

Abstract
Cholesteryl ester (CE) accumulation in macrophages represents a crucial event during foam cell formation, a hallmark of atherogenesis. Here we investigated the role of two previously described CE hydrolases, hormone-sensitive lipase (HSL) and KIAA1363, in macrophage CE hydrolysis. HSL and KIAA1363 exhibited marked differences in their abilities to hydrolyze CE, triacylglycerol (TG), diacylglycerol (DG), and 2-acetyl monoalkylglycerol ether (AcMAGE), a precursor for biosynthesis of platelet-activating factor (PAF). HSL efficiently cleaved all four substrates, whereas KIAA1363 hydrolyzed only AcMAGE. This contradicts previous studies suggesting that KIAA1363 is a neutral CE hydrolase. Macrophages of KIAA1363−/− and wild-type mice exhibited identical neutral CE hydrolase activity, which was almost abolished in tissues and macrophages of HSL−/− mice. Conversely, AcMAGE hydrolase activity was diminished in macrophages and some tissues of KIAA1363−/− but unchanged in HSL−/− mice. CE turnover was unaffected in macrophages lacking KIAA1363 and HSL, whereas cAMP-dependent cholesterol efflux was influenced by HSL but not by KIAA1363. Despite decreased CE hydrolase activities, HSL−/− macrophages exhibited CE accumulation similar to wild-type (WT) macrophages. We conclude that additional enzymes must exist that cooperate with HSL to regulate CE levels in macrophages. KIAA1363 affects AcMAGE hydrolase activity but is of minor importance as a direct CE hydrolase in macrophages.
Keywords: 2-acetyl monoalkylglycerol ether hydrolase, cholesterol metabolism, lipase, lipids, triacylglycerol
Mobilization of cholesterol and fatty acids from cholesteryl ester (CE) and triacylglycerol (TG) stores of all cells and tissues requires lipolytic enzymes. Dysfunctional cellular hydrolysis affects lipid homeostasis, causes lipid storage disorders, and may contribute to the pathogenesis of atherosclerosis and obesity. Human and murine macrophages express cell surface scavenger receptors that bind and subsequently internalize modified LDL. An excessive and uncontrolled uptake of cholesterol results in foam cell formation, which is a hallmark of atherosclerosis. Cholesterol is esterified by acyl-CoA:cholesterol acyltransferase (ACAT)1 and stored as CE in lipid droplets. Conversely, CE hydrolases are required for the hydrolysis of lipid droplet-associated CE (1, 2).
CE hydrolysis in macrophages has been studied for more than 40 years (3). In 1989, it was first reported that the neutral CE hydrolase activity in the mouse macrophage cell line WEH1 is catalyzed by hormone-sensitive lipase (HSL) (4). In RAW264.7 macrophages, neutral CE hydrolase activity was nearly completely blocked with an anti-HSL antibody, whereas overexpression of HSL increased CE hydrolysis in these cells (5). Adenoviral-mediated overexpression of HSL in human THP-1 macrophages resulted in complete elimination of CE stores (6). Enhanced CE hydrolysis, specifically in macrophages of transgenic mice overexpressing HSL, led to accumulation of free cholesterol (FC) and an increased incidence and severity of aortic fatty lesions (7). These results suggested that HSL plays a major role in the hydrolysis of CE stores in macrophages. However, these findings were challenged by unchanged CE hydrolase activities and HSL-independent CE hydrolysis in mouse peritoneal macrophages (MPM) of HSL-deficient mice (8, 9), arguing against a dominant role of HSL as a CE hydrolase in macrophages. Very recently, however, these authors reported on reduced neutral CE hydrolase activity in HSL−/− MPM (10).
Evidence was provided for neutral CE hydrolase activity by monocyte/macrophage serine esterase 1 (CES1) in human THP-1 monocytes and macrophages (11). This enzyme is related to the neutral CE hydrolase expressed in liver, and its overexpression causes cytoplasmic CE mobilization from ACAT1 stably expressing CHO cells (12). Its mouse ortholog, named carboxylesterase 3 (TG hydrolase, TGH), is expressed in macrophages at a low level, primarily in white adipose tissue, where it showed little TG and only detectable CE hydrolase activity (13). Finally, a pancreatic CE hydrolase, [cholesterol esterase, bile salt-dependent lipase (CEL)] was reported to be also expressed in macrophages in aorta (14, 15). Since CEL is expressed in human but not in murine macrophages, it was proposed that CEL plays a similar role in human macrophages as does HSL in mouse macrophages (16).
Recently, the serine hydrolase KIAA1363 was identified as a new CE hydrolase by in silico searches of databases for proteins showing homology to known lipases (17). Okazaki et al. (17) renamed KIAA1363 as neutral cholesterol ester hydrolase (NCEH) and suggested NCEH to be the key enzyme responsible for neutral CE hydrolase activity in murine macrophages (10, 17). Originally, Jessani et al. had characterized KIAA1363 as a serine hydrolase highly elevated in aggressive human cancer cells (18) and showed that this enzyme was also enriched in primary human breast tumors (19). Nomura et al. (20) described KIAA1363 as a significant detoxifying enzyme for organophosphorus nerve poisons in brain. Most recently, KIAA1363 has been shown to regulate an ether lipid metabolic network in cancer cells, where the enzyme serves as a principal 2-acetyl monoalkylglycerol ether (AcMAGE) hydrolase (20, 21). AcMAGE is the penultimate precursor in the de novo biosynthesis of platelet activating factor (PAF) (22), a potent lipid mediator playing inflammatory and physiological roles in a variety of cells. Inhibition of KIAA1363 disrupted this metabolic pathway in tumor cells and impaired their invasion and growth in vivo (21). The analysis of KIAA1363−/− mice confirmed that this enzyme is the primary AcMAGE hydrolase in brain, lung, heart, and kidney and was highly sensitive to inactivation by chlorpyrifos oxon, the bioactivated metabolite of the major insecticide chlorpyrifos (22).
The aim of the present study was to compare CE hydrolase activities of HSL and KIAA1363 to reveal the importance of these enzymes as neutral CE hydrolases in murine macrophages and tissues. Our results demonstrate the functional presence of HSL as a neutral CE hydrolase in murine macrophages. In the absence of HSL in macrophages, a so far unknown mechanism compensates for the loss of HSL, resulting in unchanged cellular CE concentrations. In our hands, KIAA1363 significantly contributes to hydrolysis of AcMAGE but not to CE hydrolysis in macrophages, which contradicts data published recently (10, 17).
MATERIALS AND METHODS
Animals and diets
Animal experiments were performed in accordance with the standards established by the Austrian Federal Ministry of Education, Science and Culture, Division of Genetic Engineering and Animal Experiments (Vienna, Austria). HSL−/− (23, 24) and KIAA1363−/− mice (22, 25) (supplementary Fig. I) as well as wild-type (WT) littermates, all on a C57Bl/6 background, were obtained from inhouse breeding and maintained in a clean environment on a regular light-dark cycle (14 h light, 10 h dark). They received a standard mouse chow diet (Ssniff R/M H; ssniff, Soest, Germany) or Western-type diet (WTD; TD88137 mod, containing 21% fat and 0.2% cholesterol; ssniff, Soest, Germany). All mice have been backcrossed onto a C57Bl/6 background.
