

Abstract
Endothelial progenitor cells (EPCs) repair damaged endothelium and promote capillary formation, processes involving receptor tyrosine kinases (RTKs) and heme oxygenase-1 (HO-1). Because estradiol augments vascular repair, we hypothesize that estradiol increases EPC proliferation and capillary formation via RTK activation and induction of HO-1. Physiological concentrations of estradiol (10nmol/L) increased EPC-induced capillarysprout and lumenformation in matrigel/fibrin/collagensystems. Propyl-pyrazole-triol [PPT; 100nmol/L; estrogen receptor (ER)-α agonist], but not by diarylpropionitrile (ER-β agonist), mimicked the stimulatory effects of estradiol on capillary formation, and methyl-piperidino-pyrazole (ER-α antagonist) abolished the effects of estradiol and PPT. Three different RTK activators [vascular endothelial growth factor (VEGF), hepatocyte growth factor and stromal derived growth factor-1] mimicked the capillary-stimulating effects of estradiol and PPT. SU5416 (RTK inhibitor)blocked the stimulatory effects of estradiol and PPT on capillary formation. Estradiol increased HO-1 expressionby 2–3 fold, an effect blocked by SU5416, and PPT mimicked the effects of estradiol on HO-1. The ability of estradiol to enhance capillary formation, increaseexpressionof HO-1, and augment phosphorylation ofERK1/2, Akt, and VEGF receptor-2 were mimicked by its cell-impermeable analog BSA-estradiol. Actinomycin (transcription inhibitor) did not alter the effects of estradiol on RTK activity or VEGF secretion. We conclude that estradiol via ER-α promotes EPC-mediated capillary formation by a mechanism that involves non-genomic activation of RTKs and HO-1 activation. Estradiol in particular and ER-αagonists in general may promote healing of injured vascular beds by promoting EPC activity leading to more rapid endothelial recovery and capillary formation following injury.
Keywords: Hormone replacement therapy, estradiol, vascular remodeling, cardiovascular disease, estrogen receptors, endothelial progenitor cells
INTRODUCTION
Evidence from epidemiological studies suggests that endogenous human estrogens and estrogen replacement therapy protect women from progression of cardiovascular disease1,2. Also, multiple animal studies and some small clinical trials support a cardioprotective action of estrogens1,2. In contrast, two large randomized clinical trials [Heart and Estrogen/progestin Replacement Study (HERS) and Women’s Health Initiative (WHI)] fail to demonstrate that exogenous estrogens protect against cardiovascular disease1–4. Although the reasons for these discordant findings remain unclear, a re-evaluation of data from HERS and WHI suggest that in older participants with established cardiovascular pathology estrogen replacement therapy is ineffective, whereas in younger healthy women estrogen therapy is protective3,4. Similarly, Hodis et al. report that estradiol inhibits age-associated increases in intimal thickening in women5. These findings have generated a renewed interest in the mechanisms by which estrogens influence the cardiovascular system. In order to correctly interpret the results of completed and future clinical studies, it is critical to elucidate the mechanisms by which estrogens influence the vessel wall and to identify the independent variables that may influence the vascular actions of estrogens.
Although estrogens induce protective effects on the cardiovascular system via multiple mechanisms6, the effects of estrogens on endothelial cell growth and function may playan important role. For example, estradiol promotes endothelial cell growth, protects endothelial cells against damage by oxidants and cholesterol and induces the generation of endothelial-derived vasodilators such as nitric oxide and prostaglandins6–8. Because estradiol promotes endothelial function and because recent studies suggest that circulating bone marrow-derived endothelial progenitor cells (EPCs) contribute to tissue repair by inducing angiogenesis and neovasculogenesis9, we hypothesize that estradiol may promote endothelial repair in part by stimulating EPC-induced capillary formation.
Estradiol influences cellular growth and differentiation in a variety of tissues in part via estrogen receptors (ERs) -α and -β1,6. Within the cardiovascular system, both ER-α and ER-β mediate the protective actions of estradiol; however, these receptors do not necessarily engage common mechanisms. Indeed, ER-α mediates estradiol-induced NO release, inhibits vascular smooth muscle cell growth and lesion formation and induces endothelial cell growth1,6–10. Arterial blood pressure is increased in ER-β knockout mice11, and in Japanese postmenopausal women a specific polymorphism in the ER-β gene is associated with hypertension12 suggesting that ER-β plays a critical role in lowering blood pressure. These findings provide evidence that ER-α and ER-β may perform distinct functions and that the endogenous regulation of the expression of these two ER subtypes may be important in defining the actions of estradiol on the vessel wall. Because ER-α regulates cell growth, we hypothesize that estradiol may activate EPC-induced capillary formation via ER-α.
