

Abstract
MicroRNAs (miRNAs) are a class of short noncoding RNAs that regulate protein-coding genes posttranscriptionally. In animals, most known miRNA targeting occurs within the 3′UTR of mRNAs, but the extent of biologically relevant targeting in the ORF or 5′UTR of mRNAs remains unknown. Here, we develop an algorithm (MinoTar—miRNA ORF Targets) to identify conserved regulatory motifs within protein-coding regions and use it to estimate the number of preferentially conserved miRNA-target sites in ORFs. We show that, in Drosophila, preferentially conserved miRNA targeting in ORFs is as widespread as it is in 3′UTRs and that, while far less abundant, conserved targets in Drosophila 5′UTRs number in the hundreds. Using our algorithm, we predicted a set of high-confidence ORF targets and selected seven miRNA-target pairs from among these for experimental validation. We observed down-regulation by the miRNA in five out of seven cases, indicating our approach can recover functional sites with high confidence. Additionally, we observed additive targeting by multiple sites within a single ORF. Altogether, our results demonstrate that the scale of biologically important miRNA targeting in ORFs is extensive and that computational tools such as ours can aid in the identification of such targets. Further evidence suggests that our results extend to mammals, but that the extent of ORF and 5′UTR targeting relative to 3′UTR targeting may be greater in Drosophila.
Keywords: comparative genomics, target prediction
In the past ten years, microRNAs (miRNAs) have emerged as an extensive class of regulators conserved across nearly all eukaryotes and with an influence on a wide variety of biological processes (1, 2). Yet one of the most important goals in the miRNA field, the identification of genes targeted by miRNAs, remains a significant challenge. Most known miRNA targets in animals have followed a canonical pattern, where target genes contain 7–8 bases in their 3′UTRs with perfect complementarity to the so-called seed region at the 5′ end of a miRNA (3). A number of target prediction tools have been designed to aid in the identification of such 3′UTR targets, incorporating additional information beyond seed matches, such as conservation, to form a set of most likely targets (see refs. 4–7 among many others). Such tools have proven to be an invaluable resource for miRNA researchers.
The extent of targeting that does not fit such a canonical pattern, and in particular the extent of biologically relevant targeting outside of 3′UTRs, remains unknown. Large-scale miRNA overexpression and knockout studies in mammals have provided evidence for ORF targeting, though of a weaker effect than 3′UTR targeting (8, 9). However, recent results from cross-linking immunoprecipitation experiments have indicated that binding of Argonaute proteins is nearly as widespread in ORFs as in 3′UTRs (10, 11). Evidence has suggested that the translation machinery can be refractory to miRNA targeting within ORFs (12) and in a short region past the stop codon (13), suggesting a mechanistic basis for the weaker effect of targeting in ORFs. However, a small number of cases of targets in ORFs have been verified (14–19) [as well as a small number of targets in 5′UTRs (20, 21)]. Together these results suggest that miRNA targeting in ORFs, although generally weaker than targeting in 3′UTRs, may still be widely important.
We sought to use a conservation-based approach to analyze miRNA targeting in ORFs. Our goals for this approach were twofold: (i) to compare the extent of conserved targeting in ORFs to that in 3′UTRs and (ii) to provide a tool to guide researchers in identifying the most likely ORF targets. Preferential conservation of miRNA seed sites has previously been observed in ORFs in both vertebrates (22) and Drosophila (23), but it has been difficult to fully analyze the extent of conserved ORF targeting and to provide confident predictions of individual ORF targets. The main difficulty encountered in such an effort is that traditional techniques based on conservation are not designed for application to coding DNA. Coding DNA is already under strong selective pressure at the amino acid level, which results in high and strongly biased conservation at the nucleotide level. We therefore developed an algorithm, MinoTar (www.minotar.csail.mit.edu), to identify conserved regulatory motifs specifically within protein-coding regions. Our approach shares some similarity with that taken recently in vertebrates by Forman et al. (18, 24), but additionally allows for the scoring of sites not perfectly conserved across all species in the alignment. Because a significant majority (70% or more) of highly preferentially conserved sites are not perfectly conserved, this allows for the confident prediction of a substantially increased set of targets and a more comprehensive comparison to the extent of targeting in 3′UTRs.
Using our algorithm, we show evidence for extensive conserved miRNA targeting in Drosophila ORFs at the scale of conserved targeting in 3′UTRs. Using a reporter assay to test a number of predicted targets, we demonstrate that our approach can recover functional target sites with high confidence. Analysis of functional annotations of our predicted targets and comparison to predicted targets in the 3′UTR provides further evidence that ORF targeting is involved in important biological processes. Further, we extend our analysis to humans and find that the scale of conserved miRNA targeting in ORFs is extensive, but that the extent of ORF and 5′UTR targeting relative to 3′UTR targeting may be greater in Drosophila.
