

Abstract
Biliary atresia is an obstructive cholangiopathy of unknown etiology. Although the adaptive immune system has been shown to regulate the obstruction of bile ducts in a rotavirus-induced mouse model, little is known about the virus-induced inflammatory response. Here, we hypothesized that cholangiocytes secrete chemoattractants in response to rotavirus. To test this hypothesis, we infected cholangiocyte and macrophage cell lines with rhesus rotavirus type A (RRV), quantified cytokines, and chemokines and measured the migration of splenocytes. We also used PCR and immunostaining to search for new cellular targets of RRV in the liver. We found that RRV-infected cholangiocytes induced the mRNA expression for chemokines, but conditioned media failed to promote chemotaxis of splenocytes. Analyzing livers after viral challenge, we detected RRV in hepatic macrophages and demonstrated that media from RRV-infected macrophages have high concentrations of cytokines and chemokines, and induced chemotaxis of neutrophils. Most notably, addition of anti-Mip2/Cxcl2 antibodies depleted this chemokine in the conditioned media and completely prevented neutrophil chemotaxis. In conclusion, infected cholangiocytes did not promote chemotaxis of inflammatory cells. Investigating alternate cellular targets of RRV, we detected the virus in hepatic macrophages, and found that infected macrophages promoted neutrophil chemotaxis by release of Mip2/Cxcl2 in response to RRV.
Biliary atresia, the most common cause of neonatal cholestasis, results from an inflammatory and fibrosing obstruction of extrahepatic bile ducts. The etiology is unknown, but studies in an experimental mouse model of rotavirus-induced biliary atresia indicate that pathogenic mechanisms of disease begin with an injury to the biliary epithelium, followed by a robust inflammatory infiltration of the wall of extrahepatic bile ducts, obstruction of the lumen by inflammatory cells, and final progression to fibrosis (1). These pathological changes in the liver and extrahepatic bile ducts closely resemble human biliary atresia (2,3). Studies of livers of infants at the time of diagnosis have long recognized an activation of inflammatory cells (4), and a broad analysis of the hepatic gene expression profile identified a prominent pro-inflammatory signature (5). Disruption of this signature by loss of interferon-gamma (IFNγ) or the loss of CD8+ lymphocytes in the mouse model largely prevented duct obstruction and the phenotype of experimental biliary atresia (6,7). Interestingly, in vivo depletion of CD4+ lymphocytes or the genetic loss of interleukin-12, or the depletion of tumor necrosis factor-alpha (TNFα) later in the cause of biliary injury did not alter the progression to biliary atresia phenotype (7–9), which supported the co-existence of accessory pathways regulating the pathology of extrahepatic bile ducts. Despite the progress in deciphering key elements regulating duct obstruction and atresia, little is known about molecular circuits that are activated in early stages of the disease.
Cellular localization studies have shown that cholangiocytes are cellular targets in early stages of disease following rotavirus administration in newborn mice (6,10). This infection induces the creation of an environment rich in chemokines, some of which probably derive from cholangiocytes, as suggested by increased expression of monocyte chemotactic protein 1 (Mcp1), regulated upon activation, normal T expressed and secreted (Rantes), KC/Cxcl1, macrophage inflammatory protein 2 (Mip2/Cxcl2), and thymus and activation regulated chemokine (Tarc) by a cholangiocyte cell line infected with RRV (11). Further, when primary cholangiocytes were submitted to flow cytometry, they were reported to express the surface markers major histocompatibility complex-I and II and CD40, but they did not function as competent antigen-presenting cells (12). Based on its role as a cellular target of RRV and as a source of inflammatory mediators, we hypothesized that cholangiocytes secrete chemoattractants to mononuclear cells in response to RRV. Testing this hypothesis, we found that conditioned media of RRV-infected cholangiocytes did not induce chemotaxis to mononuclear cells. Exploring alternative cellular sources, we found that hepatic macrophages were targeted by RRV and that conditioned media from RRV-infected macrophages was rich in Mip2/Cxcl2, attracted neutrophils, and lost chemoattractant properties after depletion of Mip2/Cxcl2.
METHODS
Cell culture and viral infection
The murine cholangiocyte cell line mCL, a SV40-large T antigen-transformed cell from Balb/c mice (13), was cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Cellgro, Herndon, VA, USA) containing 10% heat-inactivated fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin (Invitrogen, Carlsbad, CA, USA), and 1% L-glutamine (Invitrogen) at 37°C in 5% CO2-humidified air. Raw 264.7 cells were obtained from American Type Culture Collection (Manassas, VA, USA) and cultured under the conditions described in the protocol PP0159 of the Alliance for Cellular Signaling (www.signaling-gateway.org). For viral infection, mCL and Raw 264.7 cells were plated in a 12-well dish at a density of 0.2×106 (for mCL) or 0.5×106 (for Raw 264.7) cells/well. After 2 days of culture, cells were washed twice and cultured for 1 hour in Earle’s Balanced Salt Solution-Ca++ (EBSS, Sigma-Aldrich, St Louis, USA) with 4 μg/mL of trypsin containing live virus at a multiplicity of infection of 100 to ensure high infection rate of plated cells. Cultured cells were then washed twice, and further incubated in serum-free DMEM for variable intervals. At defined time points, the conditioned medium was aspirated and stored at −20°C until the time of analysis and cells were used for RNA isolation. For chemotaxis assays, the conditioned medium was transferred freshly from the culture wells to the chemotaxis chambers.
Chemotaxis assay
Wedge chemotactic response was based on 48-well microchemotaxis chamber (Neuro Probe, Gaithersburg, MD USA) as described previously(14). In brief, the lower well contained 27 μl of conditioned media from RRV-infected or naïve mCL or Raw 264.7 cells. The upper chamber was filled with 50 μl of splenocytes or neutrophils (final concentration of 2×106 cells per mL) isolated from normal adult Balb/c mice. The two well and chamber were separated by a 3.0 μm-pore size polycarbonate filter. The assay chambers were incubated at 37°C in 5% CO2 for 45 min, at which time the chambers were disassembled, the membranes were washed, fixed in methanol, and stained with Diff-Quik staining kit (Fisher Scientific, Pittsburgh, PA, USA). The cells on the inferior surface of the membranes were counted from 5 fields of each membrane at 400x magnification using a Nikon Labophot 2 Microscope (Nikon, Melville, NY, USA), and the mean cell number was calculated for individual wells; each experiment was performed in quadruplicate. fMLP 1μM (Sigma-Aldrich) was added to separate wells as a positive control. All experiments were repeated 2–3 times unless otherwise stated, and the results are expressed as mean cells/high power field ± standard error (S.E.).
Animal model and cell isolation
Balb/c mice were injected with 0.9% saline solution (controls) or 1.5×106 focus-forming units (ffu) of RRV intraperitoneally within 24 hours of birth. Each group of RRV-or saline-injected mice contained 3–5 newborn mice per time point, unless otherwise specified in the results or in figure legends. All mice were examined daily and the diagnosis of experimental biliary atresia was determined by the presence of jaundice in non fur-covered skin and acholic stools, as well as by the direct finding of lumenal obstruction in serial sections of extrahepatic bile ducts harvested at 7 or 14 days after RRV infection and stained with hematoxylin-eosin, as described by us in a previous publication (6); the phenotype cannot be ascertained 3 days after RRV infection because the first signs of cholestasis emerge at days 5–6. At the time of sacrifice, livers were snap frozen in liquid N2 for analysis of mRNA expression or used for isolation of mononuclear cells as described previously with minor modifications (8). In brief, livers were minced, passed through a nylon mesh, layered on 3 mL of histopaque (Sigma, St Louis, USA), and centrifuged at 270 g. Freshly isolated mononuclear cells were used in immunofluorescence assays or processed for RNA isolation (see below). The Institutional Animal Care and Use Committee of the Cincinnati Children’s Research Foundation approved all animal protocols.
Immunofluorescence assay
For immunofluorescence, primary liver suspensions of mononuclear cells obtained 7 days after RRV infection were centrifuged, the supernatant was removed, and the cell pellet was cytospun onto a glass slide, fixed in cold 80% acetone × 10 min, and then in cold methanol for 10 min. Fixed cells were blocked with 2% normal goat serum, incubated with anti-rotavirus and anti-F4/80 primary antibodies overnight at 4°C, washed, then incubated with Texas Red- and FITC-conjugated secondary antibodies. The cells/slides were washed twice and mounted using permount/DAPI.
Chemokine and cytokine ELISAs
The concentration of chemokines and cytokines was determined in conditioned media using commercially available ELISA Quantikine sandwich enzyme immunoassay kits for TNFα, IFNγ, IFNα, IFNβ, IL1β, Mip2/Cxcl2, and KC/Cxcl1 (R&D Systems, Minneapolis, MN, USA), according to the manufacturer’s instructions. A range of 50–100 μL of media was used for each well. The optical density was determined using the Spectramax Plus microplate reader (Molecular Devices Corp, Sunnyvale, CA, USA), and the concentration was calculated with the Softmax Pro v2.2.1 software (Molecular Devices Corp).
Chemokine immunodepletion
Mip2/Cxcl2 was depleted from conditioned media by incubation with 50 μg of mouse antibody against Mip2/Cxcl2 (R&D Systems) followed by incubation with CNBr-activated protein G sepharose overnight at 4°C, and washed to remove unbound products. Supernatants of RRV-infected or naïve macrophages in culture (1 mL of supernatant collected after 12 h of culture) were incubated with the protein G-coupled antibody mixture and incubated for 2 hours at room temperature. Immune complexes were pelleted at 2500 g for 3 minutes and the supernatant was collected for chemotaxis assay. The immunodepletion protocol was also applied to supernatants without anti-Mip2/Cxcl2 antibody to serve as controls. The efficiency of Mip2/Cxcl2 immunodepletion was determined by ELISA (as described by above) by measuring the concentration of the chemokine in supernatants before and after immunodepletion.
Real-time PCR
RNA from the whole liver and mCL cells was isolated using Trizol (Invitrogen), reverse transcribed with the MMLV Reverse-Transcription kit from Invitrogen, and subjected to real-time PCR in a Mx3000P (Stratagene, La Jolla, CA USA) as described previously (6,7) using specific primers for individual genes (Table 1).
Table 1.