Capillary formation (angiogenesis or neovascularization) is a dynamic process regulated by growth factors and signaling pathways. Activators of receptor tyrosine kinases (RTKs), such as vascular endothelial growth factor (VEGF), hepatocyte growth factor (HGF) and stromal derived growth factor-1 (SDF-1) induce EPCs to form capillaries13. Also, angiogenesis appears to be dependent on heme oxygenase-1(HO-1), a 32-kD stress-inducible enzyme which catalyzes the rate limiting step in the degradation of heme, resulting in the liberation of iron, CO and bilverdin13. Growth factors and cytokines induce HO-1 as an adaptive and beneficial response to tissue injury14. Pro-angiogenic factors such as VEGF increase HO-1 expression in endothelial cells in vitro, and inhibition of HO-1 blocks angiogenesis in vivo15. Because estradiol is known to induce VEGF synthesis and endothelial cell growth2,4,6, we hypothesize that estradiol may induce EPC-induced capillary formation via sequential activation of RTKs and HO-1.
The main purpose of the present study was to investigate whether estradiol promotes EPC-induced capillary formation and whether these effects are mediated via ER-α, ER-β or both. Because EPC-induced capillary formation likely is regulated by sequential activation of RTKs and HO-1, a second aim of our study was to investigate their roles in mediating the stimulatory effects of estradiol on capillary formation.
METHODS
Isolation and Culture of Endothelial Progenitor Cells (EPCs)
EPCs were isolated, cultured and characterized as previously described9. For detailed Materials and Methods please see the expanded data in Supplemental Data and table-S1 available online at http://hyper.ahajournals.org.
Capillary Formation Studies
Well established gel (matrigel, collagen and Cytodex-3 beads in fibrin) based assays were employed to study capillary, lumen and sprout formation by EPC derived endothelial cells. Capillary, lumen and sprout formation was assessed microscopically by measuring the length of capillaries or counting lumens and sprout formations. For details see the expanded methods in Supplemental Data available online at http://hyper.ahajournals.org.
Protein Expression Studies
Western blotting, ELISA assays and flow cytometry were employed to assess the role of various proteins in mediating the angiogenic effects of estradiol on EPCs. For details see the expanded methods in Supplemental Data available online at http://hyper.ahajournals.org.
Statistics
Data were analyzed by analysis of variance, and statistical significance (p<0.05) calculated using Fisher’s LSD test.
RESULTS
Cell Characterization
A homogeneous population of CD34 positive cells were isolated following antibody based magnetic separation (Figure-1A). The purity of EPCs was also confirmed by staining with AC133, a marker for stem cell glycoprotein selectively expressed in CD34 positive progenitor cells (Figure-1A). To assess whether progenitor cell-derived endothelial cells were phenotypically similar to endothelial cells, the cells were immunostained for endothelial cell specific markers. As shown in the photomicrographs (Figures-1B, 1C and 1D), EPCs stained positive forvon Willebrand factor, PECAM-1 and MCAM-1. Human aortic smooth muscle cells were used as negative controls and did not show any positive staining. To assess whether EPCs express ERs, EPC lysates were analyzed by Western blotting. Both ER-α and ER-β were highly expressed in EPCs (Figure-1E). To assess whether EPCs were able to form capillaries on matrigel, EPCs were plated on matrigel-coated slides and treated with 10% steroid-free serum. Treatment with serum resulted in robust capillary formation by EPCs (Figure-1F).
Figure 1.
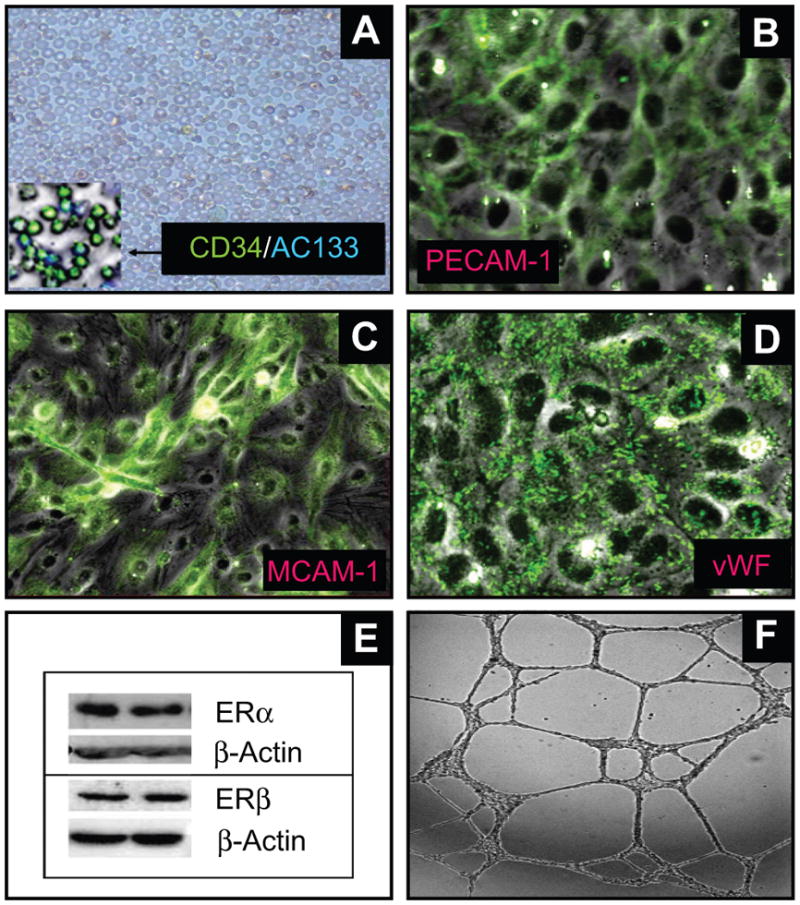
Representative photomicrographs and Western blot showing that circulating CD34+ progenitor cells can transform into endothelial cells and express estrogen receptors. (A) depicts the CD34+ cells isolated following antibody based magnetic separation. The inset depicts the purity of the cells stained with CD34 and AC133 antibodies; Panels B-D depicts that progenitor cell-derived endothelial cells express the endothelial cell markers: platelet endothelial cell adhesion molecule-1 (PECAM-1; B); melanoma cell adhesion molecule-1 (MCAM-1; C) and von Willebrand factor (vWF; D). (E) Representative Western blots demonstrating that EPCs express both estrogen receptors (ERs) α and β. Photomicrograph in panel F depicts the functional capability of EPCs to form capillaries in response to serum.
Capillary Formation
Treatment of EPCs with estradiol increased capillary formation from 168 ±16 μm in vehicle-treated controlcells to 487 ± 24μm in cells treated with 10nmol/L estradiol (p<0.05). The stimulatory effects of estradiol on capillary formation were mimicked by the ER-α agonist propyl-pyrazole-triol (PPT; 100nmol/L) which stimulated capillary formation by 330±23% (p<0.05 versus untreated control; Figure-2A). In contrast to estradiol and PPT, treatment of EPCs with 100 nmol/L of the ER-β agonist diarylpropionitrile (DPN) failed to induce capillary formation (Figure-2A). The stimulatory effects of estradiol on capillary formation were abrogated in EPCs co- treated with the ER-α antagonist methyl-piperidino-pyrazole (MPP; 1μmol/L). MPP also blocked the stimulatory effects of the ER-α agonist PPT on capillary formation (p<0.05 vs EPCs treated with estradiol or PPT alone; Figure-2A). Similar to increasing capillary length, estradiol induced capillary junction formation via ER-α (please see supplemental figure-S1 http://hyper.ahajournals.org). Moreover, estradiol promoted lumen (Figure-2B) and sprout (Figure-2C) formation in EPCs cultured for 24 hours in collagen gels or Cytodex beads for 7 days in fibrin gels, respectively.
Figure 2.

(A) Bar graph and representative photomicrographs showing the effects of estradiol (Est; 10nmol/L), PPT (ER-α agonist; 100nmol/L) and DPN (100nmol/L) on capillary formation by EPCs in the presence and absence of the ER-α antagonist MPP (1μmol/L). Serum starved EPCs were plated at a density of 50,000 cells/200μl/well on matrigel coated 8-well multichamber slides. After 4 hours the capillary formation was assessed by microscopically measuring the length of capillaries at 5 random locations (4x-magnification). Values represent means±SEM. (B) EPC-EC were seeded into collagen gels (3.75mg/ml) in EGM2 medium and incubated for 24 hours as follows: (a) control (Cont); (b) VEGF (40ng/ml); (c) estradiol (Est,10nmol/L); or (d), positive control[phorbol myristate actetate (PMA;50ng/ml) + VEGF]. Cultures were subsequently fixed in 2% paraformaldehyde, stained with toluidine blue and photographed. Shown are mean number of lumens and SD; *(P<0.05), **(P<0.01) relative to control, by Student t test. (C) EPC-EC-coated Cytodex3 beads in fibrin gels were cultured in EGM2 medium as follows: (a) without VEGF; (b) with VEGF (15ng/ml); (c) with estradiol (Est,10nmol/L); or (d) with VEGF+E2. Gels were photographed and the number of sprouts counted after 7 days. Shown are means and SD; *(P<0.05), **(P<0.01) relative to control, by Student t test.
Similar to estradiol and PPT, treatment of EPCs with 100ng/ml of VEGF, HGF or SDF-1 stimulated EPC-induced capillary formation (Figure-3A). The stimulatory effects of estradiol on lumen and sprout formation were also mimicked by VEGF and phorbol myristate acetate (Figure-2B, 2C). The efficacies of the RTK activators on capillary formation were comparable to those of estradiol and PPT. Also, the stimulatory effects of VEGF, HGF and SDF-1 on capillary formation were abolished when EPCs were co-treated with 5μmol/L of the RTK inhibitor SU5416 (Figure-3A). Treatment of EPCs with SU5416 also abrogated the stimulatory effects of estradiol and PPT on EPC-induced capillary formation by 93% (p<0.05 versus EPCs treated with estradiol or PPT alone; Figure-3B). Similar to the effects on capillary length, estradiol and PPT induced junction formation and these effects were mimicked by RTK activators and blocked by SU5416 (please see supplemental figure-S2 http://hyper.ahajournals.org).