Results
miRNA Seed Sites in Coding Regions Are Highly Conserved.
To assess the evidence for selection on putative miRNA binding sites within coding regions, we developed an algorithm (MinoTar) to score sequences within coding DNA for evidence of preferential conservation. Searching for preferentially conserved regulatory sequences within coding regions presents a unique challenge: Coding DNA is already highly conserved due to selective pressures at the protein level, and such selection may in general be far stronger than any additional selection at the nucleotide level. Furthermore, selection at the protein level causes highly biased conservation patterns, influenced both by the form of the genetic code and by codon bias.
In order to handle this challenge, our approach automatically accounts for and removes bias introduced by conservation at the protein level to find sequences under strong selection at the nucleotide level (for details, see Methods). In brief, our algorithm begins with a multiple species alignment of coding genes from which we remove all overlapping noncoding features that could confound analysis (including overlapping 3′UTRs and 5′UTRs from any transcript). Using this alignment, our algorithm first computes the empirical conservation rates of all codons and partial codons, conditioned on the conservation of the amino acid encoded. The algorithm then uses these empirical rates to build a background model for the nucleotide conservation of any k-mer within coding DNA. By using such conditional conservation rates, the background model automatically takes into account the amino acid context of the k-mer and the observed amino acid conservation, as well as the effects of codon bias. In order to also account for and remove gene-region specific variations in conservation rates, the algorithm first bins regions of coding DNA by conservation level and learns a different background model for each bin. Using this background model, the algorithm then produces a p value for every instance of a k-mer in the alignment, giving the probability under the background model that this k-mer would be conserved in as many species as observed. The algorithm also calculates the minimum possible p value, giving the probability that the k-mer would be conserved in as many species as is possible given the amino acid sequence. This second score gives a measure of the limit of information that can be inferred from conservation at that location and allows for the exclusion of sites where nothing can be inferred: for example, when there is no freedom of codon choice and nucleotide conservation is completely accounted for by amino acid conservation.
We began by running our algorithm on every instance of all 8-mers within Drosophila protein-coding genes and found that miRNA seeds accounted for the majority of the most highly conserved 8-mers. To evaluate this, we formed a conservation score for each 8-mer, given by the fraction of instances with p value below 0.05. Most 8-mers had scores close to 0.05 expected by chance, but a small group showed significantly more conservation (Fig. 1A). By grouping the top 26 most conserved 8-mers into 10 motifs, we observed that 8 of these 10 motifs correspond to miRNA seeds and 2 to other unknown motifs (Table S1).
Fig. 1.

MicroRNA seed sites are among the most highly conserved motifs in coding regions. (A) Histogram of conservation scores for all 65,536 8-mers. Nearly all of the top-conserved 8-mers correspond to miRNA seed sites. (B) Cumulative plot of scores for different sets of 8-mers. Shown are all 8-mers (black), conserved Drosophila miRNA seeds (red), reverse complements of these seeds (green), 8-mers with identical dinucleotide content to these seeds (cyan), nonconserved Drosophila miRNA seeds (blue), and conserved human miRNA seeds (magenta). (C) Imposing increasingly stringent conservation cutoff results in higher signal-to-background ratios for the set of Drosophila conserved miRNA seeds, whereas control sets behave as background at all cutoffs. (D) Conservation of seed sites accompanied by 3′ base-pairing to the miRNA starting at different positions within the miRNA, as compared to the background conservation of seed sites.
We next verified that the increased conservation observed for miRNA seeds could not be explained by other sequence characteristics of these seeds. To do this, we formed five sets of 8-mers: seeds for “conserved” miRNAs (those miRNAs largely present across all 12 Drosophila species), and four control sets, (i) reverse complements of these seeds, (ii) 8-mers with identical dinucleotide content as these seeds, (iii) seeds for “nonconserved” miRNAs (those not found beyond the melanogaster subgroup), and (iv) seeds for human miRNAs. We plotted the cumulative distribution of conservation scores for these five sets, as well as the set of all 8-mers (Fig. 1B). Although seeds for conserved miRNAs showed a very significant bias to be highly conserved, the four control sets all behaved similarly to the set of all 8-mers, providing further evidence that the increased conservation seen was indeed evidence of selection on miRNA-target sites.
