

Abstract
We explored whether modulation of the estrogen receptor (ER) signaling is possible through an aryl hydrocarbon receptor nuclear translocator (Arnt)-dependent mechanism. We utilized the Arnt-interacting protein 2 (Ainp2) to examine whether the presence of Ainp2 in MCF-7 cells would interfere with the Arnt-mediated ER signaling. We found that Arnt increased the 17 beta-estradiol (E2)-dependent luciferase activity and Ainp2 significantly suppressed this Arnt-mediated luciferase activity. Ainp2 significantly suppressed 25% of the E2- and Arnt-dependent upregulation of the GREB1 message. No suppression of the ER target gene expression by Ainp2 was detected in Arnt-knockdown MCF-7 cells and in Arnt-independent ER signaling. Although Ainp2 did not interact with ER alpha and ER beta, it suppressed the ER alpha::Arnt interaction and reduced the E2-driven recruitment of Arnt to the GREB1 promoter. We concluded that Ainp2 suppresses the ER signaling by not allowing Arnt to participate in the ER-dependent, Arnt-mediated activation of gene transcription.
Keywords: estrogen receptor, Arnt, Ainp2, Arnt-interacting protein
1. Introduction
Arnt belongs to the basic-helix-loop-helix-PAS protein family and functions as an essential dimerization partner in the aryl hydrocarbon receptor (AhR) [1] and the hypoxia inducible factor-1α (HIF-1α) [2] signaling pathways. Upon ligand binding, AhR translocates into the nucleus, dimerizes with Arnt to form the AhR::Arnt heterodimer and in turn up-regulates the transcription of drug metabolizing genes such as CYP1A1 and CYP1A2 [3]. Similarly under hypoxia, HIF-1α dimerizes with Arnt and binding of this heterodimer to the HIF-1 response element triggers the upregulation of the transcription of genes such as VEGF [4], EPO [5] and several other glycolytic enzymes [6]. Our laboratory is interested in studying the Arnt-dependent signaling pathways. In an effort to identify essential proteins that play a role in these pathways, we used an Arnt construct as the bait to screen for interacting peptides from a human liver cDNA library using the phage display strategy and subsequently identified a novel Arnt-interacting protein Ainp2 [7]. Ainp2 contains 77 amino acids and is expressed predominantly in human liver and Jurkat cells and it enhances the 3-methylchloranphrene (3-MC)-induced expression of CYP1A1 in HepG2 cells [7].
The estrogen receptor (ER) is a member of the steroid receptors which belong to the superfamily of nuclear receptors [8]. These steroid receptors share three homologous domains: the N-terminal transactivation domain, the central DNA-binding domain and the C-terminal ligand-binding domain [9]. Upon treatment of the ligand 17β-estradiol (E2), the activated ER binds to the estrogen response elements (ERE), followed by activation of target genes such as pS2 [10], GREB1 [11], c-fos [12], adrenomedullin [13], and cathepsin D [14]. The overall transcriptional activation of ER-responsive genes requires receptor dimerization, conformational change in the ligand binding domain [15] and recruitment of coactivators such as the p160 coactivators (SRC-1, SRC-2, and SRC-3) [16-18], PPARBP [19], and the SWI/SNF chromatin remodeling complex [20]. This activation process is understandably complex; abnormal signaling may lead to induction and progression of different cancers including the ER-positive breast cancer [21, 22].
Mechanisms for the cross-talk between AhR and ER signaling pathways are complex as well. AhR ligands have been shown to affect the ER signaling through AhR dependent mechanisms [23, 24]. However, activation of the ER signaling by 3-MC– an AhR ligand – has been reported to be AhR independent [25] and it appears that 3-MC is also an ERα ligand [26]. One of these cross-talk mechanisms that involves Arnt can be observed at the pS2 promoter: Binding of liganded AhR::Arnt heterodimer to the DRE-like motif at −527 to −514 inhibits the ER signaling, possibly by interfering with the binding of the ER homodimer to the ERE [27]. Interestingly, Arnt acts as a potent coactivator for the ER signaling and it interacts with ER through the E2-activated ligand binding domain [28]. The function of Arnt in the ER signaling may involve the interactions with some other coactivators including p300 [24] and two coiled coil coactivators (TRIP230 and CoCoA) [29]. Here we showed that Ainp2 suppresses both the E2-dependent ERE-driven luciferase activity and the up-regulation of the GREB1 gene through an Arnt-dependent mechanism in MCF-7 cells. This Arnt-mediated mechanism reveals a novel approach for developing agents that might be effective for the treatment of the ER-positive breast cancer.
2. Materials and methods
2.1 Cell line and reagents
MCF-7 cells were grown in the media of either Advanced DMEM/F12 (Gibco, Carlsbad, CA) plus 5% HyClone FBS (Thermo Scientific, Waltham, MA) or DMEM (Sigma, St. Louis, MO) plus 10% FBS (Tissue Culture Biologicals, Tulare, CA) which is supplemented with 0.2mM L-glutaMAX, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. All cell culture reagents were purchased from Invitrogen (Carlsbad, CA) if not specified. Lipofectamine 2000 was purchased from Invitrogen (Carlsbad, CA). Fugene HD was purchased from Roche (Indianapolis, IN). The β-galactosidase plasmid pCH110 was purchased from Amersham Pharmacia (Piscataway, NJ). The pCMV-Tag4 plasmid was purchased from Stratagene (La Jolla, CA). Charcoal dextran-stripped FBS was purchased from Gemini (West Sacramento, CA). The Dual-Light luciferase assay kit was purchased from Applied Biosystems (Foster City, CA). Anti-Arnt rabbit IgG (H-172), anti-ERα mouse IgG (ER1D5), anti-His mouse IgG (sc-8036) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit IgG was purchased from Sigma (St. Louis, MO). Dynabeads protein G and DynaMag-2 magnet were purchased from Invitrogen (Carlsbad, CA). Odyssey blocking buffer and IRDye 800CW conjugated goat anti-mouse IgG were purchased from LI-COR Biosciences (Lincoln, NE). The pCMV-Tag4-Ainp2 plasmid was generated by cloning the Ainp2 cDNA into the BamHI/XhoI sites of the pCMV-Tag4 plasmid. The pCMV-GST plasmid was generated by amplifying the GST cDNA from the pET42b plasmid (EMD Biosciences Novagen, Gibbstown, NJ) and then inserting the cDNA into the EcoRI/HindIII sites of the pCMV-Tag4 plasmid. The pCMV-Tag4-GST-Ainp2 plasmid was generated by amplifying the Ainp2 cDNA and then inserting the cDNA into the BamHI/XhoI sites of the pCMV-Tag4-GST plasmid. Protein concentrations were determined by the BCA assay (Pierce, Rockford, IL). IRDye700 conjugated ERE (OL489, 5′-IRD700/GGATCTAGGTCACTGTGACCCCGGATC-3′; OL490, 5′-IRD700/GATCCGGGGTCACAGTGACCTAGATCC-3′) and IRDye700 conjugated DRE (OL439, 5′-IRD700/TCGAGTAGATCACGCAATGGGCCCAGC-3′; OL440, 5′-IRD700/TCGAGCTGGGCCCATTGCGTGATCTAC-3′) were purchased from Integrated DNA Technologies (San Diego, CA).
2.2 Luciferase assay
Either Fugene HD or Lipofectamine 2000 was used in our transfection experiments. For studies using Lipofectamine 2000, MCF-7 cells were grown to 90% confluence in a 24-well plate. For each well, the cells were incubated with a transfection mixture of 200 μl of OPTI-MEM containing Lipofectamine 2000 (2 μl/well) and various plasmids (0.2 μg of pVit-ERE-Luc, 25 ng of pCH110, 1 μg of pCMV-Tag4 or pCMV-Tag4-Ainp2) at 37 °C. After 5 h, the wells were washed HyQ HBSS twice and then incubated with 1 ml of phenol red-free DMEM/F12 supplemented with 10% charcoal dextran-stripped FBS and 0.2 mM L-glutaMAX in the presence of 10 nM E2 or vehicle (80% ethanol/20% DMSO) at 37 °C for 18 h. The luciferase assay was then performed using the Dual-Light kit as described previously [7]. For studies using Fugene HD, MCF-7 cells were pre-treated for 48 h in phenol red-free DMEM complete media with 10% charcoal dextran-stripped FBS before being seeded in a 24-well plate (105 cells/well). On the next day, cells (about 85% confluent) were transfected with 1 μg of plasmids/2.5 μl Fugene HD/well. Plasmids contained 50 ng of the pVit-ERE-Luc reporter plasmid, 25ng of the β-gal plasmid pCH110, and 925 ng of various plasmid combinations. After 24 h, cells were treated with either 10 nM E2 or vehicle (80% ethanol/ 20% DMSO) in phenol red-free media with no FBS for another 24 h before being harvested for luciferase assay using the Dual-Light kit. Each of the luciferase activity was normalized by the corresponding β-galactosidase activity.
2.3 Real-time qPCR
MCF-7 cells were grown to 90% confluence in a 6-well plate. A transfection mixture of 500 μl of OPTI-MEM containing Lipofectamine 2000 (6 μl) and a plasmid (4 μg, pCMV-Tag4, pCMV-Tag4-Ainp2, pSport or pSport-Arnt) was added to each well, followed by an incubation at 37 °C for 5 h. After that, the wells were washed with HyQ HBSS twice and then incubated with 2 ml of phenol red-free DMEM/F12 supplemented with 10% charcoal dextran-stripped FBS and 0.2 mM L-glutamate in the presence of 10 nM E2 or vehicle (80% ethanol/20% DMSO) at 37 °C for 18 h. After induction, the total RNA was isolated using the MasterPure kit (Epicentre, Madison, WI). Reverse transcription was performed using 1 μg of RNA and MMLV reverse transcriptase (Epicentre, Madison, WI) in a 50 μl reaction where 2 μl of that reaction mix was then used for real-time qPCR using the SYBR Green FailSafe kit (Epicentre, Madison, WI) and an Opticon II Real-time PCR Detection System (MJ Research) as previously published [7]. The PCR conditions were as follows (see Table I for primer sequences): for pS2/18S (35 cycles): 30 sec at 94 °C, 30 sec at 49 °C for pS2 and 55 °C for 18S, then 45 sec at 70 °C; for GREB1/18S (35 cycles): 30 sec at 94 °C, 30 sec at 54 °C for pS2 and 55 °C for 18S, then 45 sec at 70 °C. The final PCR products were also analyzed by agarose gel electrophoresis. Relative abundance of the pS2 or GREB1 transcript was determined by the cycle threshold (CT) according to the equation and normalized to the 18S standard.
Table 1.
Sequence-specific PCR primers used for SYBR Green real-time qPCR and chromatin immunoprecipitation experiments.
| Arnt sense (OL 144) | 5′-GAATTGGACATGGTACCAGG-3′ |
| Arnt antisense (OL 145) | 5′-AAGCTGATGGCTGGACAATG-3′ |
| GREB1 sense (OL213) | 5′-CAAAGAATAACCTGTTGGCCCTGC-3′ |
| GREB1 antisense (OL214) | 5′-GACATGCCTGCGCTCTCATACTTA-3′ |
| pS2 sense (OL 196) | 5′-GCCCAGACAGAGACGTGTACA-3′ |
| pS2 antisense (OL 197) | 5′-TCACACTCCTCTTCTGGAGGG-3′ |
| 18S sense (OL96) | 5′-CGCCCCCTCGATGCTCTTAG -3′ |
| 18S antisense (OL97) | 5′-CGGCGGGTCATGGGAATAAC-3′ |
| ERE3 sense (OL437) | 5′-TGTGCTCAGTGACCCTTGTG-3′ |
| ERE3 antisense (OL438) | 5′CTGCCCCAACAACTGAAAGA-3′ |
| GST-Ainp2 sense (OL480) | 5′-TGGCAAGCCACGTTTGGT-3′ |
| GST-Ainp2 antisense (OL481) | 5′-CTGAAAGCAAGATTTCAG-3′ |
2.4 TALON co-precipitation assay
Full-length cDNA sequences of ERα and ERβ were amplified by PCR from the plasmids pACsk12CMV5-ERα and pACsk12CMV5-ERβ, respectively, and then cloned in the pCMV-Tag4 plasmid for the 35S expression of ERα and ERβ in the rabbit reticulocyte lysate (Promega, Madison, WI). The reticulocyte lysate expressed 35S-ERα, 35S-ERβ or 35S-Arnt (10 μl) was rotated in 800 μl of the co-precipitation buffer (50 mM HEPES, pH 7.4, containing 10% glycerol and 150 mM KCl) with 30 μl of the TALON resin (BD Biosciences Clontech, Mountain View, CA) and 1 mg of BSA in the presence or absence of 150 μl (15 μg) of the TALON-purified baculovirus expressed 6His-Ainp2 [7] at 32 rpm at 4 °C for 2 h. After incubation, the resin was washed four times with 1 ml of the wash buffer (50 mM HEPES at pH 7.4 containing 10% glycerol, 150 mM KCl, 10 mM β-mercaptoethanol and 5 mM imidazole) and then eluted with 30 μl of the SDS-PAGE sample buffer. The samples were separated on a 10% SDS-PAGE gel. After the gel was dried, the 35S-labeled proteins were analyzed by autoradiography.
2.5 Gel shift assay
MCF-7 cells were pre-treated for 48 h in phenol red-free DMEM complete media with 10% charcoal dextran-stripped FBS, followed by either E2 or vehicle treatment for 45 min before harvest. Nuclear extracts were prepared according to a published protocol [30]. Gel shift assay was performed as follows: Gel shift buffer (25 mM HEPES, pH 7.4, 1 mM EDTA, 1 mM DTT, 10% glycerol, 0.1 M KCl) was added to the MCF-7 nuclear extracts (3 μg) in the presence or absence of additional proteins (e.g. baculovirus expressed Arnt or Ainp2) to a final volume of 13 μl. Poly-dIdC (3 μg, 1 μl) was then added. After 20 min at room temperature, IRDye700-conjugated ERE (0.5 pmol, 2 μl) was added. In some cases, IgG was added before the ERE probe addition, followed by an additional 10 min incubation at room temperature. After 20 min at room temperature, 10X orange loading dye (LI-COR, Lincoln, NE) was added before the samples was resolved on a 5% native acrylamide gel (1XTBE) at 4 °C (185 volts) for 2 h. After that, the gel was immediately analyzed using a LI-COR Odyssey imaging system. AhR gel shift assay using baculovirus expressed AhR and Arnt were performed as described previously [Luu, 2008 #1644], except that IRDye700-conjugated DRE (0.5 pmol, 2 μl) was used instead of the 32P probe. For lanes 1-4 in Fig. 3C, Dynabeads protein G (2 μl, 10 min on ice) was added after the IgG incubation step to remove the IgG from the gel shift samples before addition of the IRDye700 ERE probe.
Fig. 3. The Ainp2 effect is mediated through Arnt, but not ERα and AhR.
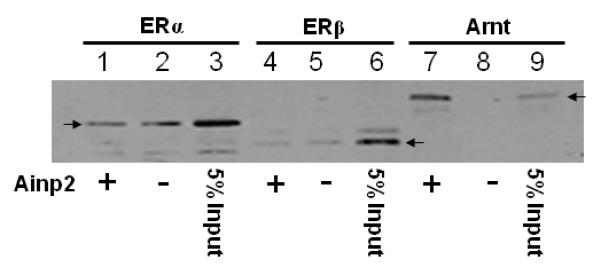
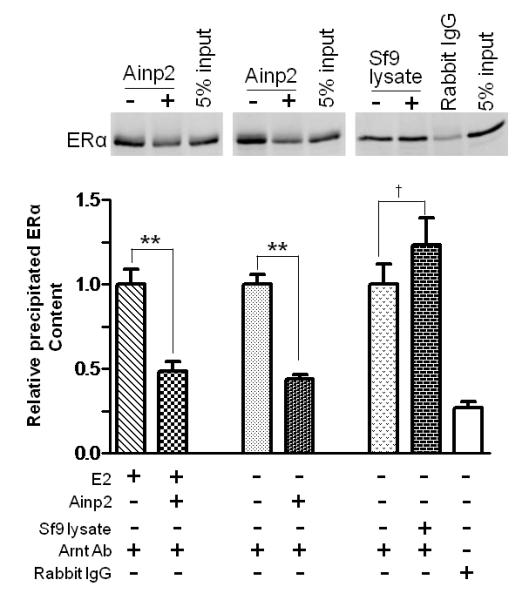
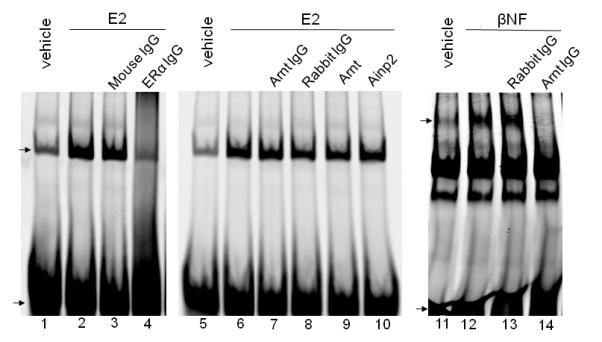
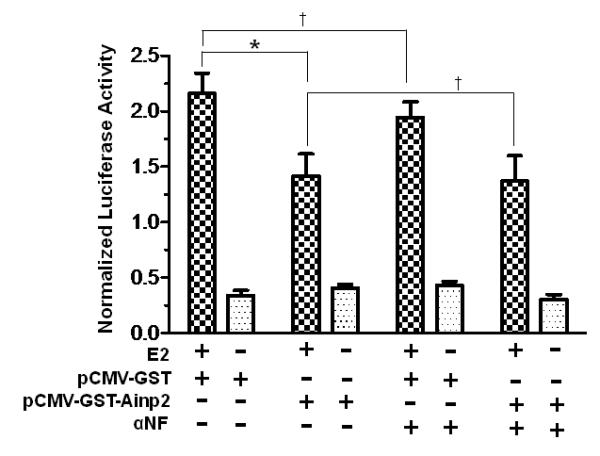
A. TALON co-precipitation assay showing that Ainp2 interacted with Arnt but not ERα and ERβ. The arrows indicate the 35S-ERα (lanes 1-3), 35S-ERβ (lanes 4-6) and 35S-Arnt (lanes 7-9) precipitated in the presence (+) or absence (−) of the baculovirus expressed 6His-Ainp2. This experiment was repeated once with similar results. B. Ainp2 suppressed the co-immunoprecipitation of ERα using anti-Arnt rabbit IgG (H-172) in MCF-7 whole cell lysates (WCL). The co-precipitated samples were reconstituted ± baculovirus expressed 6His-Ainp2 ± Sf9 lysate. Relative precipitated ERα content was calculated by normalizing with the 5% of the starting MCF-7 WCL (5% input). The condition of rabbit IgG in place of anti-Arnt IgG was the negative control. The above images were a representative of the triplicate data. Error bars represent the standard deviation of the means (n = 3, means ± SD). ** p ≤ 0.005, † p > 0.05 (not significant). This experiment was repeated at least once with similar results. C. Ainp2 did not affect the ER/ERE gel shift complex formation. MCF-7 nuclear extract (3 μg) treated with E2 (lanes 2-4, 6-10) or vehicle (lanes 1 and 5) was used. Lanes 1-2 and 5-6 were identical conditions ± E2 of two different gel shift experiments, except that lanes 1-2 were treated with Dynabeads protein G. Lanes 11-14 contained baculovirus expressed AhR (1 μl) and Arnt (0.1 μl) and Sf9 soluble extract (5 μg) ± 10 μM β-napthoflavone (βNF). Different additions to the gel shift samples are as follows: lane 3, mouse anti-His IgG (5 μl); lane 4, mouse anti-ERα IgG (5 μl); lane 7, rabbit anti-Arnt IgG (1 μl); lane 8, rabbit IgG (1 μl); lane 9, baculovirus expressed TALON-purified Arnt (1μl); lane 10, baculovirus expressed TALON-purified Ainp2 (1 μl); lane 13, rabbit IgG (1 μl); lane 14, rabbit anti-Arnt IgG (1 μl). The upper arrow shows the ER/ERE complex or AhR/Arnt/DRE complex whereas the lower arrow show the unbound IRDye700-conjugated ERE or DRE probe. Three gel shift experiments were combined in this figure (lanes 1-4, 5-10, 11-14). These gel shift assays were performed multiple times with similar results. D. Transient transfection results showing that α-napthoflavone did not alter the ER-dependent luciferase experiment ± Ainp2. Samples, ± 10nM E2 treatment, were transfected with the same amount of plasmids of different combinations (pCMV-Tag4-GST/pCMV-Tag4-GST-Ainp2 plasmid of 925 ng total). Transient transfection was performed using Fugene HD. Error bars represent the standard deviation of the means (n = 3, means ± SD). * p ≤ 0.05, † p > 0.05 (not significant).
2.6 RNA interference studies
MCF-7 cells were grown in a 6-well plate to 30-50% confluence before the transfection. Each well was transfected with 160 pmol of human Arnt siRNA (sc-29733, Santa Cruz Biotechnology, Santa Cruz, CA) using Lipofectamine 2000 (6 μl) according to the manufacturer’s protocol. After 48 h, the total RNAs were extracted and subsequent RT-qPCR was performed as described above. PCR reactions were performed as follows: 35 cycles of 30 sec at 94 °C, 30 sec at 50 °C for Arnt and 55 °C for 18S, then 45 sec at 70 °C. To study the effects of Ainp2 under the Arnt-knockdown condition, MCF-7 cells were grown to 50% confluence in a 24-well plate. To each well, a transfection mixture which contained 200 μl of OPTI-MEM I containing Lipofectamine 2000 (2 μl), various plasmids (0.2 μg of pERE-Luc, 25 ng of pCH110, and 1 μg of pCMV-Tag4 or pCMV-Tag4-Ainp2) and 20 pmol of human Arnt siRNA or human integrin α4 siRNA (sc-35685, Santa Cruz Biotechnology, Santa Cruz, CA) was added. After 5 h, cells were washed with HyQ HBSS twice and then incubated with 1 ml of phenol red-free D-MEM/F-12 supplemented with 5% charcoal dextran-stripped FBS and 0.2 mM L-glutamate. After 25 h, cells were treated with 10 nM E2 or vehicle (80% ethanol/20% DMSO) for another 18 h at 37 °C, followed by the luciferase assay as described above.
2.7 Co-immunoprecipitation assay
MCF-7 cells were pre-treated for 48 h in phenol red-free DMEM supplemented with 10% charcoal dextran-stripped FBS. After that, cells were treated with 10 nM E2 or vehicle (80% ethanol/ 20% DMSO) for 24 h at 37 °C. Cells were harvested by resusupending the cells in the lysis buffer (25 mM HEPES, pH 7.4, 1 mM EDTA, 1 mM DTT, 10% glycerol, 0.4 M KCl, 2 μg/ml of leupeptin, and 1 mM PMSF). After three freeze-thaw cycles, the samples were centrifuged at 16,000g for 10 min at 4 °C. The KCl concentration was adjusted to 0.2 M final concentration. The supernatants were the whole cell lysates (WCL) and used for co-immunoprecipitation experiments as follows: MCF-7 WCL (100 μg) were incubated in the presence or absence of other proteins (e.g. 50 μg of TALON-purified baculovirus expressed Ainp2 [7] or Sf9 lysate) at 30 °C for 30 min. Anti-Arnt rabbit IgG H-172 (10 μl) or rabbit IgG was then added. After 30 min at room temperature, 10 μl of Dynabeads protein G was added to the samples. The resulting suspension was rotated at 4 °C for 1 h. With the use of a DynaMag-2 magnet, supernatants were separated and discarded. The beads were then washed with 0.6 ml of the wash buffer (25mM HEPES, pH 7.4, 1 mM EDTA, 1 mM DTT, 10% glycerol and 0.1% Tween-20) four times with 5 min rotation at 4 °C between each wash. After washing, proteins were eluted from the beads using 20 μl of SDS-PAGE sample buffer. Quantitative Western analysis using a LI-COR Odyssey near infrared imaging system was performed to quantify the co-immunoprecipitated ERα protein.
2.8 Quantitative Western analysis
After SDS-PAGE, protein was transferred onto a nitrocellulose membrane using Bio-Rad mini trans-Blot cell for 1.5 h at 4 °C. Membrane was blocked with the Odyssey blocking buffer for 1 h and incubated with anti-ERα mouse IgG (ER1D5, 1:500) overnight at 4 °C in the blocking buffer containing 0.1% Tween-20. The membrane was then washed four times with 1XPBS plus 0.1% Tween-20 (5 min per wash) at room temperature. The washed membrane was then incubated with IRDye 800CW conjugated goat anti-mouse (1:5,000) in the blocking buffer containing 0.1% Tween-20 for 1 h at room temperature in the dark. The membrane was washed four times as described above and then analyzed by a LI-COR Odyssey near infrared imaging system.
2.9 Chromatin Immunoprecipitation assay
MCF-7 cells were grown in phenol red-free DMEM containing 10% of charcoal dextran-stripped FBS for 48 h before experiment. The plasmid (pCMV-GST or pCMV-GST-Ainp2, 20 μg) was introduced into MCF-7 cells (4×106 cells in 0.4 ml of DMEM containing 2.5% of charcoal dextran-stripped FBS) using a BTX ECM 830 electroporator which was set at LV mode, 160 volts for 70 msec. After 48 h on a 100 mm plate at 37 °C, cells were treated with either 10 nM E2 or vehicle (80% ethanol/20% DMSO) for 45 min at 37 °C. After that, cells were cross-linked with 1% formaldehyde in phenol red-free DMEM for 10 min at room temperature. Glycine (125 mM final concentration) was added to quench the reaction. After 10 min at room temperature, cell pellet was harvested, resuspended in lysis buffer (120 μl, 50 mM Tris, pH 8.1, containing 10 mM EDTA, 1% SDS) containing protease inhibitors PMSF (1 mM) and leupeptin (2 μg/ml), and then sonicated in a circulating ice water bath using a Misonix S4000 sonicator equipped with a cuphorn. The sonication setting was as follows: 40% output, 10 sec on/15 sec off for 15 times. The sonicated cell lysis was centrifuged at 16,000g for 10 min at 4 °C. The resulting supernatant was diluted ten times into the dilution buffer (20 mM Tris, pH 8.1, containing 2 mM EDTA, 1% Triton X-100, 150 mM NaCl) containing protease inhibitors (1 mM PMSF and 2 μg/ml of leupeptin) to become the sample for chromatin co-immunoprecipitation assay and an aliquot (50 μl) was set aside as the input. Each sample (1 ml) was precleared with Dynabeads Protein G (3 μl) containing sheared salmon sperm DNA (1 μg) and rabbit serum (2.5 μl, Sigma R9133) by rotating for 2 h at 4 °C. The precleared supernatant was transferred to a tube containing Dynabeads Protein G (10 μl) pre-incubated with anti-Arnt IgG (0.5 μl) for 1 h at 4 °C. After rotating overnight at 4 °C, the pellet was obtained using a DynaMag-2 magnet and then washed sequentially with Buffer I (dilution buffer plus 0.1% SDS), Buffer II (Buffer I except 500 mM NaCl), Buffer III (10 mM Tris, pH 8.1, 1 mM EDTA, 250 mM LiCl, 1% NP40 and 1% deoxycholate), and two times with 10 mM Tris, pH 8, and 1 mM EDTA. Each wash involved 10 min rotation at 4 °C. The washed pellet was resuspended into the elution buffer (50 μl, 0.1 M NaHCO3 and 1% SDS). The resuspended sample (and also the input) was heated at 65 °C for 7 h to reverse cross-linking. The resulting sample was treated with RNaseA (0.5 μl of 10 mg/ml) for 30 min at 37 °C, followed by proteinase K treatment (0.15 μl of 50 mg/ml of proteinase K, 0.65 μl of 0.5 M EDTA, and 1.3 μl of 1M Tris, pH 6.5) for 2 h at 45 °C. DNA was then purified using a Promega Wizard SV gel clean-up kit. PCR was performed using an ERE3-specific primer set ([31], see Table I for primer sequences) and a Perkin Elmer GeneAmp 2400 cycler with the following protocol: 94 °C for 2 min, followed by 40 cycles of 94 °C for 30 sec, 58 °C for 30 sec, and 72 °C for 30 sec. PCR products were visualized and quantified using a LI-COR Odyssey near infrared imaging system after staining the DNA with Syto 60 (LI-COR Biosciences, Lincoln, NE).
2.10 Extract preparation
To determine the Arnt content in nuclear extracts, MCF-7 cells were grown to 90% confluence in a 6-well plate before harvest. The washed cell pellets were resuspended into 100 μl of the cell lysis buffer (25 mM HEPES at pH 7.4 containing 1 mM EDTA, 1 mM DTT, 10% glycerol, 2 μg/ml of leupeptin and 0.5 mM PMSF). After three times of the freeze/thaw cycle, the cell suspension was centrifuged at 16,000g for 30 min. The pellet was then resuspended in 100 μl of the same lysis buffer containing 0.4 M KCl. After on ice for 30 min, the cell suspension was centrifuged at 16,000g for 30 min and the supernatant was the nuclear extract. Soluble extracts from Jurkat and MCF-7 cells were prepared using the CytoBuster reagent (Novagen) according to our published protocol [7]. Lumi-Phos WB chemiluminescent substrate (Pierce, Rockford, IL) was used to detect the Western signals.
2.10 Statistic analysis
We preformed unpaired two-tailed t test using the GraphPad Prism 5 software. Statistically significant data were highlighted by the symbol * (p ≤ 0.05) or ** (p ≤ 0.005) whereas statistically insignificant data were highlighted by the symbol † (p > 0.05).
3. Results
3.1 Effect of Arnt and Ainp2 on the E2-induced ERE-driven luciferase activity
After 10 nM E2 treatment, Arnt increased the luciferase activity by up to 5-fold in a dose-dependent manner (Fig. 1A). This observed enhancement by Arnt was statistically significant and was consistent with the literature that Arnt is a coactivator in the ER signaling [28]. To examine whether Ainp2 would affect this Arnt effect on the ER signaling, we transiently co-transfected the Ainp2 plasmid with the Arnt plasmid into MCF-7 cells and observed the change in the luciferase activity. The results showed that the enhancement effect of Arnt on the E2-dependent luciferase activity was significantly suppressed in the presence of Ainp2 (Fig. 1B). We noticed that the fold-increase by the Arnt transfection was less in Fig. 1B when compared to the data in Fig. 1A. We suspected that the differences were likely contributed by having different combinations of plasmids transfected. In the absence of the transfected Arnt, Ainp2 also suppressed the E2-driven luciferase activity significantly (about 50%) when 1 μg of the Ainp2 plasmid was transfected (Fig. 1C), suggesting that the endogenous Arnt may play a significant role in the E2-driven luciferase expression in MCF-7 cells. In contrast, the luciferase activities in the presence of the Ainp2 plasmid were more than that of the plasmid control (Fig. 1B). This observation was probably due to inadequate Ainp2 expression to suppress the Arnt effect from transient transfection.
Fig. 1.
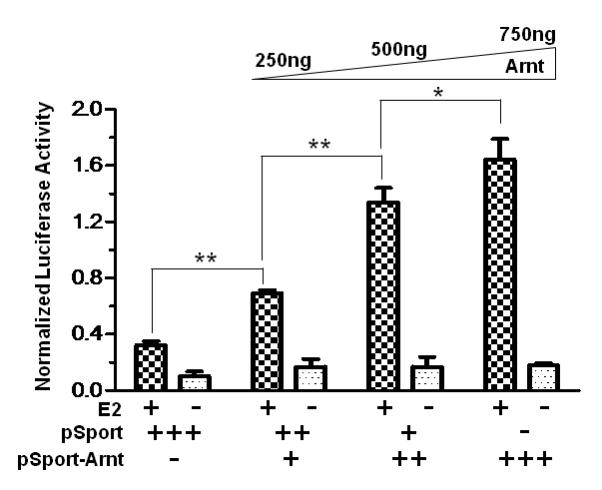
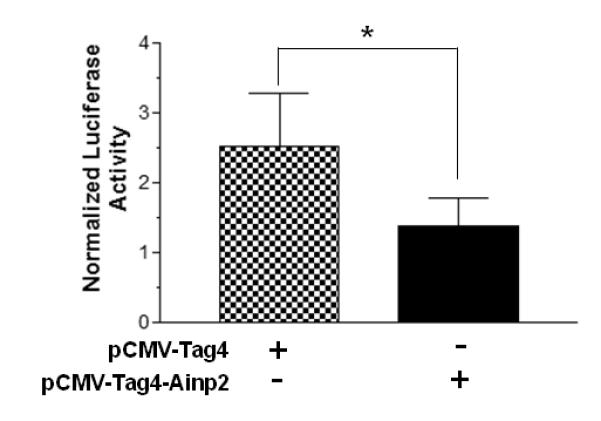
Effect of Arnt and Ainp2 on the ER-mediated luciferase activity in MCF-7 cells. A, Dose-dependent effect of Arnt. Transient transfection was performed using Fugene HD. Each condition was ± 10 nM E2 and the same amount of transfected plasmid (pSport and/or pSport-Arnt). Error bars represent the standard deviation of the means (n = 3, means ± SD). This experiment was repeated at least once with similar results. B. Ainp2 suppressed the Arnt-dependent luciferase activity. Transient transfection was performed using Fugene HD. Each condition was ± 10 nM E2 and the same amount of transfected plasmids of different combinations (pSport, pSport-Arnt, pCMV-Tag4, pCMV-Tag4-Ainp2). Error bars represent the standard deviation of the means (n = 3, means ± SD). This experiment was repeated at least once with similar results. C. Ainp2 suppressed the 10 nM E2-driven luciferase activity. Transient transfection was performed using Lipofectamine 2000. 1 μg of pCMV-Tag4 or pCMV-Tag4-Ainp2 plasmid was transfected. Error bars represent the standard deviation of the means (n = 6, means ± SD). * p ≤ 0.05, ** p ≤ 0.005.
3.2 Effect of Ainp2 on ER target gene expression
We examined the effect of Arnt and Ainp2 on the E2 induction of two ER target genes (GREB1 and pS2) in MCF-7 cells. We found that treatment of 10 nM E2 strongly increased the GREB1 message by about 9-fold; in the presence of Arnt, this induction was further enhanced significantly by 52% and Ainp2 significantly suppressed the E2-induced GREB1 message by 25% (Fig. 2A and B). In the case of the pS2 regulation, however, treatment of 10 nM E2 only upregulated the pS2 message by 1.8-2.4 fold in MCF-7 cells, depending on which empty plasmid was transfected. Moreover, we failed to observe any change in pS2 expression when cells were transfected with either Arnt or Ainp2 (Fig. 2C and D), suggesting that the Ainp2 effect on the ER signaling is Arnt-mediated.
Fig. 2.
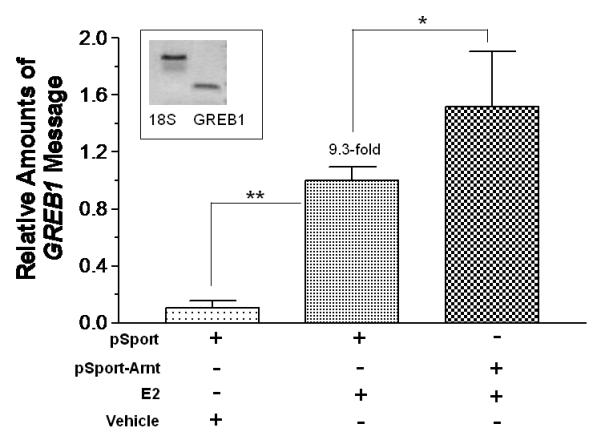
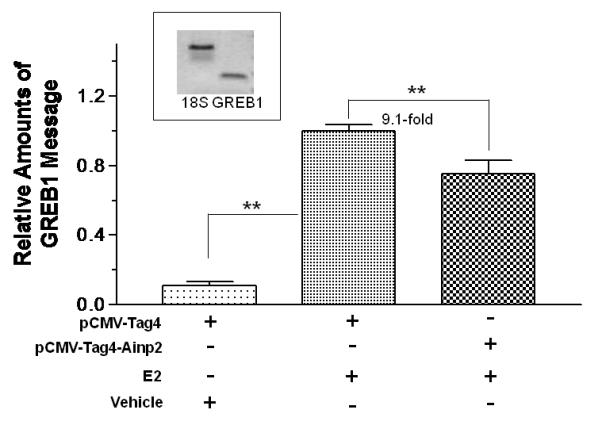
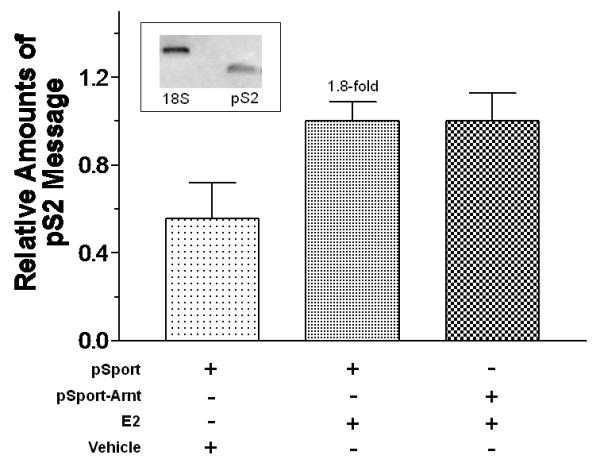
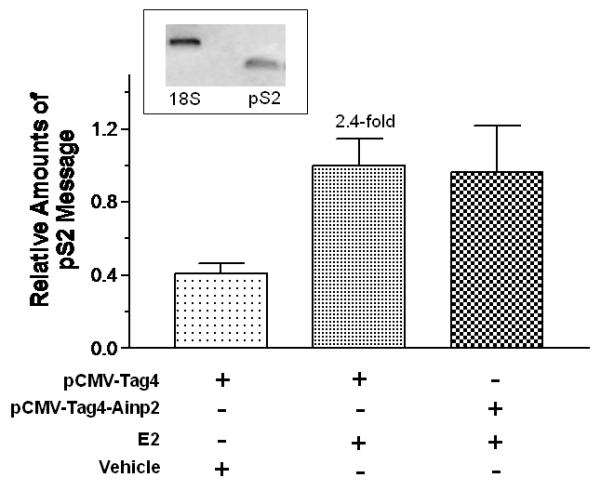
Effects of Arnt and Ainp2 on the E2 induction of the GREB1 and pS2 messages in MCF-7 cells. RT/real-time qPCR showing that Arnt up-regulated (A) and Ainp2 down-regulated (B) the E2-induced GREB1 message and Arnt (C) and Ainp2 (D) did not affect the E2-induced pS2 message. All the messages were normalized by the internal standard 18S. A plasmid (pSport, pSport-Arnt, pCMV-Tag4 or pCMV-Tag4-Ainp2) was transfected into the MCF-7 cells. Error bars represent the standard deviation of the means (A and B, n = 4; C and D, n = 2; means ± SD). All experiments in this figure were performed in 6-well plates and the final PCR products of 18S (376 bp), GREB1 (172 bp) and pS2 (173 bp) are shown. * p ≤ 0.05, ** p ≤ 0.005.
3.3 Ainp2 interacts with Arnt but not ERα and ERβ
To examine whether the suppressive effect of Ainp2 on the ER signaling is mediated through the Ainp2::ER interaction, we investigated whether Ainp2 would interact directly with ERα and ERβ. TALON co-precipitation experiments using the TALON-purified baculovirus expressed 6His-Ainp2 as the bait were performed with in vitro expressed 35S-labeled ERα, ERβ, and Arnt. 6His-Ainp2 co-precipitated the 35S-labeled Arnt (8.5% of the starting 35S-Arnt content) whereas no Arnt was detected in the absence of the bait (Fig. 3A, lanes 7-9). These results were consistent with our previous data that Ainp2 interacts with an Arnt construct CΔ418 in vitro and in HepG2 cells [7]. In contrast, only minimal amounts of 35S-ERα and 35S-ERβ were detected in the precipitated samples and these precipitated ER bands were not Ainp2-dependent (Fig. 3A, lanes 1-6), showing that Ainp2 does not directly interact with ERα and ERβ.
3.4 Ainp2 interferes with the Arnt::ER interaction
Next, we examined whether Ainp2 would interfere with the interaction between Arnt and ERα, realizing that both ERα and Ainp2 interact with Arnt. By performing co-immunoprecipitation experiments, we found that Ainp2 suppressed about 50% of the interaction between Arnt and ERα in the presence or absence of E2 (Fig. 3B). Since Ainp2 used in these experiments was the baculovirus 6His-Ainp2 which was affinity purified using the TALON resin from Sf9 lysate, many Sf9 proteins were co-purified with the Ainp2 protein. To rule out the possibility that the interference of the Arnt::ERα interaction was caused by Sf9 proteins, we performed the co-immunoprecipitation experiment in the presence of the Sf9 lysate. We found that the Sf9 lysate did not interfere with the Arnt::ERα interaction. Rabbit IgG co-precipitated ERα to a much lesser extent, suggesting that the co-precipitation of ERα was Arnt-specific. We concluded that Ainp2 competes with ERα for Arnt binding and the presence of Ainp2 suppresses the interaction between ERα and Arnt.
3.5 Ainp2 does not alter the binding of ERα to ERE in vitro
We performed non-radioactive gel shift assay using near infrared dye (IRDye700) conjugated probe to examine whether Ainp2 would affect the binding of the ERα homodimer to the ERE. We prepared MCF-7 nuclear extracts and observed the E2-dependent gel shift complex (Fig. 3C, lanes 1-2, 5-6). This complex contained ERα but not Arnt because the mouse anti-ERα IgG abolished this complex whereas the rabbit anti-Arnt IgG did not have any effect (lanes 2-4, 6-8). We showed that the same amount of the rabbit anti-Arnt IgG abolished the Arnt-containing AhR gel shift complex (lanes 12-14), confirming that there must not be any Arnt presence in the ER/ERE gel shift complex. Addition of Arnt (10-fold of the amount used for the AhR gel shift complex formation) to the ER gel shift sample did not alter significantly the complex intensity, suggesting that Arnt does not involve in the formation of the ERα/ERE complex (lanes 6, 9). In addition, Ainp2 did not change the ERα/ERE complex (lanes 6, 10), suggesting that Ainp2 does not affect the binding of ERα to the ERE enhancer.
3.6 Suppression of the ER-dependent luciferase expression by Ainp2 is not AhR-dependent
We used α-napthoflavone (αNF) to block the AhR function in MCF-7 cells to address whether AhR is involved in the Ainp2 effect. Transient transfection results showed that 10 μM αNF did not significantly change the E2-activated, ERE-driven luciferase activity (Fig. 3D). In the presence of Ainp2 (925 ng of the transfected Ainp2 plasmid), the luciferase activity was suppressed significantly by 36%, regardless of the presence or absence of αNF, suggesting that AhR does not play a role in the observed suppression of the ER activity by Ainp2.
3.7 Suppression of the ER signaling by Ainp2 is Arnt-dependent
We employed RNA interference to decrease the endogenous Arnt message in MCF-7 cells and re-assessed the effect of Ainp2 on the ER signaling in transient transfection studies. MCF-7 cells were transfected with the siRNA specific to the human Arnt, followed by RT/real-time qPCR and Western analysis to examine the Arnt expression. As we compared the Arnt message between the wild-type and Arnt-knockdown MCF-7 cells, the Arnt message in the Arnt-knockdown cells was decreased by 46% (Fig. 4A) whereas the Arnt protein was undetectable by our chemiluminescent Western analysis (Fig. 4B). The human integrin α4 siRNA was used as the negative control since the integrin α4 message was not detectable in the wild type MCF-7 cells by RT-PCR (data not shown). Data from the luciferase assay showed that treatment with the Arnt siRNA reduced the E2-activated luciferase activity significantly by about 64% (Fig. 4C, lanes 1 and 3). This effect should be Arnt-specific since there was no significant decrease of the luciferase activity when the integrin α4 siRNA was used (Fig. 4C, lanes 1 and 2). When we transfected the Ainp2 plasmid (1 μg) into the wild-type cells, the luciferase activity was suppressed by 42% (Fig. 4C, lanes 1 and 4). When we transfected the Ainp2 plasmid into the Arnt-knockdown cells, Ainp2 could no longer cause a statistically significant change in the luciferase activity (Fig. 4C, lanes 3 and 5). In addition, suppression of the luciferase activity by Ainp2 was essentially the same between the wild-type and Arnt-knockdown cells (Fig. 4C, lanes 4 and 5). These data supported that this Ainp2 effect is Arnt-dependent.
Fig. 4.
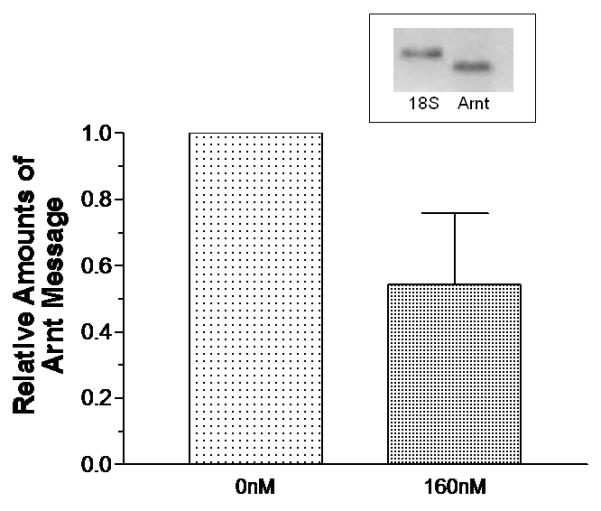
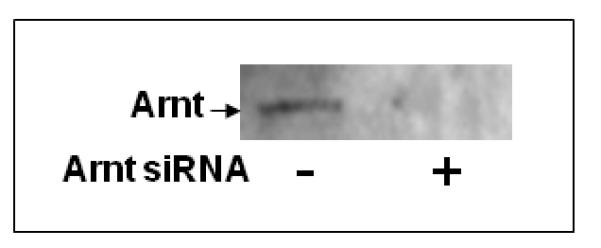
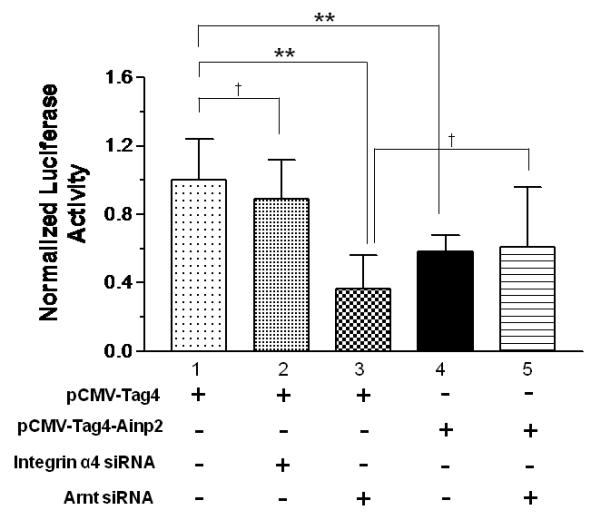
Effect of Ainp2 on the E2-activated, ER-mediated luciferase activity in Arnt knockdown MCF-7 cells. A. RT/real-time qPCR showing a decrease of the endogenous Arnt message after transfection of 160 nM Arnt siRNA. Error bar represents the standard deviation of the means (n = 2, means ± SD) of the amount of Arnt normalized to the corresponding 18S. B. Chemiluminescent Western analysis showing the reduced expression of the Arnt protein after gene silencing. 10 μg of nuclear extracts was separated on a 10% SDS-PAGE gel, followed by the Western blot using the anti-Arnt antibodies (sc-8077, Santa Cruz, 1:500). The arrow indicates the Arnt protein. C. Luciferase assay showing that the Ainp2 effect in the E2-activated ER signaling was Arnt-dependent. Transient transfection was performed using Lipofectamine 2000. All conditions contained 10 nM E2 and the same amount of transfected plasmids of different combinations (pCMV-Tag4 or pCMV-Tag4-Ainp2). The normalized luciferase activity of the empty vector (pCMV-Tag4) was arbitrarily set as 1 and used to calculate the relative luciferase activity. All conditions were performed in six replicates (n =6) except for the control (n = 12). ** p ≤ 0.005, † p > 0.05 (not significant).
3.8 Ainp2 suppresses the recruitment of Arnt to the GREB1 promoter
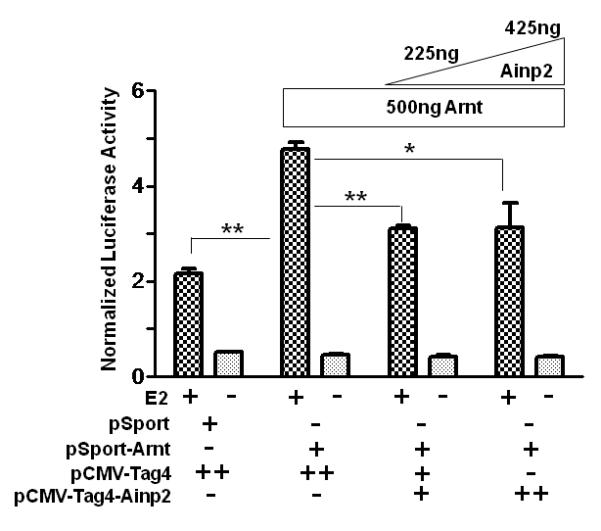
It has been reported that Arnt is recruited to the ERE promoter after E2 treatment and acts as a coactivator [28]. This recruitment likely involves the interaction between ER and Arnt. Realizing that Ainp2 suppresses the Arnt::ERα interaction, we examined whether Ainp2 would inhibit the recruitment of Arnt to an ER-dependent promoter. We used GST-Ainp2 for this study because GST can be used as the negative control, which should be expressed similarly as the GST-Ainp2 fusion protein in MCF-7 cells after transfection. First, we confirmed that GST-Ainp2 was capable of suppressing the Arnt-mediated, E2-driven luciferase activity. We found that GST-Ainp2 suppressed the Arnt-mediated luciferase activity even more so than that of Ainp2, although the fold induction by E2 was less in the presence of GST (Fig. 5A and 1B). Next, we performed chromatin immunoprecipitation experiments using MCF-7 cells that were either wild type or had undergone transient transfection with a plasmid expressing GST-Ainp2 or GST. Using a primer set which selectively amplifies the GST-Ainp2 junction, we observed the expression of the GST-Ainp2 message in MCF-7 cells after transient transfection (Fig. 5B). The chromatin immunoprecipitation results showed that Arnt recruitment by E2 to the ERE3 region of the GREB1 promoter was significantly suppressed by about 50% in the presence of GST-Ainp2 but not GST (Fig. 5C), strongly suggesting that suppression of the Arnt recruitment by Ainp2 leads to reduction of the ER-dependent activation of the GREB1 gene transcription.
Fig. 5.
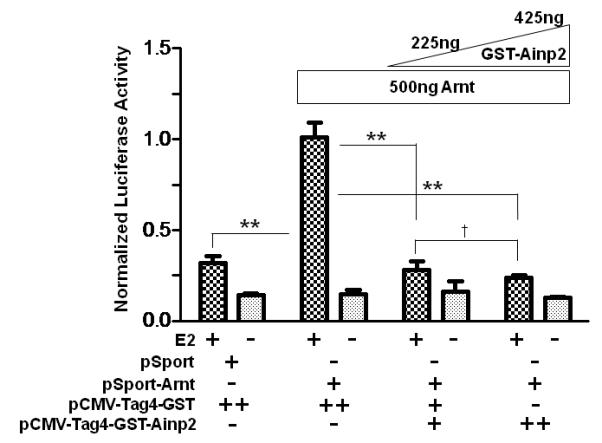
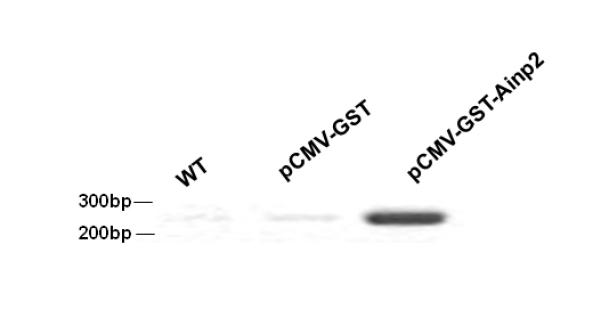
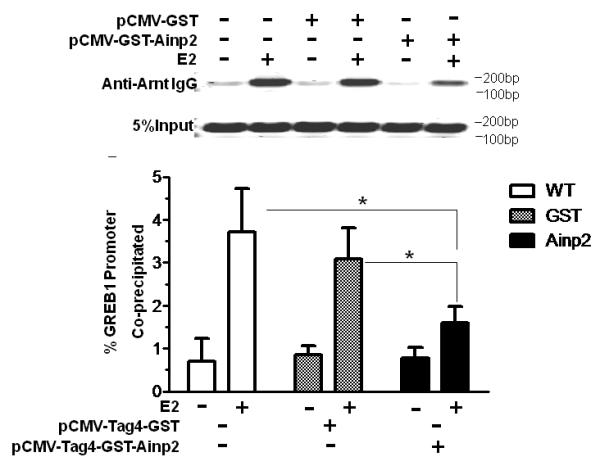
Effect of GST-Ainp2 on the E2-mediated recruitment of Arnt to the GREB1 promoter in MCF-7 cells. A. Luciferase activity of samples ± E2 treatment and transfected with the same amount of plasmids of different combinations (pSport/pSport-Arnt plasmid of 500 ng total and pCMV-Tag4-GST/pCMV-Tag4-GST-Ainp2 plasmid of 425 ng total). Transient transfection was performed using Fugene HD. Error bars represent the standard deviation of the means (n = 3, means ± SD). ** p ≤ 0.005, † p > 0.05 (not significant). B. Agarose gel images of the RT-PCR data showing the presence of the GST-Ainp2 transcript in pCMV-Tag4-GST-Ainp2 transfected (right) MCF-7 cells but not in wild type (WT, left) and pCMV-Tag4-GST transfected (middle) MCF-7 cells. PCR primers (OL480 and 481) were used to amplify a region spanning the GFP-Ainp2 junction (240 bp). C. ChIP data using the wild type and transfected MCF-7 cells (as shown in B) to show the suppression of the E2-driven Arnt recruitment to the GREB1 promoter by GST-Ainp2. The upper part is an example of agarose gel images showing the amplified ERE3 region of the GREB1 promoter (168 bp) under various conditions. 5% input represents the PCR product using 5% of the starting lysate as the template. Error bars represent the standard deviation of the means (n = 3, means ± SD). * p ≤ 0.05.
4. Discussion and Conclusions
Prior to testing out our hypothesis that Ainp2 may modulate the ER signaling via an Arnt dependent mechanism, we made certain that Arnt is involved in the ER signaling in MCF-7 cells. When we transfected Arnt into MCF-7 cells, we observed up to 5-fold increase in the E2-dependent ERE-driven luciferase activity. This luciferase activity was suppressed by more than 50% when the Arnt gene was silenced. These data revealed that Arnt plays a role in the ER-dependent activation of gene transcription in MCF-7 cells, consistent with the data in the literature showing that Arnt is a coactivator in the ER signaling pathway. This Arnt effect appears to be gene-specific: Although Arnt was not recruited to the pS2 and c-fos promoters in MCF-7 cells [24], Arnt was capable of up-regulating the GREB1 message in these cells (Fig. 2A). Moreover, the recruitment of Arnt to the ER promoter regions appears to be cell-specific: Arnt was found in the pS2 promoter of T47 [28] but not MCF-7 cells [24]. Since Arnt is not recruited to the pS2 promoter in MCF-7 cells, it is not surprising that transfection of Arnt into this cell line did not alter the pS2 message (Fig. 2C).
We hypothesized that since Ainp2 interacts with Arnt, it should affect the Arnt function in the ER signaling. This hypothesis is consistent with the literature that activation of Arnt-dependent pathways (such as AhR and HIF-1α) and down-regulation of the Arnt protein expression suppress the ER-mediated transcriptional activity [32], although the nuclear Arnt content seems abundant in mouse and rat hepatoma cells [33]. Indeed the transfected Ainp2 suppressed the E2-activated, ERE-driven luciferase activity in MCF-7 cells. Our mechanistic studies supported that this Ainp2 effect is mediated through Arnt. Events that are not Arnt-dependent, such as binding of the ERα homodimer to the ERE and upregulation of the pS2 message by E2, were not affected by Ainp2. In principle, the ligand-bound AhR and HIF-1α in MCF-7 cells may play a role in this Ainp2 effect since they all interact with Arnt. Interestingly, the ligand-bound AhR, which is part of the cullin 4B ubiquitin ligase complex, promotes the polyubiquination of ERα and in turn reduces the ERα protein level [34]; this AhR action potentially synergizes the Ainp2 suppression of the ERα function. In this paper, we have strategically addressed the Ainp2 effect on the ER signaling under conditions whereby the amount of the liganded AhR and HIF-1α should be minimal – that is, no AhR ligand was used and accumulation of the HIF-1α protein was not triggered in normoxic MCF-7 cells which were used for all Ainp2 studies. AhR does not seem to contribute to the Ainp2 effect since α-napthoflavone, which is an AhR antagonist, had no effect on the E2-activated, ERE-driven luciferase expression regardless of the presence or absence of Ainp2. However, this Ainp2 suppression on the ER signaling appears to be Arnt-mediated because (1) Ainp2 suppressed the Arnt-dependent luciferase activity; (2) the E2-activated GREB1 expression, which should be Arnt-dependent, was also suppressed significantly by Ainp2; (3) Ainp2 had no effect on the ERE-driven pS2 expression, which appeared to be not an Arnt-dependent event in MCF-7 cells; (4) Ainp2 interacted with Arnt but not ERα and ERβ; (5) Ainp2 suppressed the interaction between ERα and Arnt; (6) Ainp2 suppressed the recruitment of Arnt to the ERE3 region of the GREB1 promoter and (7) Ainp2 did not affect the luciferase activity in the Arnt-knockdown cells. Collectively, these observations strongly supported that Arnt is essential for the Ainp2 suppression of the ER function in MCF-7 cells.
Although the Ainp2 cDNA sequence was isolated from a human liver cDNA library, we could not detect its message in two human hepatoma cells (HepG2 and Hep3B). We were unable to detect the Ainp2 protein in primary human hepatocytes (BD Biosciences, San Jose, CA) (data not shown). However, the Ainp2 protein was detected in human fetal and adult liver extracts (BD Biosciences, San Jose, CA) (Fig. 6), suggesting that Ainp2 is expressed in normal liver but its expression may be sensitive to the carcinogenic process and in vitro manipulations. We know that Ainp2 can be post-translationally modified since our baculovirus expressed Ainp2 has an additional band at around 25 KDa and the transfected Ainp2 in MCF-7 cells is larger than the bacterially expressed Ainp2 [7]. Therefore, different sizes of the Ainp2 protein observed in different extracts could be due to the cell-specific post-translational modifications. There were multiple bands in the Jurkat extracts; we believe that the top band is Ainp2 since this band was absent when the Ainp2 antibodies were pre-cleared with the bacterially expressed Ainp2 before the Western blot analysis [7]. Although the endogenous function of Ainp2 is unclear, the cellular Ainp2 function is likely to be Arnt-dependent since it interacts with Arnt. We do not expect that Ainp2 plays any role in the ER signaling in MCF-7 cells since we were not able to detect the Ainp2 protein in the MCF-7 extract (Fig. 6).
Fig. 6.
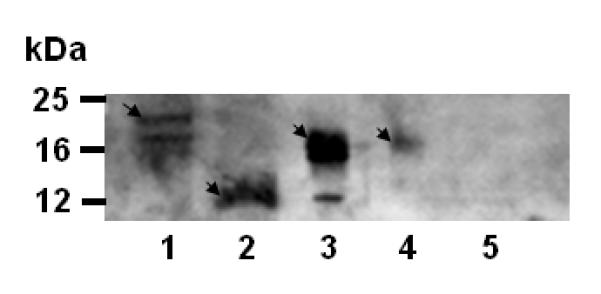
Chemiluminescent Western analysis showing the expression of Ainp2 in Jurkat and human liver extracts. Lane 1, Jurkat soluble extract (40 μg); lane 2, bacterially expressed purified 6His-Ainp2 (1 μg); lane 3, human fetal liver extract (10 μg); lane 4, human adult liver extract (10 μg); lane 5, MCF-7 soluble extract (40 μg). Arrows indicate the Ainp2 protein. The minor (lower) band in lane 3 could be the unmodified Ainp2. Samples were separated on a 15% tricine SDS-PAGE gel and the chicken Ainp2 C634FB (1:500), which was generated using the bacterially expressed 6His-Ainp2 as the antigen, was used as the primary antibodies.
The idea that the ER signaling can be affected by an Arnt-interacting molecule such as Ainp2 is certainly intriguing. Currently about 70% of women with breast cancer are ER-positive and these women most likely take either the ERα antagonist tamoxifen or the aromatase inhibitor letrozole as an adjuvant therapy after mastectomy [35, 36]. Realizing that both agents suppress the ER signaling primarily by reducing the binding of estrogen to ER and letrozole in particular appears to be less tolerable when taken chronically [37], the Arnt-mediated mechanism in suppressing the ER signaling is an appealing alternative for the treatment of breast cancer. Additionally, GREB1 has been reported to be one of the few genes that mediate the estrogen-dependent breast cancer growth in cell culture [38]. Thus, a strategy of using Ainp2 (or an Arnt-interacting peptide) to suppress the GREB1 gene expression is certainly a novel approach to treat the ER-positive breast cancer. Although some reports suggested that abnormal Arnt protein either in the form of the fusion protein TEL-Arnt [39] or the splice variants [40] may contribute to tumorigenesis by possibly inhibiting apoptosis mediated by the hypoxia signaling pathway [41], the underlying mechanisms and the precise role of Arnt in cancers are poorly understood and should be investigated extensively. Here we have shown evidence that this Arnt-mediated approach works and should allow us to rationally screen molecules that bind to Arnt for inhibitors of the ER signaling.
Acknowledgements
We thank Dr. Jesika Faridi (University of the Pacific) for providing the pVit-ERE-Luc plasmid, Dr. Roshanak Rahimian (University of the Pacific) for the pACsk12CMV5-ERα and pACsk12CMV5-ERβ plasmids, and Dr. Chris Bradfield (McArdle Cancer Lab) for the pSport-Arnt plasmid. This work is supported by a grant from the National Institutes of Health (R01 ES014050).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Ma Q. Induction of CYP1A1. The AhR/DRE paradigm: transcription, receptor regulation, and expanding biological roles. Curr. Drug Metab. 2001;2:149–164. doi: 10.2174/1389200013338603. [DOI] [PubMed] [Google Scholar]
- [2].Blancher C, Harris AL. The molecular basis of the hypoxia response pathway: tumour hypoxia as a therapy target. Cancer Metastasis Rev. 1998;17:187–194. doi: 10.1023/a:1006002419244. [DOI] [PubMed] [Google Scholar]
- [3].Nebert DW, Dalton TP, Okey AB, Gonzalez FJ. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J. Biol. Chem. 2004;279:23847–23850. doi: 10.1074/jbc.R400004200. [DOI] [PubMed] [Google Scholar]
- [4].Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wang GL, Semenza GL. Molecular basis of hypoxia-induced erythropoietin expression. Curr. Opin. Hematol. 1996;3:156–162. doi: 10.1097/00062752-199603020-00009. [DOI] [PubMed] [Google Scholar]
- [6].Semenza GL. Expression of hypoxia-inducible factor 1: mechanisms and consequences. Biochem. Pharmacol. 2000;59:47–53. doi: 10.1016/s0006-2952(99)00292-0. [DOI] [PubMed] [Google Scholar]
- [7].Li Y, Luu TC, Chan W. A novel Arnt-interacting protein Ainp2 enhances the aryl hydrocarbon receptor signaling. Arch. Biochem. Biophys. 2005;441:84–95. doi: 10.1016/j.abb.2005.06.026. [DOI] [PubMed] [Google Scholar]
- [8].O’Malley BW, Tsai MJ. Molecular pathways of steroid receptor action. Biol. Reprod. 1992;46:163–167. doi: 10.1095/biolreprod46.2.163. [DOI] [PubMed] [Google Scholar]
- [9].Whitfield GK, Jurutka PW, Haussler CA, Haussler MR. Steroid hormone receptors: evolution, ligands, and molecular basis of biologic function. J. Cell. Biochem. 1999;(Suppl):110–122. doi: 10.1002/(sici)1097-4644(1999)75:32+<110::aid-jcb14>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- [10].Barkhem T, Haldosen LA, Gustafsson JA, Nilsson S. Transcriptional synergism on the pS2 gene promoter between a p160 coactivator and estrogen receptor-alpha depends on the coactivator subtype, the type of estrogen response element, and the promoter context. Mol. Endocrinol. 2002;16:2571–2581. doi: 10.1210/me.2002-0051. [DOI] [PubMed] [Google Scholar]
- [11].Ghosh MG, Thompson DA, Weigel RJ. PDZK1 and GREB1 are estrogen-regulated genes expressed in hormone-responsive breast cancer. Cancer Res. 2000;60:6367–6375. [PubMed] [Google Scholar]
- [12].Lee CH, Edwards AM. Stimulation of DNA synthesis and c-fos mRNA expression in primary rat hepatocytes by estrogens. Carcinogenesis. 2001;22:1473–1481. doi: 10.1093/carcin/22.9.1473. [DOI] [PubMed] [Google Scholar]
- [13].Watanabe H, Takahashi E, Kobayashi M, Goto M, Krust A, Chambon P, Iguchi T. The estrogen-responsive adrenomedullin and receptor-modifying protein 3 gene identified by DNA microarray analysis are directly regulated by estrogen receptor. J. Mol. Endocrinol. 2006;36:81–89. doi: 10.1677/jme.1.01825. [DOI] [PubMed] [Google Scholar]
- [14].Waters KM, Safe S, Gaido KW. Differential gene expression in response to methoxychlor and estradiol through ERalpha, ERbeta, and AR in reproductive tissues of female mice. Toxicol. Sci. 2001;63:47–56. doi: 10.1093/toxsci/63.1.47. [DOI] [PubMed] [Google Scholar]
- [15].Beekman JM, Allan GF, Tsai SY, Tsai MJ, O’Malley BW. Transcriptional activation by the estrogen receptor requires a conformational change in the ligand binding domain. Mol. Endocrinol. 1993;7:1266–1274. doi: 10.1210/mend.7.10.8264659. [DOI] [PubMed] [Google Scholar]
- [16].Chen H, Lin RJ, Xie W, Wilpitz D, Evans RM. Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell. 1999;98:675–686. doi: 10.1016/s0092-8674(00)80054-9. [DOI] [PubMed] [Google Scholar]
- [17].Kim MY, Hsiao SJ, Kraus WL. A role for coactivators and histone acetylation in estrogen receptor alpha-mediated transcription initiation. EMBO J. 2001;20:6084–6094. doi: 10.1093/emboj/20.21.6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Karmakar S, Foster EA, Smith CL. Unique roles of p160 coactivators for regulation of breast cancer cell proliferation and estrogen receptor-alpha transcriptional activity. Endocrinology. 2009;150:1588–1596. doi: 10.1210/en.2008-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhu Y, Qi C, Jain S, Le Beau MM, Espinosa R, 3rd, Atkins GB, Lazar MA, Yeldandi AV, Rao MS, Reddy JK. Amplification and overexpression of peroxisome proliferator-activated receptor binding protein (PBP/PPARBP) gene in breast cancer. Proc. Natl. Acad. Sci. USA. 1999;96:10848–10853. doi: 10.1073/pnas.96.19.10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jeong KW, Lee YH, Stallcup MR. Recruitment of the SWI/SNF chromatin remodeling complex to steroid hormone-regulated promoters by nuclear receptor coactivator flightless-I. J. Biol. Chem. 2009;284:29298–29309. doi: 10.1074/jbc.M109.037010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Clarke R, Skaar T, Baumann K, Leonessa F, James M, Lippman J, Thompson EW, Freter C, Brunner N. Hormonal carcinogenesis in breast cancer: cellular and molecular studies of malignant progression. Breast Cancer Res. Treat. 1994;31:237–248. doi: 10.1007/BF00666157. [DOI] [PubMed] [Google Scholar]
- [22].McCafferty MP, McNeill RE, Miller N, Kerin MJ. Interactions between the estrogen receptor, its cofactors and microRNAs in breast cancer. Breast Cancer Res. Treat. 2009;116:425–432. doi: 10.1007/s10549-009-0429-7. [DOI] [PubMed] [Google Scholar]
- [23].Safe S, Wormke M, Samudio I. Mechanisms of inhibitory aryl hydrocarbon receptor-estrogen receptor crosstalk in human breast cancer cells. J Mammary Gland Biol. Neoplasia. 2000;5:295–306. doi: 10.1023/a:1009550912337. [DOI] [PubMed] [Google Scholar]
- [24].Ohtake F, Takeyama K, Matsumoto T, Kitagawa H, Yamamoto Y, Nohara K, Tohyama C, Krust A, Mimura J, Chambon P, Yanagisawa J, Fujii-Kuriyama Y, Kato S. Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature. 2003;423:545–550. doi: 10.1038/nature01606. [DOI] [PubMed] [Google Scholar]
- [25].Shipley JM, Waxman DJ. Aryl hydrocarbon receptor-independent activation of estrogen receptor-dependent transcription by 3-methycholanthrene. Toxicol. Appl. Pharmacol. 2006;213:87–97. doi: 10.1016/j.taap.2005.09.011. [DOI] [PubMed] [Google Scholar]
- [26].Abdelrahim M, Ariazi E, Kim K, Khan S, Barhoumi R, Burghardt R, Liu S, Hill D, Finnell R, Wlodarczyk B, Jordan VC, Safe S. 3-Methylcholanthrene and other aryl hydrocarbon receptor agonists directly activate estrogen receptor alpha. Cancer Res. 2006;66:2459–2467. doi: 10.1158/0008-5472.CAN-05-3132. [DOI] [PubMed] [Google Scholar]
- [27].Gillesby BE, Stanostefano M, Porter W, Safe S, Wu ZF, Zacharewski TR. Identification of a motif within the 5′ regulatory region of pS2 which is responsible for AP-1 binding and TCDD-mediated suppression. Biochemistry. 1997;36:6080–6089. doi: 10.1021/bi962131b. [DOI] [PubMed] [Google Scholar]
- [28].Brunnberg S, Pettersson K, Rydin E, Matthews J, Hanberg A, Pongratz I. The basic helix-loop-helix-PAS protein ARNT functions as a potent coactivator of estrogen receptor-dependent transcription. Proc. Natl. Acad. Sci. USA. 2003;100:6517–6522. doi: 10.1073/pnas.1136688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Partch CL, Card PB, Amezcua CA, Gardner KH. Molecular basis of coiled coil coactivator recruitment by the aryl hydrocarbon receptor nuclear translocator (ARNT) J. Biol. Chem. 2009;284:15184–15192. doi: 10.1074/jbc.M808479200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Graven KK, Yu Q, Pan D, Roncarati JS, Farber HW. Identification of an oxygen responsive enhancer element in the glyceraldehyde-3-phosphate dehydrogenase gene. Biochim. Biophys. Acta. 1999;1447:208–218. doi: 10.1016/s0167-4781(99)00118-9. [DOI] [PubMed] [Google Scholar]
- [31].Sun J, Nawaz Z, Slingerland JM. Long-range activation of GREB1 by estrogen receptor via three distal consensus estrogen-responsive elements in breast cancer cells. Mol. Endocrinol. 2007;21:2651–2662. doi: 10.1210/me.2007-0082. [DOI] [PubMed] [Google Scholar]
- [32].Ruegg J, Swedenborg E, Wahlstrom D, Escande A, Balaguer P, Pettersson K, Pongratz I. The transcription factor aryl hydrocarbon receptor nuclear translocator functions as an estrogen receptor beta-selective coactivator, and its recruitment to alternative pathways mediates antiestrogenic effects of dioxin. Mol. Endocrinol. 2008;22:304–316. doi: 10.1210/me.2007-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pollenz RS, Davarinos NA, Shearer TP. Analysis of aryl hydrocarbon receptor-mediated signaling during physiological hypoxia reveals lack of competition for the aryl hydrocarbon nuclear translocator transcription factor. Mol. Pharmacol. 1999;56:1127–1137. doi: 10.1124/mol.56.6.1127. [DOI] [PubMed] [Google Scholar]
- [34].Ohtake F, Babe A, Takada I, Okada M, Iwasaki K, Miki H, Takahashi S, Kouzmenko A, Nohara K, Chiba T, Fujii-Kuriyama Y, Kato S. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature. 2007;446:562–566. doi: 10.1038/nature05683. [DOI] [PubMed] [Google Scholar]
- [35].Goss PE, Ingle JN, Martino S, Robert NJ, Muss HB, Piccart MJ, Castiglione M, Tu D, Shepherd LE, Pritchard KI, Livingston RB, Davidson NE, Norton L, Perez EA, Abrams JS, Therasse P, Palmer MJ, Pater JL. A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. N. Engl. J. Med. 2003;349:1793–1802. doi: 10.1056/NEJMoa032312. [DOI] [PubMed] [Google Scholar]
- [36].Ganz PA. Breast cancer, menopause, and long-term survivorship: critical issues for the 21st century. Am. J. Med. 2005;118:136–141. doi: 10.1016/j.amjmed.2005.09.047. [DOI] [PubMed] [Google Scholar]
- [37].Fontaine C, Meulemans A, Huizing M, Collen C, Kaufman L, De Mey J, Bourgain C, Verfaillie G, Lamote J, Sacre R, Schallier D, Neyns B, Vermorken J, De Greve J. Tolerance of adjuvant letrozole outside of clinical trials. Breast. 2008;17:376–381. doi: 10.1016/j.breast.2008.02.006. [DOI] [PubMed] [Google Scholar]
- [38].Rae JM, Johnson MD, Scheys JO, Cordero KE, Larios JM, Lippman ME. GREB 1 is a critical regulator of hormone dependent breast cancer growth. Breast Cancer Res. Treat. 2005;92:141–149. doi: 10.1007/s10549-005-1483-4. [DOI] [PubMed] [Google Scholar]
- [39].Salomon-Nguyen F, Della-Valle V, Mauchauffe M, Busson-Le Coniat M, Ghysdael J, Berger R, Bernard OA. The t(1;12)(q21;p13) translocation of human acute myeloblastic leukemia results in a TEL-ARNT fusion. Proc. Natl. Acad. Sci. USA. 2000;97:6757–6762. doi: 10.1073/pnas.120162297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Qin C, Wilson C, Blancher C, Taylor M, Safe S, Harris AL. Association of ARNT splice variants with estrogen receptor-negative breast cancer, poor induction of vascular endothelial growth factor under hypoxia, and poor prognosis. Clin. Cancer Res. 2001;7:818–823. [PubMed] [Google Scholar]
- [41].Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshert E. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]