Plasma lipid parameters
Blood was taken from fed and fasted age-matched mice by retro-orbital bleeding and EDTA plasma was prepared within 20 min. Plasma concentrations of TG (DiaSys, Holzheim, Germany), total cholesterol (TC) (Greiner Diagnostics, Langenthal, Switzerland), and FC (DiaSys, Holzheim, Germany) were determined enzymatically according to the manufacturer's protocols. Plasma samples (200 µl) of fasted mice were pooled and lipoproteins were isolated by fast protein liquid chromatography (FPLC) on a Pharmacia FPLC system equipped with a Superose 6 column (Amersham Biosciences, Piscataway, NJ) as described previously (26).
Cell culture
COS-7 (ATCC CRL-1651) cells were cultivated in a humidified incubator under standard conditions (5% CO2 at 37°C) in DMEM (Gibco, Invitrogen, Lofer, Austria) containing 10% FCS (Sigma-Aldrich Chemie, Vienna, Austria), 1% L-glutamine, and 1% streptomycin/penicillin. Thioglycolate-elicited MPM were isolated with 10 ml PBS three days after peritoneal injection of 3 ml 3% thioglycolate medium. MPM were centrifuged, washed with PBS, and cultured in 6-well plates in serum-free DMEM for 2 h. Thereafter, nonadherent cells were removed. Either MPM were used immediately for determination of lipid parameters or hydrolase activities, or they were cultured overnight in DMEM/10% LPDS for efflux experiments. Human LDL was isolated by density gradient ultracentrifugation (density 1.019–1.063 g/ml) in a near-vertical rotor. LDL was acetylated (acLDL) as described (27). β-VLDL was isolated from WTD-fed apolipoprotein (apo)E−/− mice as described (28). LPDS was prepared from newborn bovine serum by ultracentrifugation. To achieve foam cell formation, MPM were incubated in the absence or presence of 100 µg acLDL/ml for 48 h and 72 h.
cDNA cloning for the expression of recombinant His-tagged HSL and KIAA1363
Coding sequences of murine HSL and KIAA1363 were amplified by PCR from MPM cDNA using Advantage® cDNA Polymerase Mix (BD Biosciences Clontech, Palo Alto, CA). Primers were designed to create endonuclease cleavage sites (underlined): HSL forward: 5′- t ggtacc tatggatttacgcacgatgacaca-3′, HSL reverse 5′-c ctcgag cgttcagtggt gcagcaggcg-3′. KIAA1363 forward: 5′-cgggatccaggtcgtcatgcgtcctact-3′, KIAA1363 reverse: 5′-ccctcgagtcacaggttttgatccagcc-3′. PCR products, containing the complete open reading frames, were ligated into the eukaryotic expression vector pcDNA4/HisMax (Invitrogen Corp., Carlsbad, CA). A control pcDNA4/HisMax vector expressing β−galactosidase (LacZ) was obtained from the manufacturer (Invitrogen Corp., Carlsbad, CA).
RNA isolation and real-time PCR analysis
Total RNA from macrophages was isolated using RNeasy Mini Kit (Qiagen, Hilden, Germany). ABCA1 and ABCG1 mRNA levels were determined on a LightCycler 480 (Roche Diagnostics, Mannheim, Germany) using the QuantifastTM SYBR®GREEN PCR Kit (Qiagen) as described (29). Data are displayed as expression ratios normalized to cyclophilin A as reference gene. Primer sequences are available upon request.
Expression of recombinant proteins in cultured cells
Before transfection, COS-7 cells were collected in the logarithmic growth phase. Cells (150,000 cells/well) were seeded in 6-well plates and cultured overnight. Transient transfection of COS-7 cells with pcDNA4/HisMax encoding His-tagged KIAA1363, HSL, and LacZ was performed using Metafectene™ (Biontex, Munich, Germany). For that purpose, 2.5 µg of purified DNA (Qiagen HiSpeed™, Qiagen, Valencia, CA) per well were incubated with 10 μl Metafectene™ for 20 min at room temperature in serum-free DMEM in a total volume of 100 μl. Subsequently, cells were incubated with the DNA/metafectene complex for 4 h in serum-free medium. Afterwards, the medium was replaced by DMEM containing 10% FCS. Then 48 h after transfection, cells were washed twice with PBS, scraped in lysis buffer (100 mM potassium phosphate, 250 mM sucrose, 1 mM EDTA, 1 mM dithiothreitol, 20 µg/ml leupeptin, 2 µg/ml antipain, 1 µg/ml pepstatin, pH 7.0), and used for lysate preparations.
Western blotting analysis
A total of 40 µg protein from various cellular fractions were separated on a 10% SDS-PAGE and transferred to a nitrocellulose membrane. For detection of HSL protein, anti-HSL polyclonal antibody (Cell Signaling Technology, Danvers, MA) was used at a dilution of 1:800. For detection of KIAA1363 protein, blots were incubated with an anti-KIAA1363 polyclonal antibody (21) at a dilution of 1:1000. Anti-ABCA1 (Abcam, Cambridge, UK) and anti-ABCG1 (Epitomics, Burlingam, CA) antibodies were diluted 1:1000 and 1:2000, respectively. Specifically bound immunoglobulin was detected in a second reaction with a horseradish peroxidase-labeled IgG conjugate and visualized by enhanced chemoluminescence detection (ECL plus, Amersham Bioscience, Piscataway, NJ) on an AGFA Curix Ultra X Ray film (Siemens, Graz, Austria).
Assays for p-nitrophenolvalerate, CE, TG, diacylglycerol, and 2-acetyl monoalkyl-glycerol ether hydrolase activities
p-Nitrophenolvalerate (PNPV) esterase activity (30) and AcMAGE hydrolase activity (22) were assayed as described. For determination of CE, TG, and diacylglycerol (DG) hydrolase activities, freshly prepared substrates were used as follows. CE substrate contained 20 nmol cholesteryl oleate/assay, cholesteryl [1-14C]oleate (50,000 cpm/nmol) (Amersham Biosciences, Piscataway, NJ), 35.5 µg mixed micelles of phosphatidylcholine (PC) and phosphatidylinositol (PI) (3:1, w:w), and 4 µM sodium taurocholate. Alternatively, CE hydrolase activities in macrophages were assayed using 35.5 µg mixed micelles of PC and PI (3:1, w:w) or 35.5 µg PC and 2 µM sodium taurocholate. TG substrate contained 25 nmol triolein/assay, 40,000 cpm/nmol of [9,10-3H(N)]triolein (NEN Life Science Products, Boston, MA), 15 µg mixed micelles of PC and PI (3:1, w:w), and 4 µM sodium taurocholate. DG substrate contained 40 nmol 1,2-diolein/assay and 120,000 cpm/nmol of dioleyl-rac-glycerol[oleoyl-1-14C] (American Radiolabeled Chemicals, St. Louis, MO), 15 µg mixed micelles of PC and PI (3:1, w:w), and 4 µM sodium taurocholate. After evaporation, substrates were sonicated (Labsonic, B. Braun, Melsungen, Germany) in 100 mM potassium phosphate buffer (pH 7.0) containing 4 µM sodium taurocholate and 2.5% fatty acid-free BSA exactly as described by Holm et al. (31) One hundred µg of cell extracts (lysates or subfractions) and 100 µl substrate were incubated in a water bath at 37°C for 1 h. The reaction was terminated by the addition of 3.25 ml of methanol/ chloroform/heptane (10:9:7) and 1 ml of 100 mM potassium carbonate, 100 mM boric acid, pH 10.5. After centrifugation (800 g, 20 min), the radioactivity in 1 ml of the upper phase was determined by liquid scintillation counting, and the release of FFA was calculated.