Oligonucleotide primers used for detection of different chemokines
| Gene | Primer Sequences | Annealing Temp. (°C) |
|---|---|---|
| KC/Cxcl1 | Forward: 5′-ACCGAAGTCATAGCCACACTC -3′ Reverse: 5′-TGGGGACACCTTTTAGCATC -3′ |
52 |
| Mip2/Cxcl2 | Forward: 5′-CCCAGACAGAAGTCATAGCCAC -3′ Reverse: 5′-GCCTTGCCTTTGTTCAGTATC -3′ |
52 |
| TNFα | Forward: 5′-AAGGGAGAGTGGTCAGTTGCC -3′ Reverse: 5′-CCTCAGGGAAGAGTCTGGAAAGG -3′ |
55 |
| Rantes | Forward: 5′-GGAGTATTTCTACACCAGCAGC -3′ Reverse: 5′-TCTTGAACCCACTTCTTCTCTG -3′ |
52 |
| Cxcl9 | Forward: 5′-GAGCTAGATAGACCTCACCAAG -3′ Reverse: 5′-CCATTAGCACCATCTCTGA -3′ |
55 |
| Cxcl10 | Forward: 5′-TCGCTCAAGTGGCTGGGATG -3′ Reverse: 5′-TAGGGAGGACAAGGAGGGTGTG -3′ |
55 |
| Gapdh | Forward: 5′-TGGTTTGACAATGAATACGGCTAC -3′ Reverse: 5′-GGTGGGTGGTCCAAGGTTTC -3′ |
55 |
| Rotavirus VP6 | Forward: 5′-GCGGTAGCGGTGTTATTTCC -3′ Reverse: 5′-TTGTTTTGCTTGCGTCGG -3′ |
55 |
| Rotavirus NSP3 | Forward: 5′-TGTCAAGAGAATACCTGGGAAATC -3′ Reverse: 5′-GGAATCATCAACTTCAACTTCACC -3′ |
54 |
| Ccl12 | Forward: 5′-CACCATCAGTCCTCAGGTAT -3′ Reverse: 5′-GGACGTGAATCTTCTGCTTA -3′ |
54 |
Temp = temperature
Statistical analysis
Values are expressed as mean ± S.E. and statistical significance was determined by 2-tailed, unpaired t test, with a significance set at P<0.05. Numbers of samples for individual experiments are described in the results or figure legends.
RESULTS
RRV infection induces the expression of chemokines by cholangiocytes
We have shown that RRV infects and replicates in biliary epithelial cells (6). To determine whether the infection induces the expression of soluble mediators that localize the inflammatory response to the duct epithelium, we quantified the mRNA expression of chemokines known to be important to the recruitment of neutrophils (Mip2/Cxcl2 and KC/Cxcl1) and T and NK cells (Rantes, Cxcl9, Cxcl10, Ccl12) following RRV infection of a cholangiocyte cell line from Balb/c mice (15–17). We found that RRV infection of cholangiocytes induces the expression of all chemokines several fold above RRV-naïve cholangiocytes (Figure 1). mRNA expression was uniformly higher at or beyond 6 hours of culture for all chemokines, with KC/Cxcl1 and Rantes mRNA also increasing as early as 1 hour after RRV challenge. Based on these data and on a previous report that these chemokines are also expressed at the protein level following RRV infection of mCL cells (11), we determined the ability of conditioned media to induce chemotaxis to splenocytes. To our surprise, conditioned media obtained after 12 and 24 hours of culture did not attract splenocytes after 15, 30, or 45 minutes of culture despite the expression of chemokines, whereas many splenocytes migrated through the inter-chamber membrane toward fMLP as a positive control (data not shown). Therefore, we explored the existence of other cellular targets of RRV that could play a role in the production of soluble chemoattractants.
Figure 1. mRNA expression for chemokines by RRV-infected cholangiocytes.

mRNA expression of chemokines in the cholangiocyte line mCL in culture at different time points after infection with RRV. mRNA levels were determined by real-time PCR and expressed as a ratio to GAPDH. *P<0.05 (between RRV-infected and naïve mCL cells); grey bars=control (saline injected mice); black bars=RRV injected mice; N=3 wells for each time point and experimental group.
Hepatic macrophages are targeted by RRV
To examine whether liver non-epithelial cells are targeted by RRV in experimental biliary atresia, we isolated hepatic mononuclear cells isolated by histopaque gradient at 3, 7, and 14 days after injection of RRV into Balb/c newborn mice within 24 hours of birth. Mice were asymptomatic 3 days after infection, but displayed jaundice and had acholic stools at 7 and 14 days, as reported previously (6). Using RNA from the hepatic mononuclear cells, we detected mRNA encoding the viral proteins NSP3 and VP6 at all time points (Figure 2A). Based on our previous report that hepatic lymphocytes do not express NSP3 or VP6 mRNA after RRV challenge (6,7), we subjected hepatic mononuclear cells isolated after 7 days of infection to dual-fluorescence immunostaining using antibodies that identify macrophages (panF4/80) or RRV. We focused on panF4/80+ cells because macrophages from gut-associated lymphoid tissue have been previously reported to display rotavirus-specific proteins after oral challenge with rotavirus (18). We found that hepatic cells positive for panF4/80+ (macrophages) also stained positive for RRV (Figure 2B). Consistent with the targeting of macrophages by RRV, immunostaining of liver sections 7 days after RRV infection detected panF4/80+ cells in portal spaces that also stained positive for RRV (Figure 2C). Altogether, these data identify hepatic macrophages as non-parenchymal cells that are targeted by RRV in experimental biliary atresia.
Figure 2. Detection of RRV in hepatic mononuclear cells and macrophages.