We next found that our algorithm could produce very high-confidence target predictions. To test this, we investigated the effect of an increasingly stringent cutoff on the confidence of predicted sites at that cutoff. We pooled seeds for all conserved miRNAs together and calculated the fraction of instances of these seeds with p values below each cutoff, repeating the same for the four control sets of 8-mers defined above. For each cutoff, we then calculated the signal-to-background ratio by dividing by the background fraction of instances reaching each cutoff (Fig. 1C). At the most stringent cutoffs, a signal-to-background ratio of over 10 (confidence > 90%) could be achieved, producing a set of hundreds of very high-confidence targets. The four control sets showed signal-to-background ratios near 1 at all cutoffs.
Finally, we evaluated the evidence for increased functionality of miRNA seed sites, when such sites are accompanied by additional 3′ pairing to the miRNA. Following ref. 13, we grouped seed sites according to the extent of additional 3′ pairing starting at different positions within the miRNA and calculated the fraction of such sites with p value below a cutoff of 0.05. Signal to background was computed by comparing this fraction to the expected fraction of seed sites reaching such a cutoff (see Methods). We found that although the majority of conserved seed sites are not accompanied by extensive additional 3′ pairing, those sites with such pairing showed some evidence of increased conservation (Fig. 1D). In particular, as has been observed in 3′UTRs (13), those seed sites with contiguous 4-mer or 5-mer base pairing beginning at positions 12–14 of the miRNA showed a statistically significant increase in conservation (number of conserved seed sites: 2,094 total; 4-mers: 178 vs. 145 expected, p < 0.002; 5-mers: 51 vs. 38 expected, p < 0.01; χ2 test).
The Extent of miRNA Targeting in ORFs, 3′UTRs, and 5′UTRs.
We next compared the extent of evidence for miRNA targeting in ORFs, 3′UTRs, and 5′UTRs. For 3′UTRs and 5′UTRs, we performed an analysis similar to that done previously in 3′UTRs (25): Conservation of a miRNA seed site was judged by the number of species within the alignment the site was conserved to, and background conservation rates were estimated by nucleotide-matched background sets (see Methods).
We characterized the level of conservation by the fraction of potentially conserved sites that showed conservation above background level (Fig. 2A). The fraction of ORF sites preferentially conserved was about 60% that in 3′UTRs, whereas the fraction of sites in 5′UTRs was about 60% that in ORFs. Though miRNA seed sites are denser in AT-rich 3′UTRs than in ORFs, because of the significantly larger size of ORFs and smaller size of 5′UTRs (roughly 2 × 107 total bases in ORFs, 7 × 106 total bases in 3′UTRs, and 2.5 × 106 total bases in 5′UTRs), the number of preferentially conserved sites in ORFs and 3′UTRs was very similar (∼7,000 sites within 3′UTRs vs. ∼6,500 in ORFs), whereas the number in 5′UTRs was significantly smaller (∼700 sites) (Fig. 2B).
Fig. 2.

The scale of conserved miRNA targeting in 3′UTRs, ORFs, and 5′UTRs. (A) Fraction of sites conserved above background for both 8-mers and 7-mers in 3′UTRs, ORFs, and 5′UTRs. (B) Number of predicted sites above background for 8-mers and 7-mers in 3′UTRs, ORFs, and 5′UTRs. Error bars show standard deviation in the estimates obtained from sampling of background sets (see Methods).
The extent of conservation of miRNA seed sites in ORFs varied considerably between different miRNAs. Those with the most conserved sites tended to be well known and highly expressed miRNAs, whereas those with few conserved sites tended to be more recently discovered and expressed at lower levels. This suggested that the levels of conservation correlate with the number of targets each miRNA has acquired, with more widely and highly expressed miRNAs tending to have acquired more targets. To provide support for this, we compared the level of conservation of individual miRNA seeds within ORFs, 3′UTRs, and 5′UTRs. If conservation levels reflect the number of acquired targets, then we surmised that the miRNAs with the most conserved targets in each region should largely agree. We looked at 8-mer seeds and chose a cutoff for ORFs and 3′UTRs that gave predictions at 60% confidence (p = 0.05 for ORFs, conservation to 8 out of 12 species for 3′UTRs). For 5′UTRs, we used the same cutoff as for 3′UTRs, as 60% confidence was not possible to achieve. For each seed, we compared the fraction of instances with conservation above background in the different regions (Fig. 3 A and B). The level of conservation in both ORFs and 5′UTRs was highly correlated to those in 3′UTRs: Mean conservation above background in ORFs and 5′UTRs was significantly higher for the top 50% most conserved miRNA seeds in the 3′UTR than for the bottom 50% (ORFs: 0.14 vs. 0.02, p < 10-7; 5′UTRs: 0.10 vs. 0.01, p < 3 × 10-4; Mann–Whitney U test). To ensure that we were not observing biases in conservation independent of selection due to miRNA targeting or conservation due to overlap with unannotated 3′UTRs, we repeated the same procedure with promoter sequences (500 nucleotides upstream of the transcription start site) (Fig. 3C). Within promoters, miRNA seed sites overall showed no preferential conservation and the top 50% most highly conserved seeds in the 3′UTR showed no tendency to be more conserved than the bottom 50% (-0.01 vs. -0.014, p = 0.32; Mann–Whitney U test).
Fig. 3.

MicroRNA seeds showing the highest level of conservation in 3′UTRs tend also to be the most conserved in ORFs and in 5′UTRs, but not in promoter regions. Shown are the fractions of 8-mer sites conserved above background at 60% confidence cutoff (for 5′UTRs and promoters the cutoff was chosen to be the same as for 3′UTRs) among (A) 3′UTRs and ORFs, (B) 3′UTRs and 5′UTRs, and (C) 3′UTRs and promoters. Dotted vertical and horizontal lines show the cutoff for conservation above background equal to the maximal amount by which any miRNA was conserved below background.
To further analyze the set of predicted ORF targets, we formed a set of genes for each miRNA that had either a 7-mer or 8-mer conserved to the 60% confidence level. We compared these target lists to the set of predicted 3′UTR targets from the TargetScan Web site (25). Although most (> 97%) of our predicted ORF target genes were not predicted by TargetScan to be targeted in their 3′UTR by the same miRNA, genes with a predicted ORF target for one miRNA were significantly more likely to contain a predicted site in their 3′UTR for that miRNA than for any other miRNA (1.7-fold more likely, p < 2 × 10-7, χ2 test), providing evidence for some degree of simultaneous targeting by sites in both the 3′UTR and ORF. For each of the predicted ORF target sets, we searched for significantly enriched Gene Ontology (GO) terms using Amigo Term Enrichment software (26). We found that 37 out of 94 miRNAs had target sets with significantly enriched terms (vs. 70 out of 94 for 3′UTRs), including 21 of the top 25 miRNAs with the most conserved ORF targets. Enrichment terms were significantly more likely to be shared by predicted targets of the same miRNA in ORFs and 3′UTRs than by two different miRNAs (1.9-fold more likely, p < 10-12 χ2 test). Target predictions are available on the MinoTar Web site (www.minotar.csail.mit.edu).
ORF Predictions Recover Functional miRNA Targets with High Confidence.
To test whether predicted ORF target sites could confer substantial down-regulation, we selected six genes with highly conserved seed sites for three different miRNAs: mir-1, mir-8, and mir-6 (a member of the K-Box family). Because one of the genes, Arp87c, was predicted to be targeted by both mir-1 and mir-8, in total we tested seven miRNA-target pairs. For each of these genes, we cloned the ORF into a reporter plasmid and measured down-regulation upon coexpression with the targeting miRNA in S2R+ cells. Briefly, each ORF was fused with one of either a Myc or FLAG epitope tag, while the same ORF with synonymous point mutations in the miRNA seed site was fused with the other tag (Fig. 4A). This allowed for the ORF with wild-type miRNA seed site (ORF-WT) and with the mutated site (ORF-Mut) to be cotransfected and simultaneously visualized on a Western blot using two different secondary antibodies. For quantification, the ratio of ORF-WT to ORF-Mut when transfected with miRNA was compared to the ratio when transfected with a control plasmid. As an additional control, all ORFs were cotransfected with nontargeting miRNAs. All experiments were repeated at least twice under each epitope tag.
Fig. 4.

Experimental verification of target predictions. (A) Illustration of the experiment. Each ORF with wild-type miRNA-target site and the same ORF with mutated site were placed under different epitope tags and coexpressed with either a control plasmid or miRNA, and then run on Western blot. Quantification of down-regulation was made by comparing the ratio of the two channels under miRNA vs. under control plasmid. In all cases, epitope tags were flipped to confirm the effect was consistent under each set of tags. Shown are the bands from a test of CG11178 targeting by mir-1. (B) Down-regulation of target genes. Shown are the fold changes of targets under targeting miRNAs (red bars) as well as under miRNAs not predicted to target the genes (blue bars). Error bars show standard deviation, asterisks denote p values (Student’s t test; **: p < 0.01, ***: p < 0.001). (C) Effect of multiple target sites. Shown is down-regulation of CG8494 by mir-1 with all eight combinations of the three predicted sites (WT sites are marked as + and mutated sites as −), averaged over three separate experiments. Error bars give standard deviation.
Down-regulation was detectable in five out of seven miRNA-target pairs, whereas no nontargeting miRNAs caused down-regulation of any of the genes (Fig. 4B). Four out of seven miRNA-target pairs showed down-regulation greater than 25%, and two out of seven showed greater than 50% down-regulation. The strongest effect was seen for one of the mir-1 targets (CG8494) that contained three seed sites for mir-1 (one 8-mer and two 7-mers). In order to observe the effect of multiple sites within a single gene, we systematically mutated away all three sites for this gene and tested the down-regulation for all eight possible mutated configurations (Fig. 4C). Regression on the observed log fold change showed that down-regulation was largely additive between the targeting sites (R2 = 0.91) with all three sites conferring significant down-regulation (site 1: 13% ± 9%; site 2: 36% ± 8%; site 3: 45% ± 6%; errors give 95% confidence intervals). This suggests that as in 3′UTRs, genes with multiple sites are more likely to be strongly regulated by a miRNA.
Predicted ORF Targets Are Preferentially Down-Regulated.
In order to examine the scale of miRNA targeting in ORFs on endogenous targets, we transfected S2R+ cells with either mir-1 or a control plasmid and compared expression levels using a whole genome microarray. Although microarray analysis allows one to observe effects only at the mRNA and not the protein level, recent results have suggested that changes at the mRNA level capture a significant portion of the effects caused by miRNA targeting (8, 9). We looked at the effect of mir-1 overexpression on four categories of genes: (i) genes with a mir-1 ORF site predicted by our algorithm, (ii) genes with any mir-1 seed site in the ORF, (iii) genes predicted to be targeted by mir-1 in the 3′UTR by TargetScan, and (iv) genes with any mir-1 seed site in the 3′UTR (Fig. 5). Predicted ORF targets were significantly down-regulated vs. the set of all genes (12%, p = 2 × 10-16; K-S test), significantly more down-regulated than genes with a nonconserved seed in their ORF (12% versus 6%, p < 2 × 10-4; K-S test) and showed mean down-regulation about half as strong as predicted 3′UTR sites (12% vs. 24%). These results suggest that, although weaker than 3′UTR targeting, targeting in ORFs is widespread and that conservation can preferentially recover functional ORF sites. Additionally, we looked at the down-regulation of genes with a mir-1 seed site in their 5′UTR. These genes showed mean down-regulation of a similar scale to those with nonconserved seed sites in their ORF (6%, p < 4 × 10-5; K-S test).
Fig. 5.

Predicted ORF sites are preferentially down-regulated. Shown is the cumulative distribution for genes with no sites (black), genes with a conserved ORF site (red), genes with any ORF site (blue), genes with a conserved 3′UTR site (magenta), and genes with any 3′UTR site (cyan). Genes with conserved ORF sites show down-regulation about twice as strong as those with any ORF site, and about half as strong as those with predicted 3′UTR sites.
Extent of Conserved miRNA Targeting in Mammalian ORFs.
We next applied our algorithm to a multiple alignment of vertebrate species with human. As in Drosophila, miRNA seed sites in ORFs accounted for most of the top-conserved 8-mers, were highly conserved overall, and highly conserved sites could be discriminated above background with high (> 90%) confidence, whereas control sets all behaved similarly to the set of all 8-mers (Fig. S1 A–C and Table S2). We also compared our predicted conservation of miRNA seed sites in human ORFs to the conservation observed in multiple species alignments of human 3′UTRs and 5′UTRs (Fig. S2 A and B). Interestingly, compared to Drosophila, there was a larger drop-off in the level of conserved targeting between 3′UTRs and ORFs and between ORFs and 5′UTRs. The fraction of conserved sites above background in ORFs was about 40% that in 3′UTRs, whereas the fraction of conserved sites above background in 5′UTRs was low, and not reliably above zero. The level of conservation was again highly correlated between miRNA sites in 3′UTRs and ORFs, and to a small extent between 3′UTRs and 5′UTRs, but sites in the promoter region showed no similar relationship (Fig. S3 A–C).
Discussion
We have shown that conserved miRNA targeting in Drosophila ORFs occurs at a similar scale to conserved targeting in 3′UTRs. As in 3′UTRs, not all of the contextual features that make some target sites more effective than others are known. However, we have seen that by seeking highly conserved seed matches, our method can recover functional ORF sites with high confidence. Additionally, our results suggest that factors indicative of stronger targeting in 3′UTRs, particularly the presence of multiple seed sites, should also be helpful in finding the most effective ORF sites. And while most predicted ORF targets are not predicted to be 3′UTR targets, the set of genes targeted in both regions show significantly more overlap than expected, suggesting that simultaneous targeting of genes in both regions occurs in some cases.
In general, targeting in ORFs appears to be weaker than 3′UTR targeting. However, given the scale of conserved ORF targeting we observe, it seems likely that a large number of important ORF targets remain to be discovered. For both 3′UTR and ORF targets, it remains an open question how to interpret the vast scale of conserved miRNA targeting observed. With tens to hundreds of preferentially conserved targets per miRNA in the 3′UTR alone, a similar number in ORFs, and the potential for species-specific (27) as well as noncanonical targets (28), the scale of targeting by miRNAs is daunting. It has been suggested that a significant fraction of target sites may impart only modest down-regulation and exist merely to finely tune expression levels (29). Given the weaker strength of targeting in ORFs compared to 3′UTRs, it seems possible that for ORFs an even greater fraction of sites may serve such a purpose.
Interestingly, comparison of our results in Drosophila and in human indicates that the relative importance of targeting in ORFs and 5′UTRs to 3′UTRs may be stronger in Drosophila. The discrepancy is particularly strong for 5′UTRs, where for humans the fraction of miRNA seed sites conserved is quite small, whereas in Drosophila the fraction is about 40% the fraction conserved in 3′UTRs. Indeed, in our mir-1 overexpression microarray experiment, genes with a 5′UTR site for mir-1 showed significant down-regulation, suggesting that many of these sites may be functional. It is still not completely clear what makes targeting in ORFs and 5′UTRs generally weaker than in 3′UTRs, though evidence suggests that miRNA targeting in ORFs can be blocked by translating ribosomes, and it has been speculated that miRNA targeting in 5′UTRs may be blocked by translation initiation factors (12, 13). If interference from the translational machinery is indeed what weakens targeting in ORFs and 5′UTRs, it may turn out that the strength of this effect differs across species. It may also be that the strength of such an effect can be modulated by cellular state, so that ORF and 5′UTR targeting are of greater strength in some contexts than in others.
An intriguing implication of our results is that a significant fraction of coding DNA may be serving noncoding regulatory functions as well. Although miRNA-target sites seem to account for a large part of such “dual-purpose” regions, our results suggest that many important regulatory signals in coding DNA unrelated to miRNAs exist. It has recently been suggested that the genetic code is, in fact, optimal for encoding additional noncoding information (30). Traditionally, genomes have been largely viewed as divided into coding and noncoding DNA and, with a small number of exceptions (for example, see refs. 31 and 32), most computational tools for analyzing regulatory signals in DNA have been designed to work within noncoding DNA. In cases where regions of DNA are serving both a coding and noncoding function, new approaches may need to be developed.
Methods
Alignments and miRNA Sequences
Multiple species alignments and genome annotations for Drosophila (12-way) and human (17-way) were downloaded from the University of California at Santa Cruz (UCSC) genome browser (http://genome.ucsc.edu/). Three fish species were excluded from analysis because of significant nonaligned sequence. For coding regions, genome annotations were used to exclude all regions overlapping a 3′UTR or 5′UTR for any transcript. In all statistical calculations, regions overlapping more than one transcript were used only once to avoid overcounting. For analysis of 5′UTRs, annotations were used to remove all regions overlapping ORFs or 3′UTRs. Alignments of 3′UTRs for both Drosophila and humans were taken from the TargetScan Web site (http://www.targetscan.org). Alignments of promoters (taken as the 500 bp upstream of Transcription Start Sites) were downloaded from the UCSC genome browser. Regions overlapping ORFs, 3′UTRs, or 5′UTRs of any transcripts were removed. Mature miRNA sequences as well as annotations of miRNA conservation were downloaded from the TargetScan Web site.
Background Model for Conservation of k-Mers in ORFs
Our approach scores conservation of k-mers under a null model where codon conservation at adjacent positions is independent, but is conditional on the observed amino acid conservation. This approach only scores nucleotide conservation level highly if the codon choice across species for a given k-mer is significantly more conserved than expected by empirically measured codon conservation rates. The null model is based on empirical codon conservation rates within an N-species alignment. We denote by σi one of the 64 possible codons, AA(σi) the amino acid encoded for by σi, by Sj one of the 2N possible subsets of the N species, and by Cons(x) the subset of species to which the feature x is conserved. For all σi, and all possible pairs of subsets of species, Sj and Sk, we first calculate the fraction of instances of codons perfectly conserved across the subset of species Sj, conditional on the encoded amino acid being conserved across the subset Sk: Prob[Cons(σi)⊃Sj|Cons(AA(σi)) = Sk]. Similarly, we record these probabilities when only a portion of the codon is considered (for instance, only the last two nucleotides rather than all three nucleotides of the codon), for which we also use the label Cons(σi) below. Given a k-mer, the probability under the null model of observing perfect conservation of the k-mer over a subset Sj of species is then given by the probability that all overlapping or partially overlapping codons were simultaneously conserved on Sj: 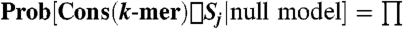 Prob[Cons(σi)⊃Sj|Cons(AA(σi))], where the product is taken over all overlapping full or partial codons. In cases where there is partial overlap with a codon, it is possible for the partial codon to be conserved even when the encoded amino acid is not. In this case the conditional probability Prob[Cons(σi)⊃Sj|Cons(AA(σi))] is taken as the product over terms for subsets of Sj within which the amino acid is fixed. From Prob[Cons(k-mer)⊃Sj] for all subsets Sj, we calculate the probability under the null model (p value) of conservation of the k-mer to M out of N species. Given a k-mer, we find the p value (p), calculated for the actual number of species to which that k-mer is conserved, and the smallest achievable p value (pmin), calculated for the maximum number of species to which the k-mer could have been conserved given the observed amino acid sequences.
Prob[Cons(σi)⊃Sj|Cons(AA(σi))], where the product is taken over all overlapping full or partial codons. In cases where there is partial overlap with a codon, it is possible for the partial codon to be conserved even when the encoded amino acid is not. In this case the conditional probability Prob[Cons(σi)⊃Sj|Cons(AA(σi))] is taken as the product over terms for subsets of Sj within which the amino acid is fixed. From Prob[Cons(k-mer)⊃Sj] for all subsets Sj, we calculate the probability under the null model (p value) of conservation of the k-mer to M out of N species. Given a k-mer, we find the p value (p), calculated for the actual number of species to which that k-mer is conserved, and the smallest achievable p value (pmin), calculated for the maximum number of species to which the k-mer could have been conserved given the observed amino acid sequences.
Binning Gene Regions by Conservation Level.
A background model for codon conservation was first trained on all ORF sequences, which was used to produce a p value at every codon instance in all genes. Every codon instance was then assigned a region conservation score given by the mean of the p values in a window of 120 nucleotides (40 amino acids) centered at that codon. Codons were then sorted by their region conservation scores and placed according to these scores into five equally spaced bins. A set of background models was relearned separately for each of these bins. When evaluating k-mer conservation, a region conservation score was evaluated for the 120 nucleotides centered at that k-mer, and the bin corresponding to that score was used as the background model.
Assessing Conservation Levels in ORFs and Noncoding Regions.
In ORFs, the conservation rate of a set of k-mers for a given cutoff value pcutoff was determined by the fraction of instances achieving p < pcutoff among those instances with pmin < pcutoff. Similarly, in noncoding regions the conservation rate of a set of k-mers was assessed by the fraction of instances conserved to at least M out of N species among those instances with aligned sequence in at least M out of N-species. In both cases, background sets of k-mers, consisting of all k-mers with identical nucleotide content as each of the k-mers in the true set, were used to judge expected background levels of conservation. Conservation above background was measured by the signal-to-background ratio, defined by Sig-Bgd = (fraction conserved true set)/(fraction conserved background set). Confidence at a given cutoff was calculated as (Sig-Bgd - 1)/Sig-Bgd. Errors for fractions and numbers of sites above background were estimated by repeating the analysis 50 times with background sets of size equal to the set of miRNA seeds.
3′ Binding to miRNAs.
miRNA seed sites were grouped based on their potential binding to the 3′ end of corresponding miRNAs following ref. 13 (defined by contiguous base-pairing starting at positions 9–16 within the miRNA, and allowing for shifts of up to two nucleotides of such matches within the mRNA). In cases of seed families with multiple miRNAs, the member with greatest potential base pairing was chosen. Signal to background was defined as the fraction of seed sites with given 3′ binding conserved to the 60% confidence threshold divided by the expected fraction conserved to this threshold. Expected conservation was found by repeating the above procedure while swapping miRNA seed sites with the 3′ ends of all other miRNAs.
GO-term enrichment.
The AmiGO GO-term enrichment tool (http://amigo.geneontology.org/cgi-bin/amigo/term_enrichment) was used to search for statistically enriched GO terms in sets of genes. Thresholds were corrected p-value of 0.05, minimum of two gene products.
Experimental Methods.
Experimental methods are described in SI Methods.
Supplementary Material
Acknowledgments.
We thank Dave Bartel, Carl Novina, and members of the Perrimon and Berger labs for helpful discussions. M.S.L. was supported by a Hertz Foundation Fellowship and an National Defense Science and Engineering fellowship. N.P. is an investigator of the Howard Hughes Medical Institute. This work was partially supported by funding from the Starr Foundation.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1006172107/-/DCSupplemental.
References
- 1.Bushati N, Cohen SM. microRNA Functions. Annu Rev Cell Dev Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- 2.He L, Hannon GJ. MicroRNAs: Small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 3.Bartel DP. microRNAs: Target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krek A, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 6.Brennecke J, Stark A, Russel RB, Cohen SM. Principles of microRNA-target recognition. PLoS Biol. 2005;3:0404–0418. doi: 10.1371/journal.pbio.0030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kertesz M, Iovino M, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007;39:1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 8.Baek D, et al. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Selbach M, et al. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 10.Hafner M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chi SW, Zang JB, Mele A, Darnell AM. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gu S, Jin L, Zhang F, Sarnow P, Kay MA. Biological basis for restriction of microRNA targets to the 3′ untranslated region in mammalian mRNAs. Nat Struct Mol Biol. 2009;16:144–150. doi: 10.1038/nsmb.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grimson AG, et al. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elcheva I, Goswami S, Noubissi FK, Spiegelman VS. CRD-BP protects the coding region of βTrCP1 mRNA from miR-183-mediated degradation. Mol Cell. 2009;35:240–246. doi: 10.1016/j.molcel.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duursma AM, Kedde M, Schrier M, Le Sage C, Agami R. miR-148 targets human DNMT3b protein coding region. RNA. 2008;14:872–877. doi: 10.1261/rna.972008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tay Y, Zhang J, Thomson AM, Lim B, Rigoutsos I. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature. 2008;455:1124–1129. doi: 10.1038/nature07299. [DOI] [PubMed] [Google Scholar]
- 17.Easow G, Teleman AA, Cohen SM. Isolation of microRNA targets by miRNP immunopurification. RNA. 2007;13:1198–1204. doi: 10.1261/rna.563707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forman JJ, Legesse-Miller A, Coller HA. A search for conserved sequences in coding regions reveals that the let-7 microRNA targets Dicer within its coding sequence. Proc Natl Acad Sci USA. 2008;105:14879–14884. doi: 10.1073/pnas.0803230105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen WF, Hu YL, Uttarwar L, Passegue E, Largman C. MicroRNA-126 regulates HOXA9 by binding to the homeobox. Mol Cell Biol. 2008;14:4609–4619. doi: 10.1128/MCB.01652-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orom UA, Nielsen FC, Lund AH. MicroRNA-10a binds the 5'UTR of ribosomal protein mRNAs and enhances their translation. Mol Cell. 2008;30:460–471. doi: 10.1016/j.molcel.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 21.Lytle JR, Yario TA, Steitz JA. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5′ UTR as in the 3′ UTR. Proc Natl Acad Sci USA. 2007;104:9667–9672. doi: 10.1073/pnas.0703820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are MicroRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 23.Stark A, et al. Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature. 2007;450:219–232. doi: 10.1038/nature06340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forman JJ, Coller HA. The code within the code: MicroRNAs target coding regions. Cell Cycle. 2010;9:1533–1541. doi: 10.4161/cc.9.8.11202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kheradpour P, Stark A, Roy S, Kellis M. Reliable prediction of regulator targets using 12 Drosophila genomes. Genome Res. 2007;17:1919–1931. doi: 10.1101/gr.7090407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carbon S, et al. AmiGO: Online access to ontology and annotation data. Bioinformatics. 2009;25:288–289. doi: 10.1093/bioinformatics/btn615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen K, Rajewsky N. Natural selection on human microRNA binding sites inferred from SNP data. Nature Genet. 2006;38:1452–1456. doi: 10.1038/ng1910. [DOI] [PubMed] [Google Scholar]
- 28.Lal A, et al. miR-24 inhibits cell proliferation by targeting E2F2, MYC, and other cell-cycle genes via binding to “seedless” 3′UTR microRNA recognition elements. Mol Cell. 2009;35:610–625. doi: 10.1016/j.molcel.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bartel DP, Chen CZ. Micromanagers of gene expression: The potentially widespread influence of metazoan microRNAs. Nat Rev Genet. 2004;5:396–400. doi: 10.1038/nrg1328. [DOI] [PubMed] [Google Scholar]
- 30.Itzkovit S, Alon U. The genetic code is nearly optimal for allowing additional information within protein-coding sequences. Genome Res. 2007;17:405–412. doi: 10.1101/gr.5987307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen H, Blanchette M. Detecting non-coding selective pressure in coding regions. BMC Evol Biol. 2007;7(Suppl 1):S9. doi: 10.1186/1471-2148-7-S1-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kural D, Ding Y, Wu JT, Korpi AM, Chuang JH. COMIT: Identification of noncoding motifs under selection in coding sequences. Genome Biol. 2009;10:R133. doi: 10.1186/gb-2009-10-11-r133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.