For lysate preparation, cells (COS-7 or MPM) were washed two times with PBS and sonicated twice for 10 s in lysis buffer (100 mM potassium phosphate, 250 mM sucrose, 1 mM EDTA, 1 mM dithiothreitol, 20 µg/ml leupeptin, 2 µg/ml antipain, 1 µg/ml pepstatin, pH 7.0). Nuclei and cell debris were removed by centrifugation at 1,000 g for 5 min (4°C). Cytosolic and membrane fractions were prepared by centrifugation at 100,000 g for 1 h (4°C). For determination of CE hydrolase activities in tissues of WT, HSL−/−, and KIAA1363−/− mice, tissues were surgically removed and washed in PBS containing 1 mM EDTA. Homogenization was performed in lysis buffer on ice using a Precellys24 homogenizer (Bertin, Montigny-le-Bretonneux, France). Lysates were centrifuged for 30 min at 20,000 g (4°C). Radioactivity corresponding to the release of FFA in 100 µg of the lipid-free infranatant was determined by liquid scintillation counting. Protein concentrations were determined using a Bradford assay (Bio-Rad Laboratories, Vienna, Austria).
Binding of the specific fluorescent activity recognition probes NBD-sn1-TGP, NBD-sn3-TGP, and NBD-CP to HSL and KIAA1363
Cytoplasmic extracts of COS-7 cells were prepared as described above. A total of 50 µg of cellular protein were incubated with 1 nmol of the specific fluorescently labeled probes 7-nitrobenz- 2-oxa-1,3-diazole (NBD) cholesteryl phosphonate (CP) and enantiomeric TG analogs (NBD-sn1-TGP and NBD-sn3-TGP) (32), and 1 mM Triton X-100 (Hoffmann La Roche, Basel, Switzerland) at 37°C for 2 h under shaking as described (33). Total protein was precipitated with 10% trichloroacetic acid for 1 h on ice, washed with acetone, and subjected to 10% SDS-PAGE. Gels were treated with 10% ethanol and 7% acetic acid, and fluorescent signals were detected on a BioRad FX Pro laser scanner (excitation 488 nm, emission 530 nm) (BioRad Laboratories, Hercules, CA).
Neutral lipid extraction
Lipids from MPM were extracted with 2 ml hexane:isopropanol (3:2, v:v) for 1 h at 4°C. Lipid extracts were dried under nitrogen, redissolved in 100 µl 1% Triton X-100 in chloroform, dried under nitrogen, and resuspended in 100 µl distilled water for 15 min at 37°C. Aliquots (30 µl) were used for enzymatic determinations of TG (DiaSys, Holzheim, Germany), TC (Greiner Diagnostics, Bahlingen, Germany), and FC (DiaSys, Holzheim, Germany) concentrations. Proteins of extracted cells were dissolved in 2 ml of 300 mM NaOH for 1 h at room temperature, and protein content was quantitated using a Bradford assay (Bio-Rad Laboratories, Vienna, Austria).
Cholesterol efflux
The amount of 50 µg of freshly prepared acLDL (not older than one week) was enriched with 1 µCi [1, 2 3H]cholesterol (Hartmann Analytik, Braunschweig, Germany). MPM from WT, HSL−/−, and KIAA1363−/− mice were seeded in 12-well plates and incubated with 50 µg 3H-cholesterol-enriched acLDL for 24 h. Afterwards, cells were equilibrated overnight in DMEM containing 0.2% fatty acid-free BSA (Sigma, Vienna, Austria). After two washing steps with PBS/0.2% fatty acid-free BSA, cells were incubated with 100 µg/ml human HDL3 or 15 µg/ml purified human apoA-I (Behring Diagnostika, Vienna, Austria) in 1 ml DMEM/0.2% BSA and 2 µg/ml ACAT-Inhibitor 58-035 (Sandoz, Kundl, Austria) in the absence or presence of 300 µM 8-bromo cAMP (Sigma, Vienna, Austria). After 1, 3, 6, and 9 h, an aliquot of medium was taken, cell debris and cholesterol crystals were precipitated at 6000 g for 10 min, and radioactivity in the supernatant was determined by liquid scintillation counting. Cells were washed with PBS, lysed in 1 ml of 300 mM NaOH, and radioactivity and protein content were determined. In all experiments, fractional efflux was corrected for the radioactivity released to DMEM in the absence of an acceptor. Cholesterol efflux was expressed as the percentage of the radioactivity released from cells into the medium relative to the total radioactivity in cells and medium.
Nile red staining and fluorescence microscopy
MPM from WT, HSL−/−, and KIAA1363−/− mice were seeded on chamber slides and loaded with 100 µg/ml acLDL for 48 h and 72 h. Cells were fixed with 4% paraformaldehyde for 30 min, and lipid droplets were visualized after Nile red staining (2.5 µg/ml) by confocal laser scanning microscopy using an LSM 510 META microscope system (Carl Zeiss, Vienna, Austria). Pictures (63×) were taken at excitation 543 nm, and signals were recorded using a 560 nm long pass filter.
Cholesteryl ester turnover experiment
Cholesterol turnover studies were performed in 24-well plates. The amount of 3 × 104 MPM per well were incubated in medium A (DMEM, 0.2% BSA, 50 µg acLDL and [3H]oleate:BSA 8:1, 1 µCi/ml) for 24 h. Thereafter, cells were washed three times with PBS and equilibrated in DMEM/0.2% BSA overnight. The turnover was assayed in DMEM/0.2% BSA in the presence of the ACAT inhibitor 58035 (5 µg/ml). At indicated time points (0, 6, 9, and 24 h), cells were washed three times with PBS. Lipids were extracted with hexane:isopropanol (3:2, v:v) for 1 h and separated by thin layer chromatography (TLC) (hexane:diethylether:acetic acid, 65:35:1, v:v:v). Bands corresponding to CE were cut, and the radioactivity was measured by liquid scintillation counting. Finally, counts per minute were normalized to protein levels.
Measurement of sterol biosynthesis
MPM (3 × 105) were seeded into 12-well plates as described above and incubated in DMEM/1× serum replacement for 8 h. Cells were washed and incubated with [3H]acetate (2 µCi, ARC) for 24 h. After treatment, total lipids were extracted as described above. Bands corresponding to FC and CE were cut, and the radioactivity was assayed by liquid scintillation counting. Proteins were dissolved in 0.3 M NaOH at room temperature, and the concentration was measured using Bradford assay.
Statistics
Statistical analyses were performed with GraphPad Prism 5.0 using Student's t-test or the one-way ANOVA analysis followed by the Bonferroni posthoc test. Significance levels were set at P < 0.05 (*), P ≤ 0.01 (**), and P ≤ 0.001 (***).
RESULTS
Hydrolytic activities of recombinant HSL and KIAA1363
For the determination of lipolytic activities of HSL and KIAA1363, we overexpressed the enzymes in COS-7 cells and used cell lysates for activity assays. Overexpression of proteins was verified by Western blot analysis (Fig. 1A) using anti-HSL (84 kDa) and anti-KIAA1363 antibodies (45 and 50 kDa). First we determined their esterase activities using PNPV as substrate. Both HSL and KIAA1363 showed a significant 9.3-fold and 2.7-fold increase in esterase activity, respectively, compared with LacZ-transfected cells (Fig. 1B).
Fig. 1.
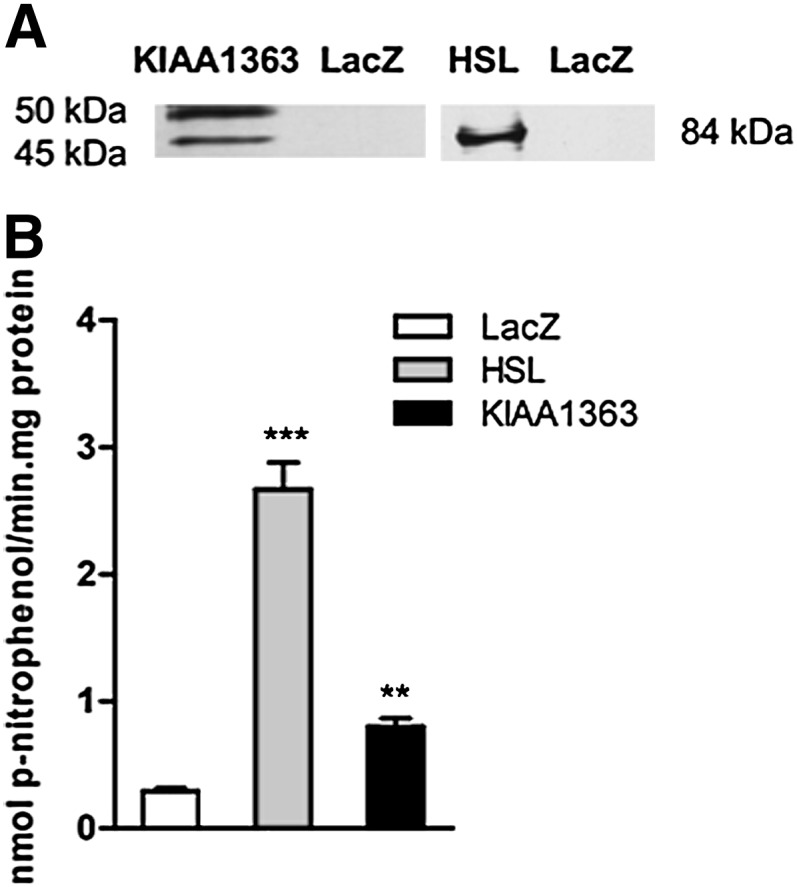
Esterase activity of recombinant HSL and KIAA1363. (A) KIAA1363 and HSL protein expressions were determined by Western blotting using anti-KIAA1363 and anti-HSL antibodies. (B) COS-7 cell lysates transiently overexpressing HSL and KIAA1363 were assayed for PNPV activity. LacZ was used as negative control. Data are presented as mean values ± SEM of three independent experiments performed in triplicate. **P ≤ 0.01; ***P ≤ 0.001. HSL, hormone-sensitive lipase; LacZ, β-galactosidase; PNPV, p-nitrophenolvalerate.
Next we measured hydrolase activities of the lysates using radiolabeled CE, TG, and DG as substrates. Overexpression of HSL in COS-7 cells resulted in markedly increased neutral CE- (17-fold) (Fig. 2A), TG- (22-fold) (Fig. 2B), and DG- (13-fold) (Fig. 2C) hydrolase activities compared with LacZ-transfected cells. No significant changes in the respective hydrolase activities were observed in cells transfected with KIAA1363 ( ). Moreover, we compared neutral CE hydrolase activities in HSL- and KIAA1363-transfected COS-7 cells using alternative mixed micelles as substrates. By using both PC/PI and PC/Na-taurocholate, we found significantly increased neutral CE hydrolase activity in cell lysates overexpressing HSL compared with LacZ-transfected cells, whereas KIAA1363-overexpressing cells lacked this activity (Fig. 2D). Finally, we preincubated extracts of transiently transfected cells with specific fluorescent TG (NBD-sn1-TGP, NBD-sn3-TGP) or CE (NBD-CP) hydrolase activity recognition probes (32), which were then subjected to SDS-PAGE analysis and fluorography. These fluorescent probes have been shown to recognize and react with enzymatically active TG or CE hydrolases (32). In HSL-transfected cells, fluorescent signals were observed with NBD-sn1-TGP and NBD-CP in the position corresponding to the expected molecular mass of HSL (84 kDa), whereas no signals were detectable in KIAA1363-transfected cells (45 and 50 kDa) (Fig. 2E). In LacZ-transfected cells, the inhibitors recognized intrinsic lipolytic and esterolytic activities of the host COS-7 cells, which resulted in multiple faint bands as described previously (32).
Fig. 2.

CE, TG, DG, and AcMAGE hydrolase activities of HSL and KIAA1363. COS-7 cells were transiently transfected with HSL, KIAA1363, or LacZ (negative control). A: CE hydrolase activities were determined in cell lysates of transfected cells by adding 35.5 µg mixed micelles of PC and PI (3:1, w:w) and 4 µM Na-taurocholate to the substrate. B: TG and (C) DG hydrolase activities were determined in cell lysates of transfected cells by adding 15 µg mixed micelles of PC and PI (3:1, w:w) and 4 µM Na-taurocholate to the respective substrates. D: CE hydrolase activities were determined in cell lysates of transfected cells by adding 35.5 µg mixed micelles of PC and PI (3:1, w:w) or 35.5 µg PC and 2 µM Na-taurocholate to the substrate. E: COS-7 cell lysates transiently overexpressing HSL and KIAA1363 were incubated with the fluorescent activity recognition probes NBD-sn1-TGP (lane 1), NBD-sn3-TGP (lane 2), and NBD-CP (lane 3) and separated by SDS-PAGE. The lack of HSL binding to NBD-sn3-TGP is consistent with previous results, demonstrating that most known lipases react poorly with that probe (32, 33). However, enantioselectivity of lipases directed to TG sn-1 or sn-3 acyl ester bonds is still under investigation as appropriately labeled substrates are needed for this purpose but are not yet commercially available. LacZ was used as negative control. Visualization was performed by laser scanning on a BioRad FY Pro laser scanner. F: AcMAGE hydrolase activities were determined in cell lysates of transfected cells. Data are presented as mean values ± SEM of four independent experiments performed in triplicate. ***P ≤ 0.001. AcMAGE, 2-acetyl monoalkylglycerol ether; CE, cholesteryl ester; DG, diacylglycerol; HSL, hormone-sensitive lipase; LacZ, β-galactosidase; NBD, 7-nitrobenz-2-oxa-1,3-diazole; NBD-sn1-TGP, enantiomeric TG analog; NBD-sn3-TGP, enantiomeric TG analog; PC, phosphatidylcholine; PI, phosphatidylinositol; TG, triacylglycerol.
KIAA1363 has been shown to hydrolyze AcMAGE in murine brain to form the hydrolysis product MAGE (22). Therefore, we measured AcMAGE hydrolase activity in lysates from HSL- and KIAA1363-transfected cells. Both lysates exhibited increased AcMAGE hydrolase activity compared with LacZ-transfected cells (1.6- and 2.0-fold, respectively) (Fig. 2F).
Effect of HSL and KIAA1363 deficiency on hydrolase activities in mouse tissues
We measured CE and AcMAGE hydrolase activities in tissues of HSL−/− and KIAA1363−/− mice. We found significantly reduced neutral CE hydrolase activities in brown and white adipose tissue, cardiac and skeletal muscle, liver, kidney, testis, and brain of HSL−/− mice (Fig. 3A) and unchanged activity in corresponding tissues of KIAA1363−/− mice (Fig. 3B) compared with WT littermates. Conversely, lack of HSL resulted in unchanged AcMAGE hydrolase activity in murine tissues (Fig. 3C), whereas KIAA1363 deficiency correlated with reduced AcMAGE hydrolase activity in brain (−76%), cardiac muscle (−76%), kidney (−25%), and testis (−50%) (Fig. 3D). We did not find any differences in liver, skeletal muscle, brown and white adipose tissue of KIAA1363−/− and WT mice.
Fig. 3.

CE and AcMAGE hydrolase activities in HSL−/− and KIAA1363−/− tissues. (A, B) Neutral CE and (C, D) AcMAGE hydrolase activities were determined in tissues isolated from HSL−/−, KIAA1363−/−, and control mice (WT). Data are presented as mean values ± SEM of three independent experiments (using three mice in each experiment). ***P ≤ 0.001. AcMAGE, 2-acetyl monoalkylglycerol ether; BAT, brown adipose tissue; CE, cholesteryl ester; CM, cardiac muscle; HSL, hormone-sensitive lipase; SM, skeletal muscle; WAT, white adipose tissue; WT, wild-type.
Effect of HSL and KIAA1363 deficiency on FC and CE concentrations in tissues
Because we found almost abolished neutral CE hydrolase activities in tissues of HSL−/− mice, we determined FC and CE content in the respective tissues (Table 1). FC concentrations were similar in all tissues of both HSL−/− and KIAA1363−/− mice compared with WT mice. CE concentrations were significantly increased only in BAT (2.2-fold), kidney (3.1-fold), and testis (3.3-fold) of HSL−/− mice. CE content in cardiac muscle, skeletal muscle, liver, and brain of HSL−/− mice and in all tissues of KIAA1363−/− mice resembled CE content of WT mice tissues.
TABLE 1.
FC and CE concentrations in tissues of HSL , KIAA1363 , and WT mice
| Tissue Lipids (mg/g tissue) |
||||
|---|---|---|---|---|
| Tissue | Genotype | N | FC | CE |
| BAT | WT | 4 | 1.10 ± 0.25 | 0.26 ± 0.14 |
| HSL | 4 | 0.97 ± 0.36 | 0.57 ± 0.12a | |
| KIAA1363 | 4 | 0.96 ± 0.18 | 0.31 ± 0.15 | |
| WAT | WT | 4 | 0.31 ± 0.11 | 0.14 ± 0.05 |
| HSL | 4 | 0.46 ± 0.13 | 0.15 ± 0.07 | |
| KIAA1363 | 4 | 0.38 ± 0.07 | 0.13 ± 0.1 | |
| CM | WT | 4 | 0.69 ± 0.06 | 0.06 ± 0.03 |
| HSL | 4 | 0.70 ± 0.06 | 0.05 ± 0.02 | |
| KIAA1363 | 4 | 0.74 ± 0.05 | 0.07 ± 0.03 | |
| SM | WT | 4 | 0.29 ± 0.04 | 0.04 ± 0.03 |
| HSL | 4 | 0.32 ± 0.03 | 0.03 ± 0.02 | |
| KIAA1363 | 4 | 0.21 ± 0.06 | 0.05 ± 0.04 | |
| Liver | WT | 4 | 2.11 ± 0.16 | 0.31 ± 0.11 |
| HSL | 4 | 2.20 ± 0.31 | 0.50 ± 0.14 | |
| KIAA1363 | 4 | 2.24 ± 0.23 | 0.28 ± 0.23 | |
| Kidney | WT | 4 | 1.12 ± 0.17 | 0.18 ± 0.04 |
| HSL | 4 | 1.08 ± 0.11 | 0.55 ± 0.19a | |
| KIAA1363 | 4 | 1.11 ± 0.48 | 0.11 ± 0.03 | |
| Testis | WT | 4 | 4.05 ± 0.33 | 0.88 ± 0.05 |
| HSL | 4 | 4.51 ± 0.21 | 2.89 ± 0.73a | |
| KIAA1363 | 4 | 3.92 ± 0.25 | 1.07 ± 0.34 | |
| Brain | WT | 4 | 0.94 ± 0.09 | 0.13 ± 0.03 |
| HSL | 4 | 0.86 ± 0.12 | 0.11 ± 0.02 | |
| KIAA1363 | 4 | 1.06 ± 0.22 | 0.12 ± 0.01 | |
FC and TC concentrations were determined enzymatically, and CE concentrations were calculated. Data are presented as mean values (n = 4) ± SEM. BAT, brown adipose tissue; CE, cholesteryl ester; CM, cardiac muscle; HSL, hormone-sensitive lipase; SM, skeletal muscle; WAT, white adipose tissue; WT, wild-type.
P < 0.05.
Plasma lipid analysis of HSLminus/minus and KIAA1363minus/minus mice
We investigated the consequences of HSL and KIAA1363 deficiency with regard to plasma lipid levels in fed and overnight-fasted mice (Table 2). Consistent with previous studies (24), plasma TG levels of HSL−/− mice were decreased by 31% after fasting. However, plasma TC levels were increased independently of the dietary status. Absence of KIAA1363 had no effect on plasma lipid parameters in both states (Table 2). Upon FPLC separation of plasma lipoproteins from fasted animals, the TC concentrations of the LDL and HDL fractions were decreased in HSL−/− mice but remained unchanged in KIAA1363−/− animals compared with WT mice (supplementary Fig. II).
TABLE 2.
Plasma lipid parameters of HSL , KIAA1363 , and control (WT) mice
| Genotype | N | TG | TC | FC | |
|---|---|---|---|---|---|
| mg/dl | mg/dl | mg/dl | |||
| Fed | WT | 9 | 67.9 ± 3.5 | 76.9 ± 2.9 | 19.1 ± 1.6 |
| KIAA1363 | 9 | 66.8 ± 4.2 | 71.9 ± 2.3 | 17.4 ± 2.3 | |
| HSL | 9 | 70.1 ± 9.6 | 90.3 ± 3.2 | 21.9 ± 2.4 | |
| Fasted | WT | 9 | 53.5 ± 4.6 | 62.0 ± 2.4 | 16.1 ± 0.9 |
| KIAA1363 | 9 | 59.6 ± 3.3 | 60.4 ± 4.9 | 14.9 ± 1.4 | |
| HSL | 9 | 36.9 ± 5.7a | 76.5 ± 3.2b | 19.0 ± 1.6 |
Blood was drawn from 8–10-week-old female mice in the fed and fasted state. TG, TC, and FC concentrations were determined enzymatically. Data are presented as mean values (n = 9) ± SEM. FC, free cholesterol; HSL, hormone-sensitive lipase; TC, total cholesterol; TG, triacylglycerol; WT, wild-type.
P < 0.05.
P ≤ 0.01.
Effect of HSL and KIAA1363 deficiency on hydrolase activities in macrophages
Next we analyzed esterase activity and neutral CE, TG, and DG hydrolase activities in peritoneal macrophages of HSL−/− and KIAA1363−/− mice. Western blot analysis confirmed the absence of HSL and KIAA1363 in HSL−/− and KIAA1363−/− macrophages, respectively (Fig. 4A, F). HSL deficiency in MPM resulted in decreased esterase activity (−77%) (supplementary Fig. III) and reduced TG hydrolase activities in cell lysates (−49%), cytosolic fractions (−78%), and membrane fractions (−57%) (Fig. 4B). In addition, DG hydrolase activities were reduced in cell lysates (−42%), cytosolic fractions (−80%), and membrane fractions (−47%) (Fig. 4C). An even more pronounced reduction was observed in CE hydrolysis, which was almost completely abolished in cell lysates (−96%), cytosolic fractions (−96%), and membrane fractions (−87%) of HSL−/− MPM (Fig. 4D). MPM lacking KIAA1363 showed similar esterase activity in cell lysates (supplementary Fig. III) as well as TG- (Fig. 4G), DG- (Fig. 4H), and neutral CE- (Fig. 4I) hydrolase activities in cell lysates, cytosolic fractions, and membrane fractions as control MPM. As a previous study has reported on unchanged neutral CE hydrolase activity in HSL−/− MPM (8), we reinforced our data using alternative substrate conditions. Under all conditions, HSL−/− MPM showed a significantly decreased neutral CE hydrolase activity (−83% and −95%, respectively) compared with WT (Fig. 5A) and KIAA1363−/− MPM (Fig. 5B). In addition, we determined neutral CE hydrolase activity in foam cells of the respective mouse models. Using PC/PI as mixed micelles, we found a 93% reduction in CE hydrolase activity of HSL−/− foam cells, whereas foam cells of KIAA1363−/− mice showed unchanged CE hydrolase activity (supplementary Fig. IV).
Fig. 4.

TG, DG, and CE hydrolase activities in HSL−/− and KIAA1363−/− macrophages. Western blotting of macrophages from (A, F) HSL−/−, KIAA1363−/−, and control mice (WT), and of (E, J) cell lysate, cytosol, and membrane fractions from WT macrophages using (A, E) anti-HSL or (F,J ) anti-KIAA1363 antibodies. (B, G) TG, (C, H) DG, and (D, I) neutral CE hydrolase activities were determined in cell lysates, cytosolic, and membrane fractions of HSL−/−, KIAA1363−/−, and WT littermate macrophages by adding (B, C, G, H) 15 µg or (D, I) 35.5 µg mixed micelles of PC and PI (3:1, w:w) and 4 µM Na-taurocholate to the respective substrates. Data are presented as mean values of three independent experiments ± SEM (using three mice in each experiment). ***P ≤ 0.001. CE, cholesteryl ester; DG, diacylglycerol; HSL, hormone-sensitive lipase; LacZ, β-galactosidase; NBD, 7-nitrobenz-2-oxa-1,3-diazole; NBD-sn1-TGP, enantiomeric TG analog; NBD-sn3-TGP, enantiomeric TG analog; PC, phosphatidylcholine; PI, phosphatidylinositol; TG, triacylglycerol; WT, wild-type.
Fig. 5.

CE hydrolase activities in HSL−/− and KIAA1363−/− macrophages using alternative mixed micelles and AcMAGE hydrolase activities. (A, B) Neutral CE hydrolase activities were determined in cell lysates from (A) HSL−/− and (B) KIAA1363−/− macrophages after adding 35.5 µg mixed micelles of PC and PI (3:1, w:w) or 35.5 µg PC and 2 µM Na-taurocholate to the substrate. (C, D) AcMAGE hydrolase activities were determined in cell lysates, cytosolic, and membrane fractions of (C) HSL−/− and (D) KIAA1363−/− macrophages and compared with their WT littermates. Data are presented as mean values (n = 3) ± SEM. ***P ≤ 0.001. AcMAGE, 2-acetyl monoalkylglycerol ether; CE, cholesteryl ester; HSL, hormone-sensitive lipase; PC, phosphatidylcholine; PI, phosphatidylinositol; WT, wild-type.
We also determined AcMAGE hydrolase activities in HSL−/− and KIAA1363−/− MPM. HSL deficiency resulted in unchanged hydrolysis of AcMAGE in cell lysates, cytosolic fractions, and membrane fractions (Fig. 5C). In contrast, KIAA1363−/− MPM exhibited a marked reduction in AcMAGE hydrolase activity in cell lysates (−59%) and membrane fractions (−79%) compared with control cells. AcMAGE hydrolase activities in cytosolic fractions were unchanged (Fig. 5D). Data are shown for female mice. However, we observed similar results in MPM isolated from male mice (data not shown).
Effect of HSL and KIAA1363 deficiency on cholesterol biosynthesis and CE turnover
We then investigated the impact of HSL or KIAA1363 deficiency on cholesterol biosynthesis or CE turnover in MPM. MPM isolated from HSL+/−, HSL−/−, KIAA1363+/−, KIAA1363−/− showed unchanged cholesterol biosynthesis compared with WT cells (supplementary Fig. V). Moreover, CE turnover in HSL−/− and KIAA1363−/− macrophages was similar compared with WT MPM (Fig. 6A, B).
Fig. 6.

CE turnover in HSL−/− and KIAA1363−/− macrophages. MPM from HSL−/− and KIAA1363−/− mice and WT littermates were loaded with [3H]oleate for 24 h. After equilibration overnight, CE turnover was assayed in the presence of the ACAT inhibitor 58035 (5 µg/ml) at the indicated time points. Radioactivity in the cells before CE turnover was set at 100%. Data represent mean values (n = 8) ± SEM of two independent experiments. CE, cholesteryl ester; HSL, hormone-sensitive lipase; MPM, mouse peritoneal macrophage; WT, wild-type.
Effect of HSL and KIAA1363 deficiency on cholesterol efflux
To assess whether HSL or KIAA1363 deficiency affects reverse cholesterol transport in vitro, MPM of HSL−/− and KIAA1363−/− mice were loaded with 3H-cholesterol labeled acLDL equilibrated for 16 h and subsequently assayed for various pathways of cholesterol efflux. Absence of HSL expression markedly reduced ATP-binding cassette transporter (ABC)G1-mediated cholesterol efflux to HDL3 (29–42% after 6 h and 9 h, respectively) (Fig. 7A). ABCA1-mediated efflux to lipid-free apoA-I was also decreased by 34% and 44%, respectively, compared with WT MPM (Fig. 7C). To investigate whether these differences depend on the activation of HSL by cAMP in WT MPM, we determined cholesterol efflux in the absence of 8-bromo cAMP in the incubation medium. Without cAMP, cholesterol efflux from HSL−/− and control MPM was identical (Fig. 7E, F). KIAA1363 deficiency did not alter cholesterol efflux to HDL3 (Fig. 7B) or apoA-I (Fig. 7D), confirming that the absence of KIAA1363 does not affect cholesterol mobilization from MPM ex vivo. We found increased ABCA1 and ABCG1 mRNA and protein levels in foam cells of all genotypes in the absence and presence of cAMP compared with unloaded MPM. The expression levels in HSL−/− and KIAA1363−/− cells remained similar to WT cells (Fig. 7G-L ).
Fig. 7.

Cholesterol efflux from HSL−/− and KIAA1363−/− macrophages. Macrophages from HSL−/− and KIAA1363−/− mice and WT littermates were labeled with [3H]cholesterol and cholesterol efflux to (A, B, E) 100 µg HDL3 and (C, D, F) 15 µg apoA1 was assessed in the (A-D) presence or (E, F) absence of 8-bromo cAMP. Cholesterol efflux was expressed as the percentage (%) of [3H]cholesterol transferred from cells to medium. Data are presented as mean values ± SEM of two independent experiments of 6–10 mice per genotype performed in triplicate. (G, I) ABCA1 and (H, J) ABCG1 mRNA levels in macrophages and foam cells of (G, H) HSL−/− and (I, J) KIAA1363−/− mice and WT littermates normalized to cyclophilin A. Expression levels in WT macrophages were arbitrarily set to 1. Western blotting of macrophages and foam cells incubated in the absence or presence of cAMP from HSL−/−, KIAA1363−/−, and WT mice using (K) anti-ABCA1 and (L) anti-ABCG1 antibodies. Anti-β-actin (42 kDa) was used as a control. *P < 0.05; **P ≤ 0.01; ***P ≤ 0.001. ABC, ATP-binding cassette; apo, apolipoprotein; HSL, hormone-sensitive lipase; MPM, mouse peritoneal macrophage; WT, wild-type.
Effect of HSL and KIAA1363 deficiency on macrophage lipid content
To investigate the effects of HSL or KIAA1363 deficiency on the lipid droplet content in macrophages, we analyzed the cells after Nile red staining by fluorescence microscopy. Unexpectedly, HSL−/− and KIAA1363−/− MPM and macrophages incubated with acLDL for 48 h and 72 h showed unaltered lipid droplet content compared with WT MPM (Fig. 8). We also measured TG, FC, and CE concentrations in MPM and acLDL-loaded cells of HSL−/−, KIAA1363−/−, and WT mice. In accordance with morphological analysis, both MPM and acLDL-loaded cells of HSL−/− and KIAA1363−/− mice showed similar concentrations of TG, FC, and CE compared with WT MPM (Table 3). In addition, β-VLDL loading failed to increase CE concentration in cells from all genotypes (supplementary Fig. VI). To investigate the impact of dietary cholesterol on CE accumulation in MPM, we isolated MPM from mice fed WTD for four weeks and determined intracellular cholesterol concentrations. We found comparable FC concentrations in macrophages from all genotypes. CE levels were slightly increased in HSL−/− and KIAA1363−/− MPM compared with WT cells (supplementary Table I). Thus, neither HSL nor KIAA1363 deficiency in macrophages led to noticeable alterations in lipid content compared with control cells.
Fig. 8.

Foam cell formation of HSL−/− and KIAA1363−/− macrophages. Representative fluorescent microscopy after Nile red staining of HSL−/−, KIAA1363−/−, and WT macrophages and foam cells. Foam cell formation was achieved by loading macrophages with 100 µg acLDL for 48 h and 72 h. Images were taken on a Zeiss LSM 510 Meta microscope. HSL, hormone-sensitive lipase; MPM, mouse peritoneal macrophage; WT, wild-type.
TABLE 3.
Lipid composition of macrophages and acLDL-loaded foam cells of HSL , KIAA1363 , and control (WT) mice
| Genotype | N | TG | FC | CE | |
|---|---|---|---|---|---|
| µg/mg protein | µg/mg protein | µg/mg protein | |||
| Macrophages | WT | 4 | 15.5 ± 1.3 | 24.6 ± 1.4 | 0.24 ± 0.07 |
| HSL | 4 | 17.1 ± 1.8 | 24.8 ± 1.9 | 0.33 ± 0.09 | |
| KIAA1363 | 4 | 17.8 ± 1.1 | 26.9 ± 2.5 | 0.40 ± 0.07 | |
| Foam cells | WT | 4 | 19.2 ± 1.6 | 27.8 ± 1.7 | 34.0 ± 2.4 |
| KIAA1363 | 4 | 18.4 ± 1.2 | 26.6 ± 3.8 | 33.1 ± 6.5 |
TG, FC, and TC concentrations were determined enzymatically, and CE concentrations were calculated. Data are presented as mean values (n = 4) ± SEM. FC, free cholesterol; HSL, hormone-sensitive lipase; TC, total cholesterol; TG, triacylglycerol; WT, wild-type.
DISCUSSION
Cholesterol is an essential lipid component of the plasma membrane and the obligatory precursor for steroid hormones. Nevertheless, increased intracellular levels of FC result in cellular dysfunction due to cell toxicity. Thus, FC is esterified with long-chain fatty acids and deposited as CE in cellular lipid droplets. CE is hydrolyzed upon demand by neutral CE hydrolases. This cycle of cholesterol esterification and CE hydrolysis is particularly important in cells with high cholesterol turnover, such as steroidogenic cells or macrophages. In contrast to the enzymes catalyzing the esterification reaction (ACAT1 and ACAT2), those responsible for CE hydrolysis are not well characterized. The present study was designed to compare the activities of two enzymes that have been reported to exhibit neutral CE hydrolase activities: HSL, an “old” enzyme still causing controversies about its function as neutral CE hydrolase in macrophages (4–10, 34), and KIAA1363, which has very recently been suggested to significantly contribute to neutral CE hydrolase activity of murine macrophages (10, 17).
Consistent with previous studies, we show that overexpression of HSL in COS-7 cells resulted in highly increased CE, TG, and DG hydrolase activities in the respective cell lysates (35). In contrast, overexpression of KIAA1363 did not result in increased CE hydrolase activities, despite an induction of esterase activity toward PNPV and increased hydrolysis of the established KIAA1363 substrate AcMAGE. The lack of CE hydrolase activity for KIAA1363 in our study contradicts recently published data showing a significant neutral CE hydrolase activity in HEK293 cells overexpressing KIAA1363 (17). Therefore, to exclude methodological differences, assays were repeated with three different substrates, including the one used by Okazaki et al. (17, 36) and high titer (150) adenovirus infection (data not shown). Independent of the substrate preparation and infection efficiency, lysates of KIAA1363-overexpressing cells lacked CE hydrolase activity, whereas overexpression of HSL resulted in highly increased activities under all experimental conditions. Hence, KIAA1363 is an AcMAGE hydrolase and a nonspecific esterase for short-chain fatty acids, but it does not hydrolyze CE. The reason for the discrepancy between our results with those of Okazaki et al. (17) concerning the CE hydrolase activity of KIAA1363 is currently unclear.
To analyze the physiological consequences of HSL and KIAA1363 deficiency for the CE cycle in vivo, we determined CE and AcMAGE hydrolase activities as well as the CE content in tissues of HSL−/− and KIAA1363−/− mice. White and brown adipose tissue (8), skeletal and cardiac muscle, liver (37), kidney, testis (8, 38), and brain of HSL−/− mice almost totally lacked neutral CE hydrolase activity, indicating that HSL is present and active as CE hydrolase in these tissues. In contrast to the pronounced alterations in enzyme activity, only moderate changes in the CE content or the CE/FC ratio were observed in tissues of HSL−/− mice. We found increased CE levels in brown adipose tissue, kidney, and testis, confirming similar results from previous studies (8). Hepatic CE concentrations were also slightly increased, consistent with recently published data (37). However, no CE accumulation occurred in white adipose tissue, muscle tissues, or brain of HSL−/− mice. Thus, although CE hydrolase activity was almost abolished in all tissues analyzed, a massive CE accumulation was not observed. This argues for the presence of non-HSL CE hydrolase(s) that are not detected by the currently utilized in vitro enzyme assays.
Unlike HSL, the disruption of KIAA1363 in mice had no effect on CE hydrolase activity in any tissue investigated, and tissue CE levels were unchanged. Together with the findings from in vitro assays, these in vivo analyses provide further evidence that KIAA1363 is unlikely to be involved in cholesterol mobilization in murine tissues.
Next we investigated the role of HSL and KIAA1363 more specifically in macrophages. In accordance with our observations in other tissues and cell types, neutral CE hydrolase activity was almost abolished in cell lysates and cytosol and markedly decreased in the membrane fraction of HSL−/− MPM. This finding contradicts similar studies in macrophages of HSL−/− mice by Osuga et al. showing unchanged neutral CE hydrolases activities in the absence of HSL (8), but it is in agreement with recently published data showing a contribution of HSL to both cytosolic and membrane neutral CE hydrolase activity (10). To exclude experimental differences between our study and the report of Osuga et al. (8), we repeated the CE hydrolase activity assays with various substrates, including the one described by Osuga et al (8). Independent of substrate choice, the difference in CE hydrolase activity between HSL−/− macrophages and control cells remained highly significant. Consistent with a functional role of HSL as CE hydrolase in macrophages, a specific antibody raised against HSL was able to markedly diminish neutral CE hydrolase activity in WT MPM (supplementary Fig. VII).
To test whether the presence or absence of HSL in macrophages alters their capacity to mobilize stored CE, thereby affecting the cellular CE content, we measured cholesterol efflux rates from macrophages and cellular CE levels, respectively. After induction of CE hydrolase activity by 8-bromo-cAMP, cholesterol efflux from WT macrophages to extracellular receptors was increased compared with unstimulated cells or macrophages from HSL−/− mice. Thus, cholesterol mobilization depends on a cAMP-regulated pathway. This finding is consistent with a previous report (40) showing that the cytosolic, neutral CE hydrolase in J774 macrophages and MPM is inducible by cAMP via protein kinase A. It is well established that cAMP activates cAMP-dependent kinase, leading to phosphorylation and activation of HSL and the hydrolysis of acylglycerol lipids (4, 9, 41). Analogously, our data propose that a similar mechanism might activate the CE hydrolase activity of HSL and induce cholesterol efflux. Decreased efflux of cholesterol from HSL−/− MPM was not related to changes in ABCA1 or ABCG1 expression on the cell surface. Despite defective cholesterol efflux rates, the absence of HSL did not affect macrophage lipid droplet formation and CE and FC concentrations in unloaded or acLDL-loaded MPM. In addition, CE turnover was identical in HSL−/− and WT cells. In view of our observations, we speculate that HSL hydrolyzes CE in macrophages together with other enzyme(s). These additional and currently unknown enzymes in macrophages (and other tissues) are apparently not recorded with the assay conditions we used, which were optimized for the determination of HSL-mediated CE hydrolase activity. Therefore, measurements of CE hydrolase activities in vitro might not perfectly reflect the in vivo situation. Non-HSL CE hydrolases may require different pH or salt conditions, alternative substrate presentation, or specific coregulators. The complexity of a multi-enzyme neutral CE hydrolysis makes it difficult to accurately measure the total CE hydrolase activity in cells and tissues.
In contrast to HSL, KIAA1363 exhibited no CE hydrolase activity in macrophages. Consistent with the findings in other tissues, KIAA1363−/− macrophages and foam cells exhibited identical neutral CE hydrolase activities as WT cells and unchanged cellular CE and FC levels. These results oppose recent findings observed in KIAA1363−/− macrophages from an independent knockout mouse line (10). Although Sekiya et al. (10) generated KIAA1363−/− mice that lacked exon 4, whereas mice used in the present study lacked exon 1 (supplementary Fig. I), this difference is unlikely to explain the opposing findings. Additionally, the discrepancy in neutral CE hydrolase activity of the present report and the recently published report (10) is not explained by technical details, as we performed the assays under identical conditions. Hence, the different findings of the two studies remain unexplained. Our efflux studies also revealed unchanged cholesterol release from KIAA1363−/− compared with WT cells in the presence of cAMP. This finding agrees with a recent study showing that KIAA1363 CE hydrolase activity is not activated by cAMP (39). Taken together, we found no evidence that KIAA1363 acts as CE hydrolase or participates in cholesterol homeostasis in murine macrophages. This conclusion is also supported by the previous observation that KIAA1363 is an integral membrane protein (20, 21) with active site residues facing the lumen of the endoplasmic reticulum (39). This renders this protein unlikely to efficiently hydrolyze CE contained in cytoplasmic lipid droplets.
KIAA1363 deficiency in macrophages resulted in significantly decreased AcMAGE hydrolase activity in the membrane fraction of cell lysates, suggesting that KIAA1363 is a major AcMAGE hydrolase in macrophages. We found a reduction in AcMAGE hydrolase activity in KIAA1363−/− testis and, in accordance with published data (22), in brain and kidney, but not in the liver of KIAA1363−/− mice. KIAA1363 expression is quite low in murine liver (25), and it is suspected that hepatic AcMAGE hydrolase activity depends on the enzyme arylacetamide deacetylase (AADA). AADA shares a significant sequence homology with KIAA1363 (44% sequence identity) and is highly expressed in the liver. KIAA1363 and AADA are two distinct proteins encoded on chromosome 3. Since AcMAGE is the precursor for PAF and MAGE (22), KIAA1363 might reduce PAF levels (and inflammation) by converting AcMAGE into MAGE.
Taken together, the present study demonstrates that KIAA1363 does not contribute to neutral CE hydrolase activity but serves as the principal AcMAGE hydrolase in macrophages. HSL is a neutral CE hydrolase in macrophages, and it interacts with other, presently unknown cytosolic hydrolases to maintain cholesterol homeostasis in macrophages.
Supplementary Material
Acknowledgments
The authors thank D. Nomura for advice in the AcMAGE assay; J. G. Strauss for providing the KIAA1363 construct; S. Povoden, E. Stanzer, A. Ibovnik, K. Masuda, M. Scholler, and T. Kueznik for excellent technical assistance; and I. Hindler and A. Hermann for mice care.
Footnotes
Abbreviations:
- AcMAGE
- 2-acetyl monoalkylglycerol ether
- apo
- apolipoprotein
- CE
- cholesteryl ester
- CP
- cholesteryl phosphonate
- DG
- diacylglycerol
- FC
- free cholesterol
- HSL
- hormone-sensitive lipase
- LacZ
- β-galactosidase
- MPM
- mouse peritoneal macrophage
- NBD
- 7-nitrobenz-2-oxa-1,3-diazole
- PAF
- platelet-activating factor
- PC
- phosphatidylcholine
- PI
- phosphatidylinositol
- PNPV
- p-nitrophenolvalerate
- TC
- total cholesterol
- TGH
- TG hydrolase
- WT
- wild-type
This work was supported by the Austrian Federal Ministry of Science and Research [GEN-AU project Genomics of Lipid-associated Disorders (GOLD)]; the Austrian Science Fund FWF (P19186, SFB-LIPOTOX F30); the Molecular Medicine PhD program at the Medical University of Graz; and the National Institutes of Health (CA-087660). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health or other granting agencies.
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of seven figures and one table.
REFERENCES
- 1.Brown M. S., Ho Y. K., Goldstein J. L. 1980. The cholesteryl ester cycle in macrophage foam cells. Continual hydrolysis and re-esterification of cytoplasmic cholesteryl esters. J. Biol. Chem. 255: 9344–9352. [PubMed] [Google Scholar]
- 2.Ho Y. K., Brown M. S., Goldstein J. L. 1980. Hydrolysis and excretion of cytoplasmic cholesteryl esters by macrophages: stimulation by high density lