials. pAKT normalized to total AKT and each experiment normalized to the t = 0 pAKT/AKT ratio. Error bars are ± s.e.m.
As a more sensitive method for probing extracellular H2O2 generation and its subsequent effects on intracellular processes, we then assayed the effects of endogenous AQP3 on Nox-derived H2O2 uptake and signaling by Western blot analysis of AKT phosphorylation status. As expected, stimulation of HT29 cells with EGF causes a patent increase in AKT phosphorylation (Fig. 4D). In contrast, stimulation with EGF in the presence of extracellular catalase, which will destroy any extracellular H2O2 produced by Nox, abrogates AKT phosphorylation, demonstrating that extracellular H2O2 production is linked to intracellular signaling processes. Similarly, EGF stimulation in the presence of diphenyleneiodonium (DPI), a general Nox/flavin inhibitor, also diminishes H2O2 signaling through AKT. Finally, cells that have endogenous AQP3 levels knocked down by shRNA also show small but significant decreases in EGF-induced AKT phosphorylation when compared to control cells transfected with an AQP4 targeting shRNA (Fig. 4 E and F), as shown by quantification of the Western data. Taken together, these data establish that endogenous extracellular H2O2 produced by Nox at the cell surface is important for EGF signaling, and AQP3 plays a role in the transduction of that signal from the extracellular space to intracellular targets (Fig. 4C). By showing that membrane regulation of H2O2 in this manner can occur and is essential for intracellular signaling, these results provide a key piece of the puzzle for use of the reactive metabolite H2O2 as a physiological effector while avoiding oxidative stress pathways but open the door to many new and interesting questions. In particular, the observation that RNAi knockdown of AQP3 does not abolish AKT signaling as much as the addition of extracellular catalase suggests that other H2O2 membrane transport mechanisms may be at play, potentially either some passive membrane diffusion or regulation by alternative aquaporins or other proteins. In addition, other membrane and nonmembrane sources of H2O2 could also be implicated in these types of signaling pathways, a hypothesis that is supported by that fact that DPI abolishes AKT signaling to a greater extent compared to addition of extracellular catalase.
Conclusions
In summary, our findings challenge the commonly held view that H2O2 is freely diffusible across biological membranes and define a connection between its membrane regulation and downstream intracellular signaling. By utilizing PY1-ME, a unique H2O2-specific synthetic small-molecule dye, in conjunction with protein reporters to directly visualize differences in H2O2 uptake in various cell lines as a function of AQP levels, we have established that specific types of AQP channels can regulate the uptake of H2O2 in mammalian cells. In particular, canonical members of the aquaglyceroporin (AQP3) and unorthodox (AQP8) family of these channels mediate H2O2 uptake through mammalian cell membranes, where a member of the classical type of aquaporin (AQP1) does not. Furthermore, AQP-mediated H2O2 accumulation can amplify or diminish downstream signaling cascades that rely on this ROS as a physiological messenger. Interestingly, in this context, membrane H2O2 regulation bears more resemblance to metal Ca2+ signals that require channel entry rather than the related small-molecule signal NO that can freely diffuse through membranes.
The evidence presented in this report indicates that some, but not all, AQPs can facilitate the uptake of H2O2 in mammalian systems, and that the ability to mediate H2O2 transport may correspond to specific AQP classes (classical, aquaglyceroporin, unorthodox). This selectivity suggests that the expression profiles of AQPs could dictate the susceptibility of a particular cell or tissue to external H2O2 signaling. Moreover, the notion that H2O2 uptake is controlled at biological barriers has broad implications for AQP channels as regulators of redox signaling, with particularly tantalizing connections in the areas of cell migration and wound healing. For example, AQP3 facilitates cell migration through an EGF/EGFr dependent pathway (52) and can mediate wound healing in the epidermis via cell migration and proliferation through p38 MAPK-dependent pathways (43). In addition, several reports have suggested a relationship between ROS fluxes and cell migration, based on imaging studies using nonspecific ROS indicators at the leading edge of migrating endothelial cells (53, 54). Finally, an exciting discovery establishing the involvement of H2O2 signaling in wound repair has been reported recently, in which H2O2 generated by the Duox class of Nox proteins acts as a signaling molecule to attract leukocytes to the site of injury (44). The involvement of both H2O2 fluxes and AQP channels in cell migration and wound healing raises the possibility that AQPs may modulate H2O2 permeability as a general way to modulate its downstream effects, where regulation of AQP channels through various mechanisms, including differential expression, trafficking, or posttranslational modification may allow for discrimination between extracellularly generated ROS molecules such as H2O2 or superoxide (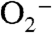 ). We are actively exploring these possibilities in a wide variety of model systems.
). We are actively exploring these possibilities in a wide variety of model systems.
Materials and Methods
Antibodies.
Rabbit anti-AQP3 polyclonal antibody was from Chemicon/Millipore. Goat anti-AQP3, rabbit anti-pAKT1/2/3, rabbit anti-AKT1/2/3, rabbit anti-AQP1, mouse anti-AQP8, goat antimouse IgG-HRP, and goat antirabbit IgG-HRP were from Santa Cruz Biotechnology. Bovine antigoat IgG-Cy3 was from Jackson Immuno Labs. Mouse antiactin was from Sigma.
Cell Culture and Transfection.
Cell lines HEK293T and HeLa (ATCC) were maintained in DMEM (Invitrogen) supplemented with 10% FBS, 1% Nonessential amino acids, and 1% Gluta-MAX (Invitrogen). The HT-29 cell line (ATCC) was maintained in DMEM supplemented with 10% FBS. HEK293T cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. HeLa cells were transfected with either Lipofectamine LTX (Invitrogen) or DreamFect Gold (OZ Biosciences) according to the manufacturer’s instructions. HT-29 cells were transfected using the Nucleofector II device (Lonza), using the high efficiency program W-017. Human AQP3 cDNA was obtained from ATCC (clone ID: BC013566) in the vector pCMV-SPORT6. The AQP3 gene was subcloned into the mammalian expression vector pNice under control of the CMV promotor using NotI and EcoRI. Human AQP1 was obtained from Origene (clone ID: M77829) in the vector pCMV6-XL4. Human AQP8 was obtained from Origene (clone ID: AB013456) in the vector pCMV6-XL4. shRNA constructs were designed against human AQP3 (NM_004925.3) using OligoEngine design tools. Three sequences were selected based on specificity and predicted strength of knockdown and incorporated into a pSUPER.retro.neo vector according to the manufacturer’s instructions:
241: 5′ ACCTGAACCCTGCCGTGAC 3′
243: 5′ CTGAACCCTGCCGTGACCT 3′
824: 5′ CAACGAGGAAGAGAATGTG 3′
Efficacy of the constructs was determined by cotransfection of the pSUPER construct along with AQP3 into HEK 293 cells (3∶1 ratio of pSUPER:AQP3 DNA), followed by analysis of AQP3 levels by Western blot, 24–48 h posttransfection. The construct targeted against 241 was selected for use in knock down studies.
Preparation of Cell Extracts and Immunoblotting.
Following transfection, cells were grown to subconfluency of 60–90% and then serum-starved in DMEM without supplements overnight (HeLa) or for 1 h (HT-29). Appropriate stimulants were added and the cells incubated at 37 °C. After stimulation, cells were cooled on ice, washed with ice-cold Dulbecco’s phosphate buffered saline (DPBS), scraped into radioimmunoprecipitation (RIPA) buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% SDS) supplemented with protease inhibitors (Roche Complete MiniTab), and 1 mM Na3VO4, lysed for 30 min on ice, and clarified by centrifugation at 13.2 × 103 rpm for 10 min at 4 °C. Protein concentration was determined by bicinchoninic acid (BCA) assay (Pierce), 4x Laemmli buffer (0.25 mM Tris, 2% SDS, 40% glycerol, 20% beta-mercaptoethanol, 0.04% bromophenol blue) was added, the samples denatured at 37 °C for 20 min (for AQP8 blots), 70 °C for 10 min (for AQP3 and AQP1 blots), or 95 °C for 5 min (pAKT/AKT blots), and equal amounts of total protein were loaded onto 12% SDS-PAGE gels. Proteins were transferred to PVDF membranes (Immobilon, Millipore), blocked for 1 h in 5% nonfat dry milk in wash buffer (10 mM Tris, pH 7.5, 100 mM NaCl, 0.1% Tween-20), and incubated overnight at 4 °C in primary antibody (1∶1000, x pAKT1/2/3, x AKT1/2/3; 1∶500 x AQP3; 1∶5000 x actin). Membranes were washed 5 times for 5 min in wash buffer, incubated 1 h at room temperature with 1∶1000 secondary antibody, washed 5 times for 5 min in wash buffer, and then visualized using enhanced chemiluminescence (Western Lightning, Perkin–Elmer) recorded on a BioRad GelDoc imaging station. Densitometic analysis was performed using QuantityOne software (BioRad). Blots were stripped using Restore stripping buffer (Fisher) or 50 mM Tris pH 6.8, 2% SDS, 100 mM 2-mercaptoethanol (BME) at 55 °C for 17 min.
Immunocytochemistry.
Cells plated on 15 mm coverslips were fixed in 4% paraformaldehyde, permeabilized in PBST (PBS + 0.3% Triton X-100), blocked in 2% FBS in phosphate buffered saline Tween-20 (PBST), incubated with 1∶1000 goat anti-AQP3 followed by bovine antigoat IgG-Cy3, and mounted before imaging using an epifluorescence microscope.
Cell Sorting.
HEK293T cells were plated at 2.5 × 105 well in a 24-well tray. One day after plating, cells were transfected with equal amounts (250 ng) of AQP expression vector or control vector and monomeric red fluorescent protein (mRFP) using Lipofectamine LTX. 48 h after transfection, cells were washed once with DPBS and incubated with 5 μM PYME in DPBS for 15 min at 37 °C. Cells were then treated with 50 μM H2O2 or buffer and incubated for 30 min at 37 °C. After treatment, cells were typsinized, resuspended in DPBS, and sorted by mRFP fluorescence. The mean PYME fluorescence of mRFP-positives in the AQP and control cells was measured.
Confocal Microscopy.
Subconfluent HEK293T cells were plated on polylysine-coated 15 mm coverslips and transfected with mRFP and AQP3 or control vector. 24–48 h posttransfection, cells were washed with DPBS, and incubated with 5 μM PYME in DPBS for 15 min at 37 °C. Following incubation, H2O2 was added to give a final concentration of 50 μM and incubated for another 30 min at 37 °C. Cells were then washed with DPBS and imaged with a Zeiss 510 Axioplan equipped with a META detector and an Achroplan 40x/0.8 NA water immersion lens. Excitation for PYME was provided at 514 nm, with emission collected from 516–569 nm. Excitation for mRFP was provided at 543 nm, with emission collected from 601–676 nm. Regions of interest were generated within mRFP+ cells and the mean PYME fluorescence from these regions of interest (ROI) were measured using ImageJ.
HyPer Imaging.
Subconfluent HEK293T or HeLa cells plated on polylysine-coated 15 mm coverslips were transfected with equal amounts of AQP3 or control vector and HyPer and allowed to grow to approximately 80–90% confluency. Prior to imaging, the coverslips were transferred to 2 mL of DPBS (no Ca2+ or Mg2+) in a 35 mm petri dish with a 14 mm glass microwell (MatTek). Images were acquired every 20 s to establish baseline fluorescence and then 1 mL of 3x H2O2 was added to the dish. The mean fluorescence intensity of HyPer-expressing cells was monitored over time using an Axiovert 200M equipped with an AxioCam MRm. The fluorescence intensity immediately before H2O2 addition was set to 1.00 and quantified using ImageJ. Excitation was provided by an X-Cite series 120 lamp using a filter cube containing a bandpass excitation filter at 470/40, a beamsplitter at 495, and a bandpass emission filter at 525/50. HT-29 imaging was done in a similar manner except that Hank’s Balanced Salt Solution (HBSS) with Ca2+ and Mg2+ was used instead of DPBS and a final concentration of 100 μM H2O2 was used instead of 10 μM H2O2. For EGF imaging, HT-29 cells were starved in DMEM for 1 h prior to imaging. As with H2O2 imaging experiments, 3x EGF (Upstate/Millipore) was added to give a final concentration of 100 ng/mL.
Supplementary Material
Acknowledgments.
This work was supported by the Packard and Sloan Foundations, the UC Berkeley Hellman Faculty Fund, Amgen, Astra Zeneca, and Novartis, and the National Institutes of Health (GM 79465). C.J.C. is an investigator with the Howard Hughes Medical Institute. B.C.D. and E.W.M. were partially supported by a Chemical Biology Training Grant from the National Instittes of Health (T32 GM066698), and E.W.M. thanks UC Berkeley for a Stauffer fellowship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1005776107/-/DCSupplemental.
References
- 1.Poole LB, Karplus PA, Claiborne A. Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol. 2004;44:325–347. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 2.Rhee SG. Cell signaling H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 3.D’Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 4.Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 5.Paulsen CE, Carroll KS. Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem Biol. 2010;5:47–62. doi: 10.1021/cb900258z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.May JM, de Haen C. Insulin-stimulated intracellular hydrogen peroxide production in rat epididymal fat cells. J Biol Chem. 1979;254:2214–2220. [PubMed] [Google Scholar]
- 7.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 8.Bae YS, et al. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem. 1997;272:217–221. [PubMed] [Google Scholar]
- 9.Zafari AM, et al. Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertension. 1998;32:488–495. doi: 10.1161/01.hyp.32.3.488. [DOI] [PubMed] [Google Scholar]
- 10.Miller EW, Tulyathan O, Isacoff EY, Chang CJ. Molecular imaging of hydrogen peroxide produced for cell signaling. Nat Chem Biol. 2007;3:263–267. doi: 10.1038/nchembio871. [DOI] [PubMed] [Google Scholar]
- 11.Suh YA, et al. Cell transformation by the superoxide-generating oxidase Mox1. Nature. 1999;401:79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- 12.Geiszt M, Kopp JB, Varnai P, Leto TL. Identification of renox, an NAD(P)H oxidase in kidney. Proc Natl Acad Sci USA. 2000;97:8010–8014. doi: 10.1073/pnas.130135897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bao L, et al. Mitochondria are the source of hydrogen peroxide for dynamic brain-cell signaling. J Neurosci. 2009;29:9002–9010. doi: 10.1523/JNEUROSCI.1706-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- 15.Lee SR, et al. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336–20342. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- 16.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 17.Woo HA, et al. Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science. 2003;300:653–656. doi: 10.1126/science.1080273. [DOI] [PubMed] [Google Scholar]
- 18.van Montfort RL, Congreve M, Tisi D, Carr R, Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature. 2003;423:773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- 19.Salmeen A, et al. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 20.Avshalumov MV, Rice ME. Activation of ATP-sensitive K+ (K(ATP)) channels by H2O2 underlies glutamate-dependent inhibition of striatal dopamine release. Proc Natl Acad Sci USA. 2003;100:11729–11734. doi: 10.1073/pnas.1834314100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwon J, et al. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci USA. 2004;101:16419–16424. doi: 10.1073/pnas.0407396101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science. 2004;304:596–600. doi: 10.1126/science.1095569. [DOI] [PubMed] [Google Scholar]
- 23.Leonard SE, Reddie KG, Carroll KS. Mining the thiol proteome for sulfenic acid modifications reveals new targets for oxidation in cells. ACS Chem Biol. 2009;4:783–799. doi: 10.1021/cb900105q. [DOI] [PubMed] [Google Scholar]
- 24.Woo HA, et al. Inactivation of Peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell. 2010;140:517–528. doi: 10.1016/j.cell.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 25.Lancaster JR. Simulation of the diffusion and reaction of endogenously produced nitric oxide. Proc Natl Acad Sci USA. 1994;91:8137–8137. doi: 10.1073/pnas.91.17.8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.King LS, Kozono D, Agre P. From structure to disease: the evolving tale of aquaporin biology. Nat Rev Mol Cell Biol. 2004;5:687–698. doi: 10.1038/nrm1469. [DOI] [PubMed] [Google Scholar]
- 27.Stroud R, Harries W, Lee J, Khademi S, Savage D. In: Structural Biology of Membrane Proteins. Grisshammer R, Buchanan S, editors. Cambridge, UK: Royal Society of Chemistry; 2006. pp. 195–211. [Google Scholar]
- 28.Ho JD, et al. Crystal structure of human aquaporin 4 at 1.8 Å and its mechanism of conductance. Proc Natl Acad Sci USA. 2009;106:7437–7442. doi: 10.1073/pnas.0902725106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bienert GP, et al. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J Biol Chem. 2007;282:1183–1192. doi: 10.1074/jbc.M603761200. [DOI] [PubMed] [Google Scholar]
- 30.Dynowski M, Schaaf G, Loque D, Moran O, Ludewig U. Plant plasma membrane water channels conduct the signalling molecule H2O2. Biochem J. 2008;414:53–61. doi: 10.1042/BJ20080287. [DOI] [PubMed] [Google Scholar]
- 31.Rojek A, Praetorius J, Frøkiaer J, Nielsen S, Fenton RA. A current view of the mammalian aquaglyceroporins. Annu Rev Physiol. 2008;70:301–327. doi: 10.1146/annurev.physiol.70.113006.100452. [DOI] [PubMed] [Google Scholar]
- 32.Hempel SL, Buettner GR, O’Malley YQ, Wessels DA, Flaherty DM. Dihydrofluorescein diacetate is superior for detecting intracellular oxidants: comparison with 2′, 7′-dichlorodihydrofluorescein diacetate, 5 (and 6)-carboxy-2′, 7′-dichlorodihydrofluorescein diacetate, and dihydrorhodamine 123-implications for intracellular measurement of reactive nitrogen species. Free Radical Bio Med. 1999;27:146–159. doi: 10.1016/s0891-5849(99)00061-1. [DOI] [PubMed] [Google Scholar]
- 33.Chang MCY, Pralle A, Isacoff EY, Chang CJ. A selective, cell-permeable optical probe for hydrogen peroxide in living cells. J Am Chem Soc. 2004;126:15392–15393. doi: 10.1021/ja0441716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller EW, Albers AE, Pralle A, Isacoff EY, Chang CJ. Boronate-based fluorescent probes for imaging cellular hydrogen peroxide. J Am Chem Soc. 2005;127:16652–16659. doi: 10.1021/ja054474f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srikun D, Miller EW, Domaille DW, Chang CJ. An ICT-based approach to ratiometric fluorescence imaging of hydrogen peroxide produced in living cells. J Am Chem Soc. 2008;130:4596–4597. doi: 10.1021/ja711480f. [DOI] [PubMed] [Google Scholar]
- 36.Dickinson BC, Chang CJ. A targetable fluorescent probe for imaging hydrogen peroxide in the mitochondria of living cells. J Am Chem Soc. 2008;130:9638–9639. doi: 10.1021/ja802355u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dickinson BC, Srikun D, Chang CJ. Mitochondrial-targeted fluorescent probes for reactive oxygen species. Curr Opin Chem Biol. 2010;14:50–56. doi: 10.1016/j.cbpa.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Srikun D, Albers AE, Nam CI, Ivarone AT, Chang CJ. Organelle-targetable fluorescent probes for imaging hydrogen peroxide in living cells via SNAP-tag protein labeling. J Am Chem Soc. 2010;132:4455–4465. doi: 10.1021/ja100117u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dickinson BC, Huynh C, Chang CJ. A palette of fluorescent probes with varying emission colors for imaging hydrogen peroxide signaling in living cells. J Am Chem Soc. 2010;132:5906–5915. doi: 10.1021/ja1014103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma T, et al. Nephrogenic diabetes insipidus in mice lacking aquaporin-3 water channels. Proc Natl Acad Sci USA. 2000;97:4386–4391. doi: 10.1073/pnas.080499597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hara M, Ma T, Verkman AS. Selectively reduced glycerol in skin of aquaporin-3-deficient mice may account for impaired skin hydration, elasticity, and barrier recovery. J Biol Chem. 2002;277:46616–46621. doi: 10.1074/jbc.M209003200. [DOI] [PubMed] [Google Scholar]
- 42.Thiagarajah JR, Zhao D, Verkman AS. Impaired enterocyte proliferation in aquaporin-3 deficiency in mouse models of colitis. Gut. 2007;56:1529–1535. doi: 10.1136/gut.2006.104620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hara-Chikuma M, Verkman A. Aquaporin-3 facilitates epidermal cell migration and proliferation during wound healing. J Mol Med. 2008;86:221–231. doi: 10.1007/s00109-007-0272-4. [DOI] [PubMed] [Google Scholar]
- 44.Niethammer P, Grabher C, Look AT, Mitchison TJ. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature. 2009;459:996–999. doi: 10.1038/nature08119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Itoh A, Tsujikawa T, Fujiyama Y, Bamba T. Enhancement of aquaporin-3 by vasoactive intestinal polypeptide in a human colonic epithelial cell line. J Gastroenterol Hepatol. 2003;18:203–210. doi: 10.1046/j.1440-1746.2003.02949.x. [DOI] [PubMed] [Google Scholar]
- 46.Perner A, Andresen L, Pedersen G, Rask-Madsen J. Superoxide production and expression of NAD(P)H oxidases by transformed and primary human colonic epithelial cells. Gut. 2003;52:231–236. doi: 10.1136/gut.52.2.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hara-Chikuma M, Verkman AS. Roles of aquaporin-3 in the epidermis. J Invest Dermatol. 2008;128:2145–2151. doi: 10.1038/jid.2008.70. [DOI] [PubMed] [Google Scholar]
- 48.Rygiel TP, Mertens AE, Strumane K, van der Kammen R, Collard JG. The Rac activator Tiam1 prevents keratinocyte apoptosis by controlling ROS-mediated ERK phosphorylation. J Cell Sci. 2008;121:1183–1192. doi: 10.1242/jcs.017194. [DOI] [PubMed] [Google Scholar]
- 49.Belousov VV, et al. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat Methods. 2006;3:281–286. doi: 10.1038/nmeth866. [DOI] [PubMed] [Google Scholar]
- 50.Manning BD, Cantley LC. AKT/PKB signaling: Navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rhee SG, Bae YS, Lee S-R, Kwon J. Hydrogen peroxide: A key messenger that modulates protein phosphorylation through cysteine oxidation. Sci STKE. 2000;2000:1–6. doi: 10.1126/stke.2000.53.pe1. [DOI] [PubMed] [Google Scholar]
- 52.Cao C, et al. EGFR-mediated expression of aquaporin-3 is involved in human skin fibroblast migration. Biochem J. 2006;400:225–234. doi: 10.1042/BJ20060816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ikeda S, et al. IQGAP1 regulates reactive oxygen species-dependent endothelial cell migration through interacting with Nox2. Arterioscler Thromb Vasc Biol. 2005;25:2295–2300. doi: 10.1161/01.ATV.0000187472.55437.af. [DOI] [PubMed] [Google Scholar]
- 54.Moldovan L, Moldovan NI, Sohn RH, Parikh SA, Goldschmidt-Clermont PJ. Redox changes of cultured endothelial cells and actin dynamics. Circ Res. 2000;86:549–557. doi: 10.1161/01.res.86.5.549. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.