Figure 3.

(A) Bar graph and representative photomicrographs showing the effects of receptor tyrosine kinase stimulators vascular endothelial growth factor (VEGF;100ng/mL), hepatocyte growth factor (HGF, 100ng/mL) and stromal cell-derived growth factor (SDF-1 100ng/mL) on capillary formation by EPCs in the presence and absence of the tyrosine kinase inhibitor SU5416 (SU; 5μmol/L). Serum starved EPCs were plated at a density of 50,000 cells/200μl/well on matrigel coated 8-well multichamber slides. After 4 hours the capillary formation was assessed by microscopically measuring the length of capillaries at 5 random locations (4x-magnification). (B) Bar graph and representative photomicrographs showing the modulatory effects of the tyrosine kinase inhibitor SU5416 (SU;5μmol/L) on estradiol (10nmol/L) and PPT (100nmol/L) induced capillary formation in EPCs. Values represent means±SEM. * p<0.05 versus vehicle treated controls.
HO-1 Expression
As shown in Figure-4A, treatment of EPCs with 10 nmol/L of estradiol induced HO-1 expression by 2-fold (from 100 ± 0.3% to 228 ± 7.8%, p<0.05 versus untreated control). Treatment of EPCs with PPT, but not DPN, mimicked the stimulatory effects of estradiol on HO-1 expression (Figure-4A). Treatment with PPT induced HO-1 expression by almost 280% (p<0.05 vs control). The ER-α antagonist MPP abolished the stimulatory effects of estradiol and PPT on HO-1 expression (Figure-4B). Moreover, the stimulatory effects of estradiol and PPT on HO-1 expression were also abrogated by the RKT inhibitor SU5416 (p<0.05 versus EPCs treated with estradiol or PPT; Figure-5A).
Figure 4.

Bar graph and representative Western blots demonstrating that estradiol induces hemeoxygenase-1 (HO-1) expression in EPCs via estrogen receptor (ER)-α. Panel A depicts the effects of estradiol (Est; 10nmol/L), PPT (ER-α agonist; 100nmol/L) and DPN ( ER-β agonist; 100nmol/L) on HO-1 expression in EPCs treated for 4 hours. Panel B depicts the effects of Est (10nmol/L) and PPT (100nmol/L) on HO-1 expression in EPCs in the presence and absence of the ER-α antagonist MPP (1μmol/L). Cells were pretreated for 15-minutes with MPP and subsequently estradiol or PPT were added for an additional 4 hours. Values represent means±SEM.
Figure 5.

(A) Bar graph and representative Western blots demonstrating that inhibition of tyrosine kinase with SU5416 (SU) abrogates the stimulatory effects of estradiol and PPT on HO-1 expression in EPCs. EPCs were pretreated with SU (5μmol/L) for 15 minutes and subsequently estradiol (Est; 10nmol/L) or PPT (100nmol/L) was added for an additional 4 hours. HO-1 expression was assayed in the lysates with β-actin as control. Bargraph depicts the densitometry analysis of the HO-1 bands, which were normalize to β-actin. * p<0.05 versus vehicle treated control.
Panel B: Depicts the modulatory effects of SU5416 (SU) on estradiol-induced ERK1/2 and Akt phosphorylation in EPCs. Cells were pretreated with SU for 15 minutes and subsequently estradiol (100nmol/L) was added for an additional 10 minutes. Cell lysates were prepared and expression of phosphorylated ERK1/2 and Akt were analysed by Western blotting. For normalization non-phosphorylated ERK1/2 and Akt were used. Bar graph depicts the densitometry analysis of the phosphorylated ERK1/2 (ERK1/2-P) and Akt (Akt-P) bands, which were normalize to ERK1/2 and Akt, respectively. * p<0.05 versus vehicle treated control. Values represent means±SEM.
(C) Bar graph showing the inhibitory effects of LY294002 (Akt inhibitor; LY; 10μmol/L) and PD98059 (ERK1/2-P inhibitor; PD; 10μmol/L) on estradiol (Est; 10nmol/L) and PPT (100nmol/L) induced capillary formation by EPCs. EPCs were pretreated for 15 minutes with either LY or PD and subsequently Est or PPT added for another 4hoursand capillary formation analyzed microscopically at 4x magnifcation. * p<0.05 vs Estradiol or PPT alone.
ERK1/2 and Akt Phosphorylation
Treatment of EPCs with estradiol (10nmol/L) significantly upregulated the expression of phosphorylated ERK1/2 and Akt (Figure-5B). Moreover these stimulatory effects were abolished by the RTK inhibitor SU5416 (Figure-5B). Treatment with the ERK1/2 pathway inhibitor PD98059 blocked the stimulatory effects of estradiol and PPT on capillary formation (Figure-5C top panel). Similarly, the Akt pathway inhibitor LY 294002blocked estradiol- and PPT-induced capillary formation (Figure-5C lower panel). Both ERK1/2 and Akt pathway inhibitors also blocked the stimulatory effects of estradiol on capillary junction formation (please see supplemental figure-S3 http://hyper.ahajournals.org).
Non-Genomic Actions on Capillary Formation
In EPCs treated with FITC labeled BSA-tagged estradiol, nuclear staining was observed in permeabilized but not intact cells (Figure-6A). Similar to estradiol, treatment of EPCs with impermeable BSA-estradiol (10nmol/L) significantly induced capillary formation (Figure-6B) and these effects were blocked by ICI (ER-α/β antagonist), MPP (ER-α antagonist), SU5416 (RTK inhibitor), PD98059 (ERK1/2 inhibitor) and LY294002 (Akt pathway inhibitor). Similar stimulatory effects of BSA-estradiol were also observed on capillary junction formation (please see supplemental figure-S4http://hyper.ahajournals.org). Moreover, like estradiol, treatment with impermeable BSA-estradiol upregulated the expression of HO-1 as well as phosphorylated ERK1/2 and Akt (Figure-6C).
Figure 6.

(A) Representative photomicrographs providing evidence that BSA-estradiol is impermeable and stains the nucleus of permeabilized, but not intact, EPCs (original magnification 20x); (B) Bar graph and representative photomicrographs showing the modulatory effects of receptor tyrosine kinase inhibitor SU5416 (SU; 5μmol/L), LY294002 (Akt pathway inhibitor; LY, 10μmol/L) and PD98059 (ERK1/2 pathway inhibitor; PD, 10 μmol/L) and MPP (ER-α antagonist, 1μmol/L) on BSA-estradiol (Est-BSA, 10nmol/L) induced capillary formation in EPCs. Serum starved EPCs were plated at a density of 50,000 cells/200μl/well on matrigel coated 8-well multichamber slides. Cells were pretreated for 15 minutes with MPP, SU, LY or PD and subsequently BSA-estradiol was added for an additional 4 hours and capillary formation was assessed by microscopically measuring the length of capillaries at 5 random locations (4x-magnification). Values represent means±SEM. §p<0.05 versus vehicle treated controls; *p<0.05 versus Est-BSA. (C) Representative Western blots demonstrating that BSA-estradiol (Est-BSA) induces the expression of hemeoxygenase-1 (HO-1), phosphorylated ERK1/2 and Akt. Cells were treated with Est-BSA (10nmol/L) for 10 minutes, lysates prepared and the expression of phosphorylatedERK1/2 and Akt analysed using Western blots. β-Actin, non-phosphorylated Akt and ERK1/2were used for normalization. (D) Bar graph showing the stimulatory effects of estradiol on tyrosine kinase activity in lysates prepared from EPCs treated for 10 minutes with estradiol (Est;10nmol/L) and estradiol+actinomycin (Actino;100nmol/L). *p<0.05 versus vehicle treated control (Cont). Values represent means±SEM.(E) Representative flow cytometry histogram and Western blots showing the effects of estradiol on VEGFR-2 expression in EPCs treated for 8 and 24 hours.(F) Bar graph showing the stimulatory effects of VEGF (100ng/ml), 10nmol/L of estradiol (Est), estradiol plus actinomycin (100nmol/L) and BSA-estradiol (Est-BSA) on phosphorylated VEGFR-2 in lysates prepared from EPCs treated for 10 minutes. *p<0.05 versus vehicle treated control(Cont). Values represent means±SEM.(G) Bar graph showing the extracellular levels of VEGF in EPCs treated for 8 hours with estradiol (10nmol/l) in presence and absence of actinomycin (100nmol/L). *p<0.05 versus vehicle treated control (Cont). Values represent means±SEM. (H) Bar graph showing the capillary forming effects of VEGF (10ng/ml) in presence and absence of estradiol (10nmol/L). Values represent means±SEM. *p<0.05 versus vehicle treated control (Cont).
Tyrosine Kinase Activation
Treatment of EPCs with estradiol (10nmol/L) for 10 minutes significantly induced tyrosine kinase activity, and these effects were not blocked by 100nmol/L of actinomycin (Figure-6D). Treatment of EPCs with estradiol (10nmol/L) did not induce VEGFR-2 expression (Figure-6E); however, the levels of phospho-VEGFR-2 were significantly induced in lysates of EPCs treated for 10 minutes with estradiol or its impermeable analog BSA-estradiol (Figure-6F), and these effects of estradiol were not blocked by actinomycin (Figure-6F). Treatment of EPCs with estradiol for 8-hours significantly induced VEGF production and this effect also was not blocked by actinomycin (Figure-6G). Estradiol did not enhance the stimulatory effects of VEGF on tube formation (Figure-6H).
DISCUSSION
This study demonstrates several important findings in human circulating EPCs: (1) Estradiol stimulates capillary formation by EPCs; (2) The capillary-stimulating effects of estradiol are mimicked by a specific agonist for ER-α (PPT), but not for ER-β (DPN); (3) MPP (ER-α specific antagonist) blocks the stimulatory effects of estradiol and PPT on capillary formation; (4) The stimulatory effects of estradiol and PPT on capillary formation are mimicked by RTK activators (VEGF, HGF and SDF-1) and blocked by the RTK inhibitor SU5416; (5) Expression of the well-known angiogenesis-stimulating enzyme HO-1 is induced by estradiol and by the ER-α agonist PPT, but not by the ER-β agonist DPN; (6) The stimulatory effects of estradiol and PPT on HO-1 expression are blocked by the ER-α antagonist MPP as well as by the RTK inhibitor SU5416; (7) Treatment with estradiol induces Akt and ERK1/2 phosphorylation, and inhibition of the ERK1/2 and Akt pathways by PD98059 and LY294002, respectively, abrogates the capillary stimulatory effects of both estradiol and PPT; and (8) Estradiol stimulates tyrosine kinase activity and phosphorylation of the RTK VEGFR-2. Together, our findings provide strong evidence that estradiol promotes EPC-induced capillary formation, that these effects are ER-α mediated and that the stimulatory effects of estradiol and PPT are mediated via activation of RTKs leading to induction of HO-1 with involvement of the Akt and ERK1/2 signal transduction pathways. Importantly, our findings that the stimulatory effects of estradiol on tube formation, HO-1 expression and phosphorylation of Akt, ERK1/2 and VEGFR-2 are mimicked by the membrane impermeable BSA-estradiol and that the stimulatory effects estradiol on tyrosine kinase activity and VEGF production are not blocked by actinomycin indicate that estradiol induces tube formation via a non-genomic mechanism involving membrane ERs.
Some of the actions of estradiol would be expected to improve vascular health. For example, estradiol induces growth of endothelial cells and speeds the recovery of denuded endothelium4,7, thus enabling the vessel wall to recover more rapidly after injury. Alternative sources of endothelial cells, for example bone marrow derived circulating EPCs9, may also participate in vascular repair, and studies show that estradiol promotes EPC growth and recruits EPCs to sites of injury. The present study shows for the first time that estradiol actually activates EPC-induced capillary formation. Our finding that estradiol promotes EPC-induced capillary formation suggests that estradiol may facilitate tissue repair in part via this mechanism.
The biological effects of estradiol are largely mediated via ER-α and ER-β1,6. Our finding that the stimulatory effect of estradiol on capillary formation is mimicked by the ERα agonist PPT, but not by the ER-β agonist DPN, provides evidence that the effects are indeed ER-α mediated. It is important to note that because selective estrogen receptor modulators (SERMs) are being developed for safer and more effective hormone replacement therapy in postmenopausal women, use of PPT as a SERM to promote endothelial and tissue recovery may be of therapeutic importance16. This concept is further supported by the facts that via ER-α estradiol inhibits injury-induced neointima formation10, induces nitric oxide release and prevents bone loss and hot flushes16.
The mechanisms involved in EPC induced capillary formation or angiogenesis remain unclear. However, it is well established that activation of the RTK pathway by ligands such as VEGF and HGF enhances EPC-induced capillary formation17. Our finding that RTK activators mimic the effects of estradiol on capillary formation and that the RTK inhibitor SU5416 blocks the effects of estradiol and PPT on capillary formation suggest that the stimulatory effects of estradiol on capillary formation are mediated by activation of RTKs. Also, our observations that estradiol induces ERK1/2 and Akt phosphorylation and that the Akt pathway inhibitor LY294002 and the ERK1/2 pathway inhibitor PD98059 attenuate the stimulatory effects of estradiol and PPT on capillary formation suggests that via ER-α estradiol can stimulate capillary formation via activation of these signal transduction mechanisms. It is likely that the Akt and ERK1/2 pathways are downstream of RTK activation because it is well-known that RTKs activate these classic signal transduction pathways17.
Previous studies implicate a role for HO-1 in angiogenesis13. Pharmacologic or genetic manipulations that increase HO-1 expression enhance proliferation and tube formation in human microvascular endothelial cells in vitro18, whereas inhibition of HO-1 decreases tube formation, a phenomenon independent of HO-218. RTKs and the Akt and ERK1/2 signal transduction pathways activate HO-119, and HO-1 mediates their effects on angiogenesis17. Our finding that estradiol and the ER-α agonist PPT, but not the ER-β agonist DPN, induce HO-1 expression links the pro- angiogenic effects of estradiol to HO-1. This notion is further supported by the fact that the stimulatory effects of estradiol and PPT on both capillary formation and HO-1 expression are blocked by the ER-α antagonist MPP and by the RTK inhibitor SU5416. Although our findings implicate HO-1 as a likely intermediate in estradiol-induced capillary formation, the mediators downstream from HO-1 remain unknown. In this regard, detailed studies are required to determine whether HO-1-derived CO or another molecule, such as VASP which is known to stimulate capillary formation by EPCs13,17, is involved in transducing the angiogenic effects of estradiol.
Estradiol induces multiple vascular actions via non-genomic mechanisms20 and increases VEGF synthesis and mobilizes the RTK VEGFR-221. Our finding that the effects of estradiol are mimicked by its impermeable analog BSA-estradiol suggests the participation of a non-genomic mechanism in estradiol-induced tube formation. The observations that estradiol induces RTK, that these effects are not blocked by actinomycin and that the membrane impermeable BSA-estradiol induces VEGFR-2 phosphorylation suggest that estradiol can activate this key pathway responsible for capillary induction. Because the effect of estradiol to increase VEGF secretion is not affected by actimomycin, it is likely that estradiol stimulates the release of VEGF from intracellular stores via a non-genomic mechanisms22. Based on the above findings it is feasible that estradiol induces capillary formation by stimulating the release of VEGF, which in turnactivates RTKs, ERK1/2, Akt and HO-1. Although our findings suggest that estradiol can stimulate capillary formation via a non-genomic mechanism, the participation of nuclear receptors or genomic activation cannot be ruled out and requires further investigated. Because the TRK family constitutes more than 58 receptors, it is feasible that receptors other than VEGFR-2 also participate in mediating estradiol’s tube forming effects.
Our observation that estradiol stimulates capillary formation viaanon-genomic mechanism suggests that estradiol may repair endothelial damage within vascular beds. Consistent with this concept, in vivo studies provide evidence that estradiol promotes accumulation of progenitor endothelial cells at sites of injury23. Our finding that estradiol promotes capillary formation within hours would imply that estradiol could facilitate tissue repair by reestablishing capillary formation to improve perfusion within the damaged vascular sites.
In summary, the present study provides evidence that estradiol induces capillary formation by human EPCs and these effects are ER-α mediated. The stimulatory effects of estradiol are likely due to RTK-mediated induction of HO-1. The Akt and ERK1/2 signal transduction pathways are also involved, perhaps as intermediates between RTK activation and HO-1 induction.
Perspective
Here we provide strong evidence that estradiol promotes EPC-mediated capillary formation via ER-α and that the stimulatory actions of estradiol are mediated via RTK and HO-1 activation. Because HO-1 induction occurs as an adaptive and beneficial response to tissue injury, estradiol in particular and ER-α agonists in general may protect the vascular system by promoting EPC activity leading to more rapid endothelial recovery and capillary formation following injury. Apart from repairing vascular endothelium, estradiol-induced capillary formation would also facilitate tissue repair in the heart following myocardial infarction by reestablishing capillaries and blood supply.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by Swiss National Science Foundation grant 32-64040.00, 320000-117998, Oncosuisse OCS-01551-08-2004, EMDO forschung, California CRCC (CCWH) and NIH grants RO1-HL60067 (CCWH), HL069846, DK068575, DK079307.
Footnotes
Disclosures: CCWH has two NIH-RO1 grants on angiogenesis and RKD one NSF-grant on estrogen metabolism.
References
- 1.Dubey RK, Jackson EK. Estrogen-induced cardiorenal protection: potential cellular, biochemical, and molecular mechanisms. Am J Physiol Renal Physiol. 2001;280:F365–F388. doi: 10.1152/ajprenal.2001.280.3.F365. [DOI] [PubMed] [Google Scholar]
- 2.Dubey RK, Imthurn B, Barton M, Jackson EK. Vascular consequences of menopause and hormone therapy: importance of timing of treatment and type of estrogen. Cardiovasc Res. 2005;66:295–306. doi: 10.1016/j.cardiores.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 3.Clarkson TB, Appt SE. Coronary artery disease and postmenopausal hormone therapy: is there a time for prevention? Gynaecol Forum. 2004;9:11–14. [Google Scholar]
- 4.Miller VM, Duckles SP. Vascular Actions of estrogens: functional implications. Pharmacol Rev. 2008;60:210–241. doi: 10.1124/pr.107.08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hodis HN, Mack WJ, Lobo RA, Shoupe D, Sevanian A, Mahrer PR, Selzer RH, Liu CrCR, Liu ChCH, Azen SP. Estrogen in the prevention of atherosclerosis. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2001;135:939–953. doi: 10.7326/0003-4819-135-11-200112040-00005. [DOI] [PubMed] [Google Scholar]
- 6.Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med. 1999;340:1801–1811. doi: 10.1056/NEJM199906103402306. [DOI] [PubMed] [Google Scholar]
- 7.Losordo DW, Inser JM. Estrogen and angiogenesis: A Review. Arterioscler Thromb Vasc Biol. 2001;21:6–12. doi: 10.1161/01.atv.21.1.6. [DOI] [PubMed] [Google Scholar]
- 8.Rosselli M, Imthurn B, Keller PJ, Jackson EK, Dubey RK. Circulating nitric oxide (nitrite/nitrate) levels in postmenopausal women substituted with 17beta-estradiol and norethisterone acetate. A two-year follow-up study. Hypertension. 1995;25:848–53. doi: 10.1161/01.hyp.25.4.848. [DOI] [PubMed] [Google Scholar]
- 9.Dzau VJ, Gnecchi M, Pachori AS, Morello F, Melo LG. Therapeutic potential of endothelial cells in cardiovascular diseases. Hypertension. 2005;46:7–18. doi: 10.1161/01.HYP.0000168923.92885.f7. [DOI] [PubMed] [Google Scholar]
- 10.Pare G, Krust A, Karas RH, Dupont S, Aronovitz M, Chambon P, Mendelsohn ME. 2002 Estrogen receptor-α mediates the protective effects of estrogen against vascular injury. Circ Res. 2002;90:1087–1092. doi: 10.1161/01.res.0000021114.92282.fa. [DOI] [PubMed] [Google Scholar]
- 11.Zhu Y, Bian Z, Lu P, Karas RH, Bao L, Cox D, Hodgin J, Shaul PW, Thorén P, Smithies O, Gustafsson JA, Mendelsohn ME. Abnormal vascular function and hypertension in mice deficient in estrogen receptor-β. Science. 2002;295:505–508. doi: 10.1126/science.1065250. [DOI] [PubMed] [Google Scholar]
- 12.Ogawa S, Emi M, Hosoi T, Ouchi Y, Inoue S. Association of estrogen receptor beta (ESR2) gene polymorphism with blood pressure. J Hum Genet. 2000;45:327–30. doi: 10.1007/s100380070002. [DOI] [PubMed] [Google Scholar]
- 13.Dulak J, Deshane J, Jozkowicz A, Agarwal A. Heme oxygenase-1 and carbon monoxide in vascular pathobiology: focus on angiogenesis. Circulation. 2008;117:231–241. doi: 10.1161/CIRCULATIONAHA.107.698316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nath KA. Heme oxygenase-1: a provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int. 2006;70:432–443. doi: 10.1038/sj.ki.5001565. [DOI] [PubMed] [Google Scholar]
- 15.Fang J, Sawa T, Akaike T, Akuta T, Sahoo SK, Khaled G, Hamada A, Maeda H. In vivo antitumor activity of pregylated zinc protoporphyrin: targeted inhibition of heme oxygenase in solid tumor. Cancer Res. 2003;63:3567–3574. [PubMed] [Google Scholar]
- 16.Harris HA, Katzenellenbogen JA, Katzenellenbogen BS. Characterization of the biological roles of the estrogen receptors, ERalpha and ERbeta, in estrogen target tissues in vivo through the use of an ERalpha-selective ligand. Endocrinology. 2002;143:4172–4177. doi: 10.1210/en.2002-220403. [DOI] [PubMed] [Google Scholar]
- 17.Deshane J, Chen S, Caballero S, Grochot-Przeczek A, Was H, Calzi SL, Lach R, Hock TD, Chen B, Hill-Kapturczak N, Siegal GP, Dulak J, Jozkowicz A, Grant MB, Agarwal A. Stromal cell-derived factor-1 promotes angiogenesis via a heme oxygenase 1- sependent mechanism. J Exp Med. 2007;204:605–618. doi: 10.1084/jem.20061609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Volti L, Sacerdoti GD, Sangras B, Vanella A, Mezentsev A, Ssapagnini G, Palck JR, Abraham NG. Carbon monoxide signalling in promoting angiogenesis in human microvessel endothelial cells. Antioxid Redox Signal. 2005;7:704–710. doi: 10.1089/ars.2005.7.704. [DOI] [PubMed] [Google Scholar]
- 19.Dulak J, Loboda A, Jozkowicz A. Effect of heme oxygenase-1 on vascular function and disease. Curr Opin Lipidol. 2008;19:505–512. doi: 10.1097/MOL.0b013e32830d81e9. [DOI] [PubMed] [Google Scholar]
- 20.Meyer MR, Haas E, Prossnitz ER, Barton M. Non-genomic regulation of vascular cell function and growth by estrogen. Mol Cell Endocrino. 2009;308:9–16. doi: 10.1016/j.mce.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garvin S, Nilsson UW, Dabrosin C. Effects of oestradiol and tamoxifen on VEGF, soluble VEGFR-1 and VEGFR-2 in breast cancer and endothelial cells. British J Cancer. 2005;93:1005–1010. doi: 10.1038/sj.bjc.6602824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koehne P, Willam C, Strauss E, Schindler R, Eckardt R-U, Bührer C. Lack of hypoxic stimulation of VEGF secretion from neutrophils and platelets. Am J Physiol Circ Physiol. 2000;279:H817–H824. doi: 10.1152/ajpheart.2000.279.2.H817. [DOI] [PubMed] [Google Scholar]
- 23.Hamada H, Kim MK, Iwakura A, Ii M, Thorne T, Qin G, Asai J, Tsutsumi Y, Sekiguchi H, Silver M, Wecker A, Bord E, Zhu Y, Kisore R, Losordo DW. Estrogen recptors αand β mediate contrinution of bone marrow-derived endothelial progenitor cells to functional recovery after myocardial infarction. Circulation. 2006;114:2261–2270. doi: 10.1161/CIRCULATIONAHA.106.631465. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.