

Abstract
Voltage-gated Na+ channels (VGSCs) have been implicated in the metastatic potential of human breast, prostate and lung cancer cells. Specifically, the SCN5A gene encoding the VGSC isotype Nav1.5 has been defined as a key driver of human cancer cell invasion. In this study, we examined the expression and function of VGSCs in a panel of colon cancer cell lines by electrophysiological recordings. Na+ channel activity and invasive potential were inhibited pharmacologically by tetrodotoxin or genetically by siRNAs specifically targeting SCN5A. Clinical relevance was established by immunohistochemistry of patient biopsies, where there was strong Nav1.5 protein staining in colon cancer specimens but little to no staining in matched-paired normal colon tissues. We explored the mechanism of VGSC-mediated invasive potential on the basis of reported links between VGSC activity and gene expression in excitable cells. Probabilistic modeling of loss-of-function screens and microarray data established an unequivocal role of VGSC SCN5A as a high level regulator of a colon cancer invasion network, involving genes that encompass Wnt signaling, cell migration, ectoderm development, response to biotic stimulus, steroid metabolic process and cell cycle control. siRNA-mediated knockdown of predicted downstream network components caused a loss of invasive behavior, demonstrating network connectivity and its function in driving colon cancer invasion.
Keywords: voltage-gated Na+ channels, invasion, colon cancer, gene network
INTRODUCTION
An increasing body of evidence is accumulating on the importance and functional contribution of ion channels, signaling molecules involved in ion transport, enzyme activity, secretion, and intercellular communication, in cancer (1–4). Voltage-gated Na+ channels (VGSCs) are most abundant in excitable cells such as neurons and cardiomyocytes where they are responsible for the depolarization phase of the action potential and are important for neurite extension and neurotransmitter release (5, 6). VGSC activation can lead to increased Na+ influx, resulting in alterations in both intracellular Ca2+ concentration and pH and additional changes in normal cellular homeostasis. Interestingly, a study performed several decades ago indicated that tumor samples had higher intracellular concentrations of Na+ compared to normal tissues, and this phenomenon was postulated to be connected with oncogenesis (7). More recent studies now implicate VGSCs in the invasive potential of prostate (8), breast (9) and lung (10) cancer cells. However, it is unclear the exact mechanism(s) by which VGSC genes and/or their functional expression confer an oncogenic advantage to cancer cells. One study has demonstrated that the activity of the VGSC α-subunit Nav1.5, encoded by the SCN5A gene and normally associated with human cardiac tissue, increases the invasiveness of human breast cancer cells possibly by providing favorable conditions for proteolytic activity on extracellular matrix proteins (11). In another study investigating melanoma cells, Nav1.8 (a paralog of the Nav1.5 isoform) has been demonstrated to facilitate podosome formation (12).
Meta-analysis of gene expression profiling data from colon cancer patient samples and oncogenic transformation of fibroblasts suggests that genes typically associated with neuronal or excitable cells, including ion channels and intracellular signaling molecules, may be `commandeered' as part of the process of cancer progression (13, 14). In addition to expression of ion channel genes, a number of neuronal molecular markers have also been described in other transformed tissues, such as small-cell lung cancer (15). Little is known about the transcriptional regulation of VGSC genes or the downstream genes regulated by VGSCs in the context of cancer progression. For example, there have been no systematic studies examining VGSC activity together with changes in gene expression and invasion potential of cancer cells. Ion channel activity stimulates a variety of intracellular signaling pathways (16, 17) and the functional state of ion channels is known to affect gene expression in neuronal and skeletal muscle cells (18, 19).
The regulatory connections among genes involved in tumor cell invasion remain rudimentary and characterization of a gene network in the invasiveness pathway is vital to fully appreciate the molecular mechanisms underlying metastasis (20). Such gene-gene network interactions can be reconstructed from the transcriptional consequences of RNA interference-mediated knockdown of network components (21). The goal of this study was to establish whether functional expression of VGSCs in colon cancer cells contributes to invasion potential through transcriptional regulation of downstream invasion/migration genes.
MATERIALS AND METHODS
Immunohistochemistry
Fresh-frozen or paraffin-embedded colon tissues were cut into 10 micron sections and processed using DakoCytomation Envision+ System − HRP (DAB) kit according to manufacturer's instructions (Dako). Sections were incubated with anti-Nav1.5 polyclonal antibody (1:100) (Alomone labs) for three hours, followed by one hour with HRP labeled polymer conjugated anti-rabbit secondary antibody. Sections were counterstained with dilute Mayer's hemotoxylin (Dako). Quantification of DAB staining was performed as described previously (22). All incubations were done at room temperature. Sections from each specimen were fixed in methanol and stained with H&E, CEA and Mak6 to confirm tissue integrity. Experiments were approved through the George Washington University Medical Center Institutional Review Board.
Cell Culture and Small Interfering RNA (siRNA) Transfections
Human colon cancer cells, HT29 (Catalog No. HTB-38), SW620 (Catalog No. CCL-227), SW480 (Catalog No. CCL-228), HEK293 (Catalog No. CRL-1573), and Caco-2 (Catalog No. HTB-37) were obtained directly from American Type Culture Collection (ATCC) and used within six months of receipt. ATCC authenticates cell lines through short tandem repeat profiling, morphology analysis, karyotyping, and isoenzyme analysis (ATCC cell line verification test recommendations, technical bulletin no. 8 (2007)). Cells were maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum at 37°C and 5% CO2. Cells were cultured for 24 hours to 50% confluence before transfection with Dharmacon On-Targetplus siRNA duplexes according to manufacturer's instructions (Thermo Fisher Scientific). Two different siRNAs were used separately for each gene. SiRNA sequences are provided in Supplementary Table S1.