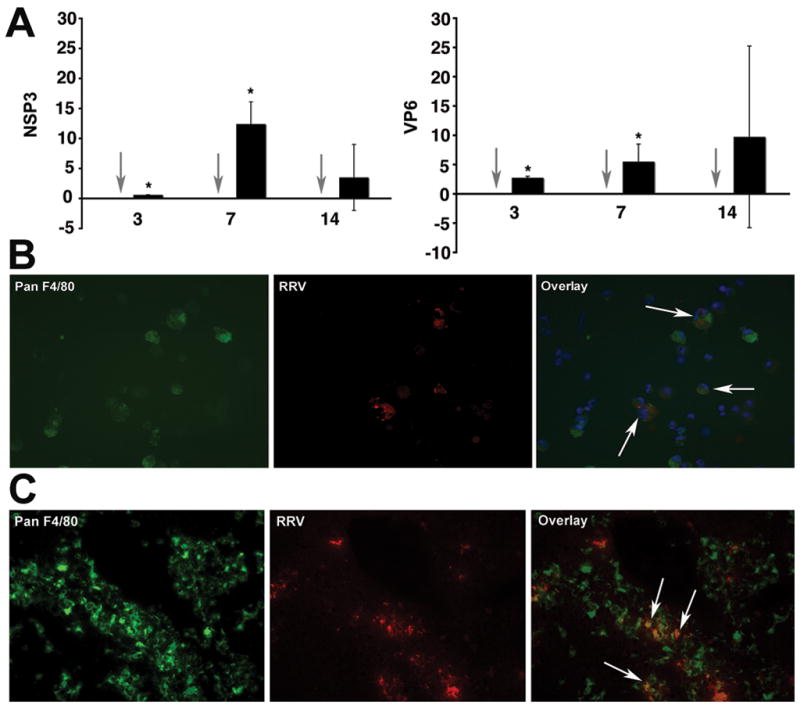
Panel A depicts the expression of mRNA encoding for the RRV proteins NSP3 and VP6 in hepatic mononuclear cells isolated from mice 7 days after RRV challenge. Grey arrows point to no detection of RRV in hepatic mononuclear cells of normal saline-injected controls; black bars=RRV injected mice; P<0.05; N=3–6 livers per time point and per experimental group. Panels B and C depict dual-immunofluorescence signals identifying RRV (red) in panF4/80+ cells (green) in purified hepatic mononuclear cells (panel B) or portal tracts (panel C) 7 days after RRV infection of newborn mice. Arrows point to double positive panF4/80+ and RRV+ cells.
Soluble mediators from RRV-infected macrophages induce chemotaxis of inflammatory cells
To investigate the role of macrophages as a source of chemoattractants following RRV infection, we made use of the macrophage cell line Raw 264.7. We did not use primary liver macrophages because of the very limited number of cells that can be isolated from very small neonatal livers. Further, Raw 264.7 cells have been used as a model system to study the biology of the rotavirus nonstructural protein NSP4 (19). In initial experiments, one-hour exposure of Raw 264.7 cells to RRV resulted in Raw cells staining positive for RRV after 24 hours (Figure 3A). Then, we collected conditioned media after 24 hours of culture of RRV-infected and naïve Raw 264.7 cells and used them in chemotaxis assays. The presence of conditioned medium from RRV-infected Raw 264.7 cells induced migration of neutrophils (and to a minor degree migration of lymphocytes) toward lower wells of chemotaxis chambers above the levels observed in chambers containing medium from RRV-naïve cells (Figure 3B). These data demonstrated that soluble mediators of chemotaxis are released from macrophages following RRV challenge, and formed a suitable experimental system to search for key inducers of chemotaxis.
Figure 3. RRV infection of Raw 264.7 cells and induction of chemotaxis by conditioned media.
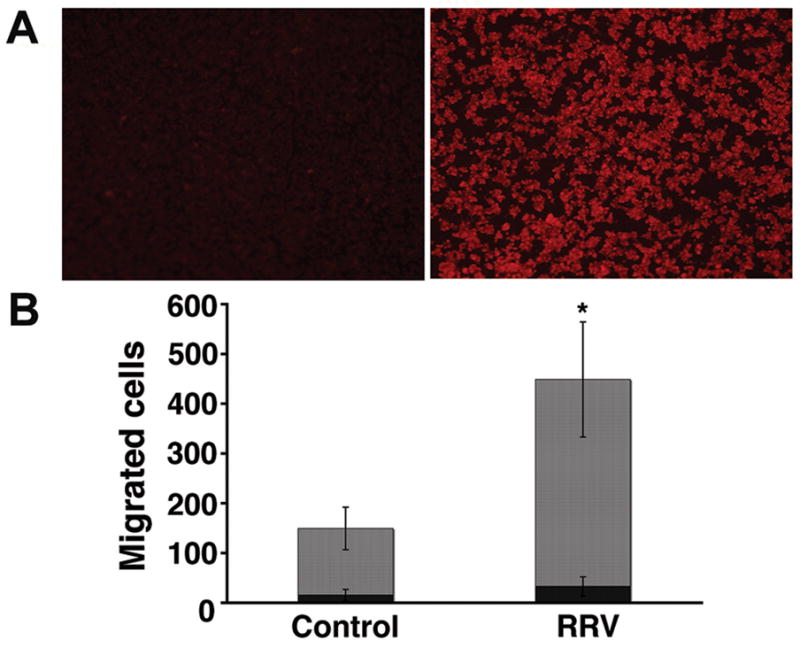
In panel A, immunofluorescence detects RRV (red signal) 24 hours after Raw 264.7 cells were exposed to RRV (right photograph); no signal is detected in cells not exposed to RRV (left photograph). Panel B depicts the numbers of neutrophils and lymphocytes that migrated to lower wells of chemotaxis chambers containing conditioned media from RRV-infected or control (RRV-naïve) Raw 264.7 cells after 45 minutes of culture. *P<0.05; N=3 chambers per group; grey bars=neutrophils; black bars=lymphocytes.
Macrophage-derived Mip2/Cxcl2 induces migration of neutrophils
Because cholangiocyte media did not induce chemotaxis despite the mRNA expression for chemokines, we determined the concentration of pro-inflammatory cytokines (TNFα, IFNγ, IFNα, IFNβ, and IL1β) and selected chemokines (Mip2/Cxcl2 and KC/Cxcl1) in media from Raw 264.7 cells and cholangiocytes. We found prominent increases in TNFα, IFNα, IFNβ, and Mip2/Cxcl2 primarily in macrophages, with undetectable levels for TNFα, IFNγ, IFNα, IFNβ, and IL1β in media from cholangiocytes (Table 2 and Figure 4). Of note, the concentration of Mip2/Cxcl2 in media from RRV-infected Raw 264.7 cells was 10 fold higher than in RRV-infected cholangiocytes at 24 hours of culture (7130±0.50 pg/mL versus 33±1 pg/mL, respectively; P<0.01). To examine whether the levels of Mip2/Cxcl2 increase during the development of experimental biliary atresia, we infected newborn mice with RRV and determined Mip2/Cxcl2 mRNA expression by real-time PCR. We found that Mip2/Cxcl2 mRNA increases at early phases of injury (3 days after RRV challenge; P<0.05), and remains variably elevated at the times of inflammatory obstruction (7 days) and atresia (14 days) of bile ducts (Figure 5A). The significant increase in Mip2/Cxcl2 mRNA at early phase of duct injury was temporally linked to the infiltration of neutrophils in portal tracts of RRV-infected mice (3 days; Figure 5B), which differs from the predominantly lymphocytic infiltration at the time of duct obstruction (7 days; Figure 5B). Combining these data with the well-established role of Mip2/Cxcl2 in chemotaxis of neutrophils (20), we hypothesized that macrophage-derived Mip2/Cxcl2 after RRV infection is a key inducer of chemotaxis.
Table 2.
Presence or absence of protein levels of cytokines and chemokines in conditioned media obtained after 24 hours of culture of RRV-infected Raw 264.7 and mCL cells. Protein levels were determined by ELISA.
| Protein | mCL cells | Raw 264.7 cells |
|---|---|---|
| TNFα | − | + |
| IFNγ | − | − |
| IFNα | − | + |
| IFNβ | − | + |
| IL1β | − | − |
| Mip2/Cxcl2 | + | + |
| KC/Cxcl1 | + | − |
“−“ Means not detectable
Figure 4. Production of cytokines and chemokines by mCL cholangiocytes and Raw 264.7 cells.

Concentrations of cytokines and chemokines in conditioned media of RRV-infected and naïve (controls) mCL cholangiocytes (panels A and B) and Raw 264.7 (panels C–F) cells at different times of culture as determined by ELISA. Arrows point to no detectable level; P<0.05 (between RRV-infected and naïve cells); grey bars=control (saline injected mice); black bars=RRV injected mice; N=3 for each time point and experimental group.
Figure 5. Hepatic expression of Mip2/Cxcl2 and inflammation of portal tracts following RRV challenge.
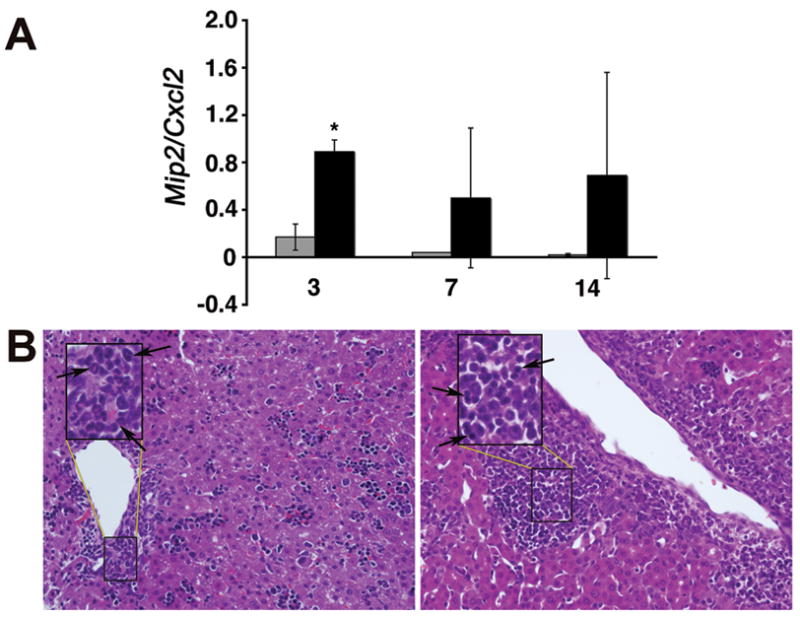
In panel A, realtime PCR shows increased hepatic expression of Mip2/Cxcl2 mRNA 3 days after RRV infection of newborn mice; P<0.05; grey bars=control (saline injected mice); black bars=RRV injected mice; N=3 at each time point and experimental group. In panel B, hematoxylin/eosin staining of liver sections shows accumulation of neutrophils at day 3 (arrows; left panel) and lymphocytes at day 7 (arrows; right panel) after RRV infection. Insets represent magnification of area within rectangles; magnification: 400x.
To test this hypothesis, we performed new chemotaxis assays using conditioned media from RRV-infected and naïve Raw 264.7 cells that were depleted of Mip2/Cxcl2. Depletion was demonstrated by the undetectable level of Mip2/Cxcl2 by ELISA after incubation of conditioned media with anti-Mip2/Cxcl2 antibody (Figure 6A). Use of Mip2/Cxcl2-depleted media in lower wells of chemotaxis chambers kept the number of migrated neutrophils at the same levels of conditioned media from RRV-naïve Raw 264.7 cells, while the Mip2/Cxcl2-containing media induced the typical surge in neutrophil migration (Figure 6B).
Figure 6. Mip2/Cxcl2-dependent chemotaxis of neutrophils.
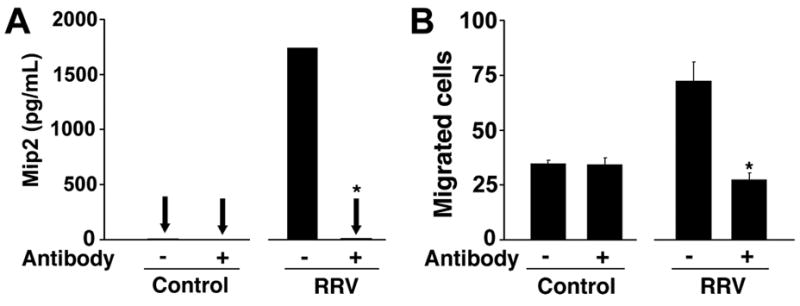
In panel A, the concentration of Mip2/Cxcl2 is determined in conditioned media from RRV-infected or RRV-naïve Raw 264.7 cells that were incubated (or not) with anti-Mip2/Cxcl2 antibody. Panel B depicts the number of neutrophils migrating to the lower wells of chemotaxis chambers containing the respective conditioned media with or without anti-Mip2/Cxcl2 antibody. *P<0.05 showing significant differences between media with or without pre-incubation with anti-Mip2/Cxcl2 antibody; N=3 for each experimental group.
DISCUSSION
We found that macrophages, rather than cholangiocytes, produce chemoattractants in response to RRV infection. Using an in vitro infection system of a cholangiocyte cell line previously shown to be susceptible to RRV and to express an array of cytokines and chemokines (6,11,12), we did not find support for a direct role of these cells in the promotion of chemotaxis of inflammatory cells. Searching for other cells with potential role in the regulation of the inflammatory response in experimental biliary atresia, we detected RRV in hepatic mononuclear cells 3–14 days after infection, and identified the virus in macrophages. Using the macrophage cell line Raw 264.7 to examine the role of these cells in production of chemoattractants to inflammatory cells, we found that they secrete pro-inflammatory cytokines previously linked to pathogenesis of biliary atresia [examples: IFNγ and TNFα, ref (6,8,21)] and high levels of the chemokine Mip2/Cxcl2. Most notably, conditioned media from RRV-infected macrophages induced prominent migration of neutrophils, which was dependent on Mip2/Cxcl2. These data identify macrophages as a new cellular target of RRV in experimental biliary atresia and point to its role as a source of soluble mediators that amplify the inflammatory population of the hepatobiliary environment.
Cholangiocytes are key epithelial targets of RRV and undergo injury by hepatic NK and CD8+ cells during pathogenesis of experimental biliary atresia (6,7,10,22); hepatocytes are also susceptible to RRV, but do so at a lower multiplicity of infection. The injury by inflammatory cells might relate to the recognition of viral epitopes in infected cells and/or to aberrant expression of MHC-associated molecules. However, recent work by another laboratory showed that RRV-exposed cholangiocytes do not appear to function as antigen-presenting cells (12). Based on the findings of increased expression of chemokines by cholangiocytes, investigators have suggested that infected cells may play a role in immunomodulation (11,12). Addressing this scenario, we first found an increase in the mRNA expression for cytokines and chemokines, but the expression at the protein level was either below the detectable levels by ELISA (for TNFα, IFNα, INFβ, INFγ, and IL1β) or still very low when compared to the levels produced by the macrophage cell line (for Mip2). The reasons for the discrepancy between the levels of mRNA expression detected by real-time PCR and the protein levels are not obvious, but may include the variable rates of translation of mRNA transcripts. Regardless of the cause for the discrepancies, we did not find support for the release of biologically sufficient amounts of chemoattractants to inflammatory cells by cholangiocytes. It is possible that the ongoing production of chemokines by cholangiocytes for expanded periods of time (beyond the 24 hours investigated in our studies) could generate a higher concentration of chemoattractants. However, we chose to stay within the constraints of 24 hours of infection in order to simulate early biological events (i.e., periductal inflammation and cholangiocyte injury) induced in the model of experimental biliary atresia (6,23). The lack of support for a role of cholangiocytes in antigen presentation or as a source of chemoattractants does not negate the possibility that cholangiocytes are critical elements of the pathogenesis of biliary atresia. For example, RRV-harboring cholangiocytes are cellular targets of NK and CD8+ cells. Further, they must maintain mucosal continuity along the intra- and extra-hepatic biliary tracts. When this continuity is disrupted by NK cell-mediated injury, duct damage occurs and the phenotype of experimental biliary atresia emerges over time. If NK cells are depleted in vivo, cholangiocyte injury is negligible and the atresia phenotype is prevented (22).
The detection of RRV proteins in hepatic macrophages expands the types of cells that are initially targeted by the virus in the neonatal liver, and formally implicates the innate immune system in pathogenesis of experimental biliary atresia. It is possible that the RRV signal observed in hepatic macrophages represents viral antigens that have been processed and are being presented on the cell surface. If this is correct, macrophages may serve as antigen-presenting cells to induce an adaptive response targeted to bile ducts. Our data adds an additional role for macrophages as producers of inflammatory mediators that recruit neutrophils to the site of infection. One of these mediators is Mip2/Cxcl2, as supported by the induction of Mip2/Cxcl2-dependent chemotaxis of neutrophils by conditioned media from RRV-infected Raw 264.7 cells and by the increased expression of Mip2/Cxcl2 mRNA in livers as early as 3 days after RRV infection. We do not know whether Mip2/Cxcl2-dependent chemoattraction of neutrophils is essential to the pathogenesis of bile duct injury in vivo, but the data reported herein form the basis for this line of future investigation.
Patient-based studies have implicated macrophages in pathogenesis of biliary atresia. The enriched expression of the lipopolisaccharide receptor CD14 in Kupffer cells and the population of the livers of children with biliary atresia by activated macrophages suggest that these cells contribute to the creation of a pro-inflammatory environment at diagnosis and/or after portoenterostomy (24,25). At least in one report, a greater population of macrophages in the affected liver was associated with poor outcome (25). Despite the lack of evidence of active viral infection at the time of diagnosis in most patients with biliary atresia, a previous exposure to viral proteins may be sufficient for the induction of pro-inflammatory signals. For example, the non-structural protein 4 (NSP4) of RRV has been shown to trigger the expression of inducible-nitric oxide synthase in ileal macrophages after RRV infection (19). While the use of cell culture systems and the experimental model of rotavirus-induced biliary atresia in newborn mice are powerful tools to understand components of operative biological processes, it is important to recognize that findings from either system require validation in tissues of affected children. Further dissection of the role of macrophages in pathogenesis of disease, either as an antigen-presenting cell or as one of the cellular targets of a viral insult, will also require the use of in vitro systems using human cells. This line of studies are likely to decipher how macrophages or macrophage-derived signals directly regulate mechanisms of disease and can be potential therapeutic targets to block progression of disease to end-stage cirrhosis.
Acknowledgments
This work was supported by funding from the National Institutes of Health, grant number DK064008, to J.A.B., and grant number DK078392, to the Integrative Morphology Core of the Digestive Disease Research Core Center at Cincinnati Children’s Hospital Medical Center.
The authors thank Dr. Pranavkumar Shivakumar for his expert assistance with chemotaxis assays and with the animal model of biliary atresia.
Abbreviations
- Mip2
macrophage inflammatory protein 2
- RRV
rhesus rotavirus type A
References
- 1.Bezerra JA. The next challenge in pediatric cholestasis: deciphering the pathogenesis of biliary atresia. J Pediatr Gastroenterol Nutr. 2006;43:S23–S29. doi: 10.1097/01.mpg.0000228197.28056.2f. [DOI] [PubMed] [Google Scholar]
- 2.Petersen C, Biermanns D, Kuske M, Schakel K, Meyer-Junghanel L, Mildenberger H. New aspects in a murine model for extrahepatic biliary atresia. J Pediatr Surg. 1997;32:1190–1195. doi: 10.1016/s0022-3468(97)90680-1. [DOI] [PubMed] [Google Scholar]
- 3.Riepenhoff-Talty M, Schaekel K, Clark HF, Mueller W, Uhnoo I, Rossi T, Fisher J, Ogra PL. Group A rotaviruses produce extrahepatic biliary obstruction in orally inoculated newborn mice. Pediatr Res. 1993;33:394–399. doi: 10.1203/00006450-199304000-00016. [DOI] [PubMed] [Google Scholar]
- 4.Mack CL, Sokol RJ. Unraveling the pathogenesis and etiology of biliary atresia. Pediatr Res. 2005;57:87R–94R. doi: 10.1203/01.PDR.0000159569.57354.47. [DOI] [PubMed] [Google Scholar]
- 5.Bezerra JA, Tiao G, Ryckman FC, Alonso M, Sabla GE, Sneider B, Sokol RJ, Aronow BJ. Genetic induction of proinflammatory immunity in children with biliary atresia. Lancet. 2002;360:1653–1659. doi: 10.1016/S0140-6736(02)11603-5. [DOI] [PubMed] [Google Scholar]
- 6.Shivakumar P, Campbell KM, Sabla GE, Miethke A, Tiao G, McNeal MM, Ward RL, Bezerra JA. Obstruction of extrahepatic bile ducts by lymphocytes is regulated by IFN-gamma in experimental biliary atresia. J Clin Invest. 2004;114:322–329. doi: 10.1172/JCI21153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shivakumar P, Sabla G, Mohanty S, McNeal M, Ward R, Stringer K, Caldwell C, Chougnet C, Bezerra JA. Effector role of neonatal hepatic CD8+ lymphocytes in epithelial injury and autoimmunity in experimental biliary atresia. Gastroenterology. 2007;133:268–277. doi: 10.1053/j.gastro.2007.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohanty SK, Shivakumar P, Sabla G, Bezerra JA. Loss of interleukin-12 modifies the pro-inflammatory response but does not prevent duct obstruction in experimental biliary atresia. BMC Gastroenterol. 2006;6:14. doi: 10.1186/1471-230X-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tucker RM, Hendrickson RJ, Mukaida N, Gill RG, Mack CL. Progressive biliary destruction is independent of a functional tumor necrosis factor-alpha pathway in a rhesus rotavirus-induced murine model of biliary atresia. Viral Immunol. 2007;20:34–43. doi: 10.1089/vim.2006.0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allen SR, Jafri M, Donnelly B, McNeal M, Witte D, Bezerra J, Ward R, Tiao GM. Effect of rotavirus strain on the murine model of biliary atresia. J Virol. 2007;81:1671–1679. doi: 10.1128/JVI.02094-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jafri M, Donnelly B, Bondoc A, Allen S, Tiao G. Cholangiocyte secretion of chemokines in experimental biliary atresia. J Pediatr Surg. 2009;44:500–550. doi: 10.1016/j.jpedsurg.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barnes BH, Tucker RM, Wehrmann F, Mack DG, Ueno Y, Mack CL. Cholangiocytes as immune modulators in rotavirus-induced murine biliary atresia. Liver Int. 2009;29:1253–1261. doi: 10.1111/j.1478-3231.2008.01921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mano Y, Ishii M, Kisara N, Kobayashi Y, Ueno Y, Kobayashi K, Hamada H, Toyota T. Duct formation by immortalized mouse cholangiocytes: an in vitro model for cholangiopathies. Lab Invest. 1998;78:1467–1468. [PubMed] [Google Scholar]
- 14.Filippi MD, Harris CE, Meller J, Gu Y, Zheng Y, Williams DA. Localization of Rac2 via the C terminus and aspartic acid 150 specifies superoxide generation, actin polarity and chemotaxis in neutrophils. Nat Immunol. 2004;5:744–751. doi: 10.1038/ni1081. [DOI] [PubMed] [Google Scholar]
- 15.Loetscher P, Seitz M, Clark-Lewis I, Baggiolini M, Moser B. Activation of NK cells by CC chemokines. Chemotaxis, Ca2+ mobilization, and enzyme release. J Immunol. 1996;156:322–327. [PubMed] [Google Scholar]
- 16.Murphy WJ, Tian ZG, Asai O, Funakoshi S, Rotter P, Henry M, Strieter RM, Kunkel SL, Longo DL, Taub DD. Chemokines and T lymphocyte activation: II. Facilitation of human T cell trafficking in severe combined immunodeficiency mice. J Immunol. 1996;156:2104–2111. [PubMed] [Google Scholar]
- 17.Taub DD, Longo DL, Murphy WJ. Human interferon-inducible protein-10 induces mononuclear cell infiltration in mice and promotes the migration of human T lymphocytes into the peripheral tissues and human peripheral blood lymphocytes-SCID mice. Blood. 1996;87:1423–1431. [PubMed] [Google Scholar]
- 18.Brown KA, Offit PA. Rotavirus-specific proteins are detected in murine macrophages in both intestinal and extraintestinal lymphoid tissues. Microb Pathog. 1998;24:327–331. doi: 10.1006/mpat.1997.0198. [DOI] [PubMed] [Google Scholar]
- 19.Borghan MA, Mori Y, El-Mahmoudy AB, Ito N, Sugiyama M, Takewaki T, Minamoto N. Induction of nitric oxide synthase by rotavirus enterotoxin NSP4: implication for rotavirus pathogenicity. J Gen Virol. 2007;88:2064–2072. doi: 10.1099/vir.0.82618-0. [DOI] [PubMed] [Google Scholar]
- 20.Haskill S, Peace A, Morris J, Sporn SA, Anisowicz A, Lee SW, Smith T, Martin G, Ralph P, Sager R. Identification of three related human GRO genes encoding cytokine functions. Proc Natl Acad Sci USA. 1990;87:7732–7736. doi: 10.1073/pnas.87.19.7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erickson N, Mohanty SK, Shivakumar P, Sabla G, Chakraborty R, Bezerra JA. Temporal-spatial activation of apoptosis and epithelial injury in murine experimental biliary atresia. Hepatology. 2008;47:1567–1577. doi: 10.1002/hep.22229. [DOI] [PubMed] [Google Scholar]
- 22.Shivakumar P, Sabla GE, Whitington P, Chougnet CA, Bezerra JA. Neonatal NK cells target the mouse duct epithelium via Nkg2d and drive tissue-specific injury in experimental biliary atresia. J Clin Invest. 2009;119:2281–2290. doi: 10.1172/JCI38879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carvalho E, Liu C, Shivakumar P, Sabla G, Aronow B, Bezerra JA. Analysis of the Biliary Transcriptome in Experimental Biliary Atresia. Gastroenterology. 2005;129:713–717. doi: 10.1016/j.gastro.2005.05.052. [DOI] [PubMed] [Google Scholar]
- 24.Ahmed AF, Nio M, Ohtani H, Nagura H, Ohi R. In situ CD14 expression in biliary atresia: comparison between early and late stages. J Pediatr Surg. 2001;36:240–243. doi: 10.1053/jpsu.2001.20063. [DOI] [PubMed] [Google Scholar]
- 25.Davenport M, Gonde C, Redkar R, Koukoulis G, Tredger M, Mieli-Vergani G, Portmann B, Howard ER. Immunohistochemistry of the liver and biliary tree in extrahepatic biliary atresia. J Pediatr Surg. 2001;36:1017–1025. doi: 10.1053/jpsu.2001.24730. [DOI] [PubMed] [Google Scholar]