Immunocytochemistry
Cells were cultured for 24 hours on glass coverslips to 30% confluence. For differentiated Caco-2 cells, culture was continued for 10 days after cells reached confluency. Samples were fixed using 4% paraformaldehyde, stained with anti-Nav1.5 polyclonal antibody (6 ug/ml) (Abcam), and detected by goat anti-rabbit Alexa 488 (1:500) (Invitrogen). Images were acquired using a Zeiss LSM510 confocal imaging system using 60× 1.4 DIC Apochromat objective. Identical acquisition settings were used for all samples.
Electrophysiology
Cells were cultured for 48 hours to 30% confluence in 35-mm dishes for electrophysiological studies. The whole-cell patch-clamp technique was used to record voltage-activated currents from individual cells. The electrode solution contained 140 mM CsCl, 2 mM MgCl2, 0.1 mM CaCl2, 1.1 mM EGTA, and 10 mM HEPES (pH 7.2). The extracellular solution contained 140 mM NaCl, 4.7 mM KCl, 1.2 mM MgCl2, 2.5 mM CaCl2, 10 mM HEPES, and 11 mM glucose (pH 7.4) with and without the indicated concentrations of TTX. Currents were recorded using an Axopatch 200B amplifier, low-pass filtered at 5 KHz, digitized at 10 KHz using a Digidata 1320A interface, and acquired using pCLAMP8 software (all from Molecular Devices, Sunnyvale, CA).
RNA Isolation, Microarrays and qRT-PCR
Total RNA was isolated using TRIzol reagent (Invitrogen) and RNeasy kit (Qiagen) according to manufacturers' instructions. Gene expression profiling and statistical analysis was performed as previously described (13, 14, 20). The hybridization data and associated normalization information can be accessed from the Gene Expression Omnibus (GEO) database under the series accession number GSE11848 and associated platform accession number GPL6978. For validation of gene knockdowns, qRT-PCR was performed as described previously (23). Housekeeping genes, PPA1 and EIFAX (Genbank Accession number NM_021129 and NM_001412, respectively), were used for normalization. Quantitation and normalization of relative gene expression were accomplished using the comparative threshold cycle method or ΔΔCT (20). Primer sequences are provided in Supplementary Table S1.
Matrigel Assay
Colon cancer cells (2.5–10 × 104) were seeded in the top well of a Matrigel coated invasion chamber (BD Biosciences) in DMEM containing 0.1% serum. The bottom well was filled with 750 μl DMEM containing 10% serum as chemoattractant. After ~48 hours, non-invading cells were scraped from the upper side using a cotton swab. Invading cells on the bottom of the insert were fixed and stained with Diff-Quik Stain (IMEB, Inc.) and counted under a light microscope. Total number of invading cells was counted for each insert.
Inference of Signaling Network
Network inferences by factor graph nested effects modeling (FG-NEM) was performed as previously described (23). Briefly, the method takes as input a matrix of expression level changes for a set of “effect” genes (E-genes) exhibiting minimal variance across gene knockdowns. Each column of the matrix represents the expression of E-genes under the knockdown of a particular signaling gene (S-gene). By searching for probabilistic nested relationships among the set of expression changes observed for the E-genes, the procedure returns network interactions among the S-genes. Additional details for FG-NEM, network bootstrap confidence determination, and network frontier expansion can be found in Supplementary Materials and Methods.
RESULTS AND DISCUSSION
SCN5A is functionally expressed in colon cancer cell lines
Quantitative real time RT-PCR (qRT-PCR) was performed on an assortment of cell lines to assess expression of VGSC isoforms (see Supplementary Table S2). The set of colon cancer cell lines SW620, SW480 and HT29 expressed multiple isoforms with the most abundant generally being SCN5A, a tetrodotoxin (TTX) resistant isoform. Protein expression and localization of Nav1.5 in colon cancer cells was confirmed immunochemically using an antibody against Nav1.5. Punctate expression of Nav1.5 protein at the plasma membrane was observed in all 3 cell lines with no nuclear localization (Fig. 1A). To our knowledge this is the first study demonstrating expression of this isoform in colon cancer cells.
Figure 1.

Nav1.5 VGSCs are functionally expressed in colon cancer cells. (A) Representative merged images demonstrating Nav1.5 immunoreactivity in SW620, SW480, and HT29 colon cancer cells. Anti-Nav1.5 conjugated to Alexa 488 in green and DAPI nuclear staining in blue. Images are representative of at least three independent experiments (Scale bar 10 μm). (B) Superimposed Na+ currents elicited by depolarizing from −80 mV to between −50 and 0 mV (top traces) and between 10 and 60 mV (bottom traces) in 10 mV increments. (C) Current-voltage (I–V) relationship of Na+ currents elicited by depolarizing from a holding potential of −80 mV to between −70 and 60 mV in 10 mV voltage steps. Current amplitudes were normalized to maximum peak current recorded from each cell. Data are averages of 36, 16, and 15 recordings from SW620 cells, SW480, and HT29 colon cancer cells, respectively. Vertical lines represent ± SEM. Error bars for SW620 and SW480 recordings are within the data symbols. (D) TTX-mediated inhibition of current after bath application of 10 μM TTX. Current is restored after subsequent wash.
To demonstrate functional expression of the VGSCs in HT29, SW480 and SW620 cells, Na+ currents were recorded by whole-cell patch clamp technique (Fig. 1B). Top traces represent currents elicited by depolarizing cells from −80 mV to between −50 and 0 mV, at 10 mV increments. Bottom traces represent currents elicited by stepping from −80 mV to between 10 and 60 mV, in 10 mV increments. The average maximum current densities for HT29, SW480, and SW620 were 7.2 ± 2.5 (n=15), 5.3 ± 1.6 (n=16) and 17.9 ± 2.2 pA/pF (n=36), respectively, compared to 57 ± 8.9 pA/pF (n=20) for SK-N-SH neuroblastoma cells. Average current voltage relationship plots demonstrate similar electrophysiological characteristics among the colon cancer cell lines with an average threshold of activation at ~−40 mV and peak current amplitude between 0 and 10 mV (Fig. 1C). The activation and inactivation curves in SW620 cells for example are very similar to those recorded previously from cardiac myocytes (24) and it appears that a small fraction of current (window current) is available at steady-state between −60 and −20 mV. (Supplementary Fig. S1A). VGSC currents were partially inhibited by 10 μM TTX treatment (Fig. 1D) and concentration-response relationships in HT29, SW480 and SW620 cells indicate IC50 values of 4, 8 and 2 μM, respectively (Supplementary Fig. S2A), suggesting functional expression of Nav1.5 which has been reported to have an IC50 value of ~5 μM (25). The absence of involvement of voltage-gated calcium channels (VGCCs) in the recorded inward currents was confirmed by the lack of measurable current in colon cancer cells but not neuronal PC12 cells, in the presence of 10 mM Ba2+ (substituting for Ca2+) and 100 μM TTX (Supplementary Fig. S3A–C). Moreover, the dihydropyridine VGCC blocker nimodipine failed to influence invasion by colon cancer cells (Supplementary Fig. S3D).
SCN5A functionally participates in the invasive potential of colon cancer cells
The contribution of Nav1.5 channels to the invasive potential of colon cancer cells was tested in a Matrigel assay following pharmacological and genetic knockdown of channel activity. In the presence of 30 μM TTX, representing a concentration 4- to 15-fold in excess of the IC50 for Na+ current inhibition and leading to >70% loss of Na+ current activity (Supplementary Fig. S2A), the total number of invading cells was significantly reduced compared to vehicle control for all 3 cell lines (Fig. 2A). In the presence of an siRNA specifically targeting the SCN5A transcript, a significant reduction was observed in the total number of invading cells compared to cells treated with a nonsense control siRNA (Fig. 2B). A loss of channel activity by gene knockdown was confirmed with whole-cell patch clamp electrophysiology where at least a 65% decrease in maximum current density was observed in all 3 lines (see Supplementary Fig. S2B for SW620 cells). Taken as a whole, our findings strongly indicate that the invasive potential of colon cancer cells is linked to the function of Nav1.5. The fact that neither pharmacologic nor genetic knockdown of SCN5A inhibited cell invasion completely may be explained by the presence of other mechanisms that contribute to cell invasion, existence of additional functional isoforms (such as Nav1.8, encoded by SCN10A, the most TTX-resistant isoform; see Supplementary Table S2), or simply incomplete gene knockdown. It is clear from this study and others that a variety of VGSC isoforms are operational in different cancer cell types. The combination of electrophysiological, molecular and Matrigel invasion assays has established a definitive role for the TTX-resistant Nav1.5 in breast (9) and colon (present study; but see discussion below pertaining to different Nav1.5 splice forms for breast and colon cancers), TTX-sensitive Nav1.7 in prostate (26) and TTX-resistant Nav1.5 and sensitive isoforms (Nav1.6 and Nav1.7) in non-small cell lung cancer cell invasiveness (10).
Figure 2.

Nav1.5 contributes to invasive potential of colon cancer cells (A) The total number of invading SW620, SW480 and HT29 colon cancer cells was significantly reduced with 30 μM TTX compared to vehicle control. (B) siRNA-mediated knockdown of SCN5A significantly reduced invasion potential of colon cancer cells compared to cells treated with a nonsense siRNA used as a control. Results are means ± SEM from at least three independent experiments *Significantly different from control (two-sided, unpaired t-Test, P < 0.05).
To further address the relationship between VGSC expression and the transformation/differentiation status of cells, we analyzed SCN5A expression and invasion potential in differentiated and undifferentiated Caco-2 colon carcinoma cells. Caco-2 cells undergo differentiation in culture, a process that is completed ~10 days post-confluency (27). SCN5A expression and invasion potential was highest in undifferentiated Caco-2 cells compared to differentiated cells (Supplementary Fig.S4A). We also analyzed Nav1.5 expression and invasion potential in low versus high passage HEK293 cells, where the latter has been shown to be tumorigenic (28). High passage cells had both significantly higher maximum current density and invasion potential compared to low passage cells (see Supplementary Fig. S4B). Taken together, these data further implicate SCN5A in an invasive phenotype.
Nav1.5 expression is restricted to the lumenal surface of human colon cancer samples
To further confirm the significance of SCN5A in colon cancer, we assessed the protein expression and localization of Nav1.5 in a panel of human colon cancer specimens by immunohistochemistry. Nav1.5 immunoreactivity was mainly confined to the plasma membrane with minimal staining in the cytoplasm. Immunostaining revealed distinct expression of this isoform in malignant cells on the lumenal surface (Fig. 3A). In contrast, normal-matched control samples showed little or no staining of colon epithelial cells (Fig. 3A). Automated digital selection (22) of DAB-labeled tissues revealed a substantial and significant higher percentage of Nav1.5 positive areas in cancer samples compared to their normal-matched controls (Fig. 3B and C). These data should be viewed in light of the report of Barshak, et al. (29) which demonstrated expression of VGSCs in normal colonic epithelial using a pan anti-Nav antibody (although isoform content remains to be delineated). Our findings suggest that functional expression of the Nav1.5 isoform may be selectively repressed in normal colon and becomes aberrantly over-expressed in colon cancer.
Figure 3.

Nav1.5 staining is significantly higher in colon cancer tissues compared to normal-matched colon tissues. (A) Nav1.5 immunoreactivity is confined primarily to the plasma membrane of malignant cells in the luminal surface (brown staining in the periphery). Images are representative of at least three independent experiments from each of seven patients (C1 – C7). Sections C1 – C4 were prepared from fresh frozen tissue specimens and C5 – C7 from formalin-fixed paraffin-embedded tissue specimens. (B) DAB+ stained areas selected from 24-bit BN image using preset threshold on ImageJ image processing software. (C) Quantification of DAB+ stained areas is displayed as percent positive pixels divided by total number of pixels. Results are the mean ± SEM. *Significantly different from normal (two-sided, paired t-Test, P < 0.05).
Of interest is the finding that the neonatal, but not the adult, splice variant form of Nav1.5 in primary breast cancer specimens strongly correlates with lymph node metastasis (3, 9). Analogously, expression of the neonatal form is associated in vitro with strong invasive behavior in the highly metastatic breast cancer cell line MDA-MB-231, while the weakly metastatic lines MDA-MB-468 and MCF-7 do not readily invade Matrigel nor do they express the neonatal variant (9). Consequently, we investigated the nature of the Nav1.5 variant expressed in both clinical specimens and the 3 colon cancer cell lines. Quantitative RT-PCR analysis revealed the presence of only the adult variant in colon cancer specimens and cell lines (data not shown), including SW620 which is derived from a lymph node metastasis (30). In conclusion, our findings suggest that the adult variant of Nav1.5 may be critical for colon cancer invasiveness while the fetal variant appears to be essential for breast cancer metastatic behavior.
Mapping of a Nav1.5-Regulated Colon Cancer Invasion Transcriptional Network
A major goal of this study is to identify a structured gene network participating in colon cancer invasion and to determine if VGSCs participate in the regulation of such a network. It is plausible that recruitment of Nav1.5 expression in colon cancers may facilitate the regulation of downstream genes involved in invasive potential, given VGSC activity is directly associated with gene expression changes in neuronal cells (18). Previously, we had mapped a rudimentary colon cancer invasion network in HT29 cells comprised of different tiers or levels of invasion genes (20). Tier 1 contained the genes ADAM21, CCR9, and CD53 while tier 2 contained GLS, RPL32, KRT20, DHX32 and transcription factor TFDP1 (20) (gene names and corresponding gene symbols can be found in Supplementary Table S1). A drawback to this early map was a lack of connectivity among the eight colon cancer invasion genes owing to the sequential nature of our earlier mapping strategy.
More recently, we have developed a probabilistic computational approach termed Factor Graph-Nested Effects Modeling (FG-NEM) to identify a richer set of connections among signaling genes or S-genes (23). FG-NEM iteratively processes data from loss-of-function screens (targeted gene knockdown by siRNA and screening for loss of invasion) and gene expression experiments profiling the downstream transcriptional effects resulting from each knockdown. A network is constructed among the knocked-down genes based on their downstream effects. FG-NEM also attaches new “effect” genes (E-genes) to the network by identifying the most likely attachment points. We refer to the collection of attached E-genes as the “network frontier.” The modeling is iterative and repeats itself as E-genes are chosen from the frontier for knockdown, loss-of-function screening and transcriptional profiling.
FG-NEM was applied to the original microarray data derived from the knockdowns of eight invasion genes (20) along with expression data from the knockdown of SCN5A in HT29 cells (this study). A total of 54 E-genes were identified and 15 were chosen for individual gene knockdown in HT29 cells (Fig. 4A). The choice of E-genes was based, in part, on the potential role of these genes in cancer invasion. For example, invasion requires degradation of the extracellular matrix, a feat accomplished by disintegrin and metalloproteinase domain-containing proteins, such as ADAM9, and matrix metalloproteinases (17, 31). UBE2L6 functions as part of the ubiquitin-proteasome pathway for protein degradation, and this pathway is intricately tied to the expression and/or activation of matrix metalloproteinases (32). Lastly, the guanine nucleotide binding protein GNAI3 has been associated with the control of cell motility and is thought to be involved in direction detection (33), while the serine/threonine kinase STK24 is a homolog of the yeast protein Ste20p, which is involved in MAPK signaling for invasive growth by yeast (34).
Figure 4.

SCN5A and predicted network genes involved in the invasive potential of HT29 cells. siRNA-mediated knockdown of individual genes proposed to be involved in the invasion network leads to a loss of invasion. (A) qRT-PCR was performed to validate mRNA knockdowns were at least 50% or greater. (B) The total number of invading cells was significantly reduced when mRNA expression was knocked-down with siRNA. Results are means ± SEM from at least three independent experiments. All depicted targeted gene knockdowns are significantly different from siNonsense control (ANOVA, post-hoc Tukey, P < 0.05). Successful knockdown of ADAM21, CCR9, CD53, DHX32, GLS, KRT20, RPL32 and TFDP1 genes and loss of invasion potential in HT29 cells have previously been described (20).
Two different siRNAs were tested for each of the 15 E-genes (as was the case for the original eight invasion genes (20) and SCN5A), whenever feasible, to ensure specificity (Fig. 4B). Successful gene knockdown was defined as a ≥50% reduction in mRNA levels as determined by qRT-PCR. Knockdown of 13 of the 15 tested E-genes led to a significant reduction in the total number of invading HT29 cells compared to cells treated with a nonsense control siRNA (Fig. 4B). It should be noted though that the less efficient knockdown of a particular gene (e.g. ADAM9) may contribute to the limited impact of the gene on invasion. The 13 E-genes empirically linked to loss-of-invasion were `promoted' to S-gene status. Expression profiling after individual knockdown of the 13 S-genes was performed for a second round of FG-NEM analysis to identify additional downstream regulatory connections, thus allowing reconstruction and expansion of the invasion network.
The second round of FG-NEM analysis on the 22 S-genes (i.e. - the 8 original invasion genes, SCN5A, and 13 downstream genes from the first round of FG-NEM) identified 1114 downstream E-genes (Fig. 5A). The resulting network was fully connected, and spanned all 22 S-genes (Fig. 5B). Confidence levels in network features were assessed by bootstrapping the data and inferring new networks for each bootstrap sample (Fig. 5C). Across virtually all bootstrap iterations, SCN5A was placed upstream of all other S-genes.
Figure 5.

Network interactions predicted from E-gene expression under S-gene knockdown. (A) Expression values of selected E-genes. Each row shows the log-ratio expression of a single E-gene under various targeted siRNA-mediated knockdowns relative to a nonsense siRNA control. (B) Inferred S-gene network and Frontier. Nodes represent S-genes (ovals), E-genes (gray boxes), and Gene Ontology categories (white boxes). Arrows indicate activation, and tees indicate repression. Mixed arrow/tee line endings indicate GO set enrichment among both activated and inhibited E-genes. For simplicity only direct interactions are shown. (C) S-gene interaction confidence. Each pixel in the heatmap corresponds to the proportion of times an S-gene interaction was recovered across bootstrap iterations. Upstream S-genes are labeled on the right; downstream on the top. Rows show upstream and columns show downstream bootstrap proportion.
The invasion network could be delineated into five sub-domains with SCN5A, STK24 and KRT20/RPL32 as gene entry points (Fig. 5B). It is intriguing that the SCN5A gene was situated as a highly confident entry point in the invasion network. Its upstream position implicates VGSCs as potential transducers of the invasion machinery, connecting membrane electrophysiology with metastatic behavior. Lending further support to this idea are studies revealing that VGSC genes are commonly mutated in both glioblastoma (N.H.L. and R.L.S., unpublished findings) and colon cancer (35, 36). Of particular interest, at least one of the mutants harbors a predicted gain-of-function mutation in the conserved voltage sensor (N.H.L. and R.L.S., unpublished findings), and such mutations are typically associated with ion channelopathies leading to sustained cationic leak (37, 38). Each network sub-domain was associated with a distinct functional theme, namely integral membrane proteins and proteases, Wnt signaling regulation, calcium signaling, MAP kinase signaling, and membrane remodeling and secretion (Fig. 5B).
Functional Enrichment of the Frontier
FG-NEM was used to expand the frontier of the 22 S-gene invasion network by predicting where new E-genes might attach. We identified 1752 E-genes attached in the frontier at a 5% FDR level. Gene Ontology (GO) enrichment analysis was performed on the E-genes at specific attachment points of the frontier (boxes in Fig. 5B). Seven main GO categories were found to be representative of the frontier at an FDR of 5%, specifically ectoderm development (24 E-genes), carboxylic acid transport (22 E-genes), negative regulation of mitotic cell cycle (9 E-genes), response to biotic stimulus (17 E-genes), steroid metabolic process (28 E-genes), cell migration (30 E-genes), and regulation of Wnt signaling (5 E-genes) (see the complete list provided as Supplementary Table S3). Moreover, the network frontier appears to be significantly enriched with colon cancer-specific genes based on meta-analysis of Oncomine datasets describing the interrogation of different cancers including colon cancer (see Supplementary Tables S4 and S5). Nine of the frontier E-genes have previously been shown to be mutated in colon cancer (i.e. LAMA4, FN1, SMAD3, COL3A1, APOB, LAMC1, TCF3, APBB2, and KRT20) (35, 36), and seven from this subset are predicted to participate in a protein-protein interactome network (39). In a separate study, two of our frontier E-genes, PPL and IFITM1, are predicted to participate in a protein interaction network in late stage human colorectal cancer (40). IFITM1, located in the `response to biotic stimulus' category, exhibited the highest attachment score of any E-gene in our network, and this gene has recently been proposed to be a molecular marker for human colorectal tumors (41). Furthermore, elevated expression of IFITM1 in head and neck squamous cell cancer (42) and in gastric cancer (43), leads to increased levels of invasive behavior; conversely, suppressing IFITM1 decreases invasive behavior.
The cell migration category contains a number of E-genes associated with colon cancer invasion. In particular, the neuronal cell adhesion molecule (NRCAM) gene had the highest attachment score. NRCAM has been previously identified in our laboratory as a gene regulated by osteopontin and CD44 signaling in Ras-transformed NIH3T3 cells, promoting invasive behavior (14). Both osteopontin and CD44 are molecular markers for metastatic colon carcinomas (44). Increased NRCAM expression has also been shown to be mediated by the Wnt signaling pathway, enhancing cell motility and tumorigenesis of colon cancer cells (45). The attachment of NRCAM to the Wnt signaling region of our network is consistent with this earlier finding. Another cell migration E-gene with a high attachment score, CCDC88A (also known as KIAA1212, GIV, or GIRDIN), is predicted to be inhibited by the invasion network. CCDC88A is an established binding partner of our S-gene GNAI3 (33), and GNAI3 has been demonstrated to be essential for leading-edge pseudopod formation and cell migration (33).
Among E-genes associated with the GO category steroid metabolic process, the colon cancer marker gene INSIG2 had the strongest connection to the invasion network and has recently been shown to promote invasive behavior when ectopically expressed in HCT116 colon cancer cells (46).
Within the regulation of Wnt signaling category, PPP2R1A had the highest attachment score. Protein phosphatase 2A (PP2A) is a heterotrimeric serine-threonine phosphatase comprised of structural subunit A (i.e. encoded by PPP2R1A or PPP2R1B), regulatory subunit B and catalytic subunit C. PP2A activity has been shown to regulate Wnt and phosphatidylinositol 3-kinase signaling (47). Mutations in both structural subunit A genes have been found in several cancers including breast, lung, and colon, and studies suggest PP2A functions as a tumor suppressor (47, 48).
The most significantly enriched GO term was Ectoderm Development. FG-NEM predicts that the invasion network represses some members of this set and activates others, mostly collagens, laminins, keratins, and regulators of these structural genes. The gene CTGF, which encodes the protein connective tissue growth factor, had the highest attachment score in this GO term, and is predicted to be inhibited by the invasion network. CTGF produces an extracellular matrix protein, and in liver has been proposed as a master regulator of the epithelial-mesenchymal transition (49).
Validation of network frontier
To validate the involvement of predicted downstream E-genes in the invasion network, the invasion potential of HT29 cells was assessed following individual siRNA-mediated knockdown of NRCAM, IFITM1, INSIG2, CTGF and PPP2R1A, typifying high attachment score genes from 5 different regions (Fig. 5B). NRCAM, IFITM1 and INSIG2 represent activated genes in the invasion network. Consequently, one might predict that suppression of these genes would lead to a loss of invasion potential. A significant loss of invasion indeed occurred with knockdowns of NRCAM, IFITM1 or INSIG2 compared to nonsense siRNA control (Fig. 6). Conversely, PPP2R1A and CTGF would be predicted to suppress invasion based on their upstream inhibitory connections (Fig. 5B). Alternatively, these inhibitory connections may represent counter-regulatory measures. A significant enhancement of the invasive potential indeed occurred upon knockdown of PPP2R1A (Fig. 6). The knockdown of CTGF was unsuccessful with 5 tested siRNAs based on qRT-PCR results. However, the specific combination of 2 siRNAs resulted in a 53% knockdown and a corresponding loss of invasion potential (Fig. 6). The combined results of siRNA knockdown and Matrigel invasion assays strongly support the regulatory connections proposed in the invasion network.
Figure 6.
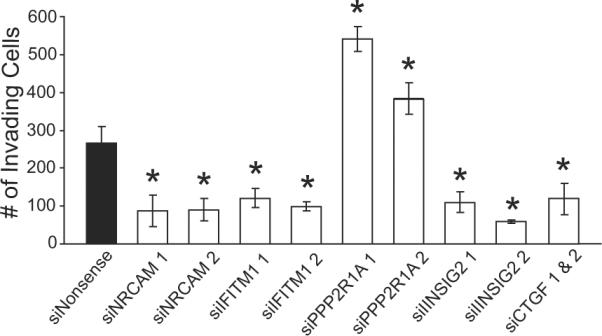
Validation of predicted downstream network interactions. Significant changes in invasion occur with siRNA-mediated knockdown of frontier E-genes predicted to be connected to the invasion network. Knockdowns were determined to be ≥50% by qRT-PCR. Results are means ± SEM from three independent experiments for each siRNA. *Significantly different from siNonsense control (ANOVA, post-hoc Tukey, P < 0.05).
In conclusion, we have reconstructed a network of gene interactions implicated in invasive colon cancer. Genes previously associated with colon cancer and those never before tied to colon cancer have been linked operationally. Our data highlight the transcriptional changes that occur with functional VGSC expression and support the hypothesis that ion channel activity leads to gene expression changes that favor an invasion phenotype. Both activation and inactivation gene links have been defined in the invasion network as would be expected in the regulation of a complex network. It should be noted that future studies are warranted to define precisely how ion channel activity leads to downstream transcriptional effects in colon cancer. A number of recent studies have associated the use of local anesthetics (blockers of VGSCs) during surgical resection of cancers with decreased reoccurrence and metastasis (50). These findings are intriguing in light of the positioning of SCN5A as an early entry point in the invasion network and immunohistological validation of aberrant up-regulation of Nav1.5 protein in clinical colon cancer specimens. Our study implicates the VGSC Nav1.5 subunit (adult splice variant) and its network constituents as potential targets for the development of new therapies for hindering colon cancer progression.
Precis.
Certain ion channel isotypes expressed on the surface of metastatic cancer cells may represent tractable new theranostic molecules for study
Supplementary Material
Acknowledgements
We thank Anastas Popratiloff for assistance with confocal microscopy.
This work was supported by National Institutes of Health grants CA120316 and 1S10RR025565-01 (N.H.L.) and by PhRMA Foundation (C.D.H.).
REFERENCES
- 1.Kunzelmann K. Ion channels and cancer. J Membr Biol. 2005;205(3):159–73. doi: 10.1007/s00232-005-0781-4. [DOI] [PubMed] [Google Scholar]
- 2.Schuller HM. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nature reviews. 2009;9(3):195–205. doi: 10.1038/nrc2590. [DOI] [PubMed] [Google Scholar]
- 3.Onkal R, Djamgoz MB. Molecular pharmacology of voltage-gated sodium channel expression in metastatic disease: clinical potential of neonatal Nav1.5 in breast cancer. European journal of pharmacology. 2009;625(1–3):206–19. doi: 10.1016/j.ejphar.2009.08.040. [DOI] [PubMed] [Google Scholar]
- 4.Roger S, Potier M, Vandier C, Besson P, Le Guennec JY. Voltage-gated sodium channels: new targets in cancer therapy? Curr Pharm Des. 2006;12(28):3681–95. doi: 10.2174/138161206778522047. [DOI] [PubMed] [Google Scholar]
- 5.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117(4):500–44. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis TH, Chen C, Isom LL. Sodium channel beta1 subunits promote neurite outgrowth in cerebellar granule neurons. The Journal of biological chemistry. 2004;279(49):51424–32. doi: 10.1074/jbc.M410830200. [DOI] [PubMed] [Google Scholar]
- 7.Cameron IL, Smith NK, Pool TB, Sparks RL. Intracellular concentration of sodium and other elements as related to mitogenesis and oncogenesis in vivo. Cancer research. 1980;40(5):1493–500. [PubMed] [Google Scholar]
- 8.Laniado ME, Lalani EN, Fraser SP, et al. Expression and functional analysis of voltage-activated Na+ channels in human prostate cancer cell lines and their contribution to invasion in vitro. Am J Pathol. 1997;150(4):1213–21. [PMC free article] [PubMed] [Google Scholar]
- 9.Fraser SP, Diss JK, Chioni AM, et al. Voltage-gated sodium channel expression and potentiation of human breast cancer metastasis. Clin Cancer Res. 2005;11(15):5381–9. doi: 10.1158/1078-0432.CCR-05-0327. [DOI] [PubMed] [Google Scholar]
- 10.Roger S, Rollin J, Barascu A, et al. Voltage-gated sodium channels potentiate the invasive capacities of human non-small-cell lung cancer cell lines. Int J Biochem Cell Biol. 2007;39(4):774–86. doi: 10.1016/j.biocel.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 11.Gillet L, Roger S, Besson P, et al. Voltage-gated Sodium Channel Activity Promotes Cysteine Cathepsin-dependent Invasiveness and Colony Growth of Human Cancer Cells. The Journal of biological chemistry. 2009;284(13):8680–91. doi: 10.1074/jbc.M806891200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carrithers MD, Chatterjee G, Carrithers LM, et al. Regulation of podosome formation in macrophages by a splice variant of the sodium channel SCN8A. The Journal of biological chemistry. 2009;284(12):8114–26. doi: 10.1074/jbc.M801892200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malek RL, Irby RB, Guo QM, et al. Identification of Src transformation fingerprint in human colon cancer. Oncogene. 2002;21(47):7256–65. doi: 10.1038/sj.onc.1205900. [DOI] [PubMed] [Google Scholar]
- 14.Teramoto H, Castellone MD, Malek RL, et al. Autocrine activation of an osteopontin-CD44-Rac pathway enhances invasion and transformation by H-RasV12. Oncogene. 2005;24(3):489–501. doi: 10.1038/sj.onc.1208209. [DOI] [PubMed] [Google Scholar]
- 15.Onganer PU, Seckl MJ, Djamgoz MB. Neuronal characteristics of small-cell lung cancer. British journal of cancer. 2005;93(11):1197–201. doi: 10.1038/sj.bjc.6602857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jull BA, Plummer HK, 3rd, Schuller HM. Nicotinic receptor-mediated activation by the tobacco-specific nitrosamine NNK of a Raf-1/MAP kinase pathway, resulting in phosphorylation of c-myc in human small cell lung carcinoma cells and pulmonary neuroendocrine cells. J Cancer Res Clin Oncol. 2001;127(12):707–17. doi: 10.1007/s004320100289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roy R, Wewer UM, Zurakowski D, Pories SE, Moses MA. ADAM 12 cleaves extracellular matrix proteins and correlates with cancer status and stage. The Journal of biological chemistry. 2004;279(49):51323–30. doi: 10.1074/jbc.M409565200. [DOI] [PubMed] [Google Scholar]
- 18.Tolon RM, Sanchez-Franco F, Lopez Fernandez J, Lorenzo MJ, Vazquez GF, Cacicedo L. Regulation of somatostatin gene expression by veratridine-induced depolarization in cultured fetal cerebrocortical cells. Brain research. 1996;35(1–2):103–10. doi: 10.1016/0169-328x(95)00188-x. [DOI] [PubMed] [Google Scholar]
- 19.Juretic N, Urzua U, Munroe DJ, Jaimovich E, Riveros N. Differential gene expression in skeletal muscle cells after membrane depolarization. Journal of cellular physiology. 2007;210(3):819–30. doi: 10.1002/jcp.20902. [DOI] [PubMed] [Google Scholar]
- 20.Irby RB, Malek RL, Bloom G, et al. Iterative microarray and RNA interference-based interrogation of the SRC-induced invasive phenotype. Cancer research. 2005;65(5):1814–21. doi: 10.1158/0008-5472.CAN-04-3609. [DOI] [PubMed] [Google Scholar]
- 21.Markowetz F, Kostka D, Troyanskaya OG, Spang R. Nested effects models for high-dimensional phenotyping screens. Bioinformatics. 2007;23(13):i305–12. doi: 10.1093/bioinformatics/btm178. [DOI] [PubMed] [Google Scholar]
- 22.Brey EM, Lalani Z, Johnston C, et al. Automated selection of DAB-labeled tissue for immunohistochemical quantification. J Histochem Cytochem. 2003;51(5):575–84. doi: 10.1177/002215540305100503. [DOI] [PubMed] [Google Scholar]
- 23.Vaske CJ, House C, Luu T, et al. A factor graph nested effects model to identify networks from genetic perturbations. PLoS Comput Biol. 2009;5(1):e1000274. doi: 10.1371/journal.pcbi.1000274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gavillet B, Rougier JS, Domenighetti AA, et al. Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circulation research. 2006;99(4):407–14. doi: 10.1161/01.RES.0000237466.13252.5e. [DOI] [PubMed] [Google Scholar]
- 25.Gellens ME, George AL, Jr., Chen LQ, et al. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(2):554–8. doi: 10.1073/pnas.89.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brackenbury WJ, Djamgoz MB. Activity-dependent regulation of voltage-gated Na+ channel expression in Mat-LyLu rat prostate cancer cell line. J Physiol. 2006;573(Pt 2):343–56. doi: 10.1113/jphysiol.2006.106906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaillard JL, Finlay BB. Effect of cell polarization and differentiation on entry of Listeria monocytogenes into the enterocyte-like Caco-2 cell line. Infection and immunity. 1996;64(4):1299–308. doi: 10.1128/iai.64.4.1299-1308.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen C, Gu M, Song C, et al. The tumorigenicity diversification in human embryonic kidney 293 cell line cultured in vitro. Biologicals. 2008;36(4):263–8. doi: 10.1016/j.biologicals.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 29.Barshack I, Levite M, Lang A, et al. Functional voltage-gated sodium channels are expressed in human intestinal epithelial cells. Digestion. 2008;77(2):108–17. doi: 10.1159/000123840. [DOI] [PubMed] [Google Scholar]
- 30.Leibovitz A, Stinson JC, McCombs WB, 3rd, McCoy CE, Mazur KC, Mabry ND. Classification of human colorectal adenocarcinoma cell lines. Cancer research. 1976;36(12):4562–9. [PubMed] [Google Scholar]
- 31.Murphy G. The ADAMs: signalling scissors in the tumour microenvironment. Nature reviews. 2008;8(12):929–41. doi: 10.1038/nrc2459. [DOI] [PubMed] [Google Scholar]
- 32.Meiners S, Hocher B, Weller A, et al. Downregulation of matrix metalloproteinases and collagens and suppression of cardiac fibrosis by inhibition of the proteasome. Hypertension. 2004;44(4):471–7. doi: 10.1161/01.HYP.0000142772.71367.65. [DOI] [PubMed] [Google Scholar]
- 33.Ghosh P, Garcia-Marcos M, Bornheimer SJ, Farquhar MG. Activation of Galphai3 triggers cell migration via regulation of GIV. J Cell Biol. 2008;182(2):381–93. doi: 10.1083/jcb.200712066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roberts RL, Fink GR. Elements of a single MAP kinase cascade in Saccharomyces cerevisiae mediate two developmental programs in the same cell type: mating and invasive growth. Genes & development. 1994;8(24):2974–85. doi: 10.1101/gad.8.24.2974. [DOI] [PubMed] [Google Scholar]
- 35.Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science (New York, NY. 2006;314(5797):268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 36.Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science (New York, NY. 2007;318(5853):1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 37.Ashcroft FM. From molecule to malady. Nature. 2006;440(7083):440–7. doi: 10.1038/nature04707. [DOI] [PubMed] [Google Scholar]
- 38.Sokolov S, Scheuer T, Catterall WA. Gating pore current in an inherited ion channelopathy. Nature. 2007;446(7131):76–8. doi: 10.1038/nature05598. [DOI] [PubMed] [Google Scholar]
- 39.Lin J, Gan CM, Zhang X, et al. A multidimensional analysis of genes mutated in breast and colorectal cancers. Genome research. 2007;17(9):1304–18. doi: 10.1101/gr.6431107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nibbe RK, Markowitz S, Myeroff L, Ewing R, Chance MR. Discovery and scoring of protein interaction subnetworks discriminative of late stage human colon cancer. Mol Cell Proteomics. 2009;8(4):827–45. doi: 10.1074/mcp.M800428-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andreu P, Colnot S, Godard C, et al. Identification of the IFITM family as a new molecular marker in human colorectal tumors. Cancer research. 2006;66(4):1949–55. doi: 10.1158/0008-5472.CAN-05-2731. [DOI] [PubMed] [Google Scholar]
- 42.Hatano H, Kudo Y, Ogawa I, et al. IFN-induced transmembrane protein 1 promotes invasion at early stage of head and neck cancer progression. Clin Cancer Res. 2008;14(19):6097–105. doi: 10.1158/1078-0432.CCR-07-4761. [DOI] [PubMed] [Google Scholar]
- 43.Yang Y, Lee JH, Kim KY, et al. The interferon-inducible 9–27 gene modulates the susceptibility to natural killer cells and the invasiveness of gastric cancer cells. Cancer Lett. 2005;221(2):191–200. doi: 10.1016/j.canlet.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 44.Irby RB, McCarthy SM, Yeatman TJ. Osteopontin regulates multiple functions contributing to human colon cancer development and progression. Clinical & experimental metastasis. 2004;21(6):515–23. doi: 10.1007/s10585-004-2873-4. [DOI] [PubMed] [Google Scholar]
- 45.Conacci-Sorrell ME, Ben-Yedidia T, Shtutman M, Feinstein E, Einat P, Ben-Ze'ev A. Nr-CAM is a target gene of the beta-catenin/LEF-1 pathway in melanoma and colon cancer and its expression enhances motility and confers tumorigenesis. Genes Dev. 2002;16(16):2058–72. doi: 10.1101/gad.227502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li CG, Gruidl M, Eschrich S, et al. Insig2 is associated with colon tumorigenesis and inhibits Bax-mediated apoptosis. Int J Cancer. 2008;123(2):273–82. doi: 10.1002/ijc.23510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sablina AA, Hahn WC. The role of PP2A A subunits in tumor suppression. Cell Adh Migr. 2007;1(3):140–1. doi: 10.4161/cam.1.3.4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Calin GA, di Iasio MG, Caprini E, et al. Low frequency of alterations of the alpha (PPP2R1A) and beta (PPP2R1B) isoforms of the subunit A of the serine-threonine phosphatase 2A in human neoplasms. Oncogene. 2000;19(9):1191–5. doi: 10.1038/sj.onc.1203389. [DOI] [PubMed] [Google Scholar]
- 49.Gressner OA, Gressner AM. Connective tissue growth factor: a fibrogenic master switch in fibrotic liver diseases. Liver Int. 2008;28(8):1065–79. doi: 10.1111/j.1478-3231.2008.01826.x. [DOI] [PubMed] [Google Scholar]
- 50.Biki B, Mascha E, Moriarty DC, Fitzpatrick JM, Sessler DI, Buggy DJ. Anesthetic technique for radical prostatectomy surgery affects cancer recurrence: a retrospective analysis. Anesthesiology. 2008;109(2):180–7. doi: 10.1097/ALN.0b013e31817f5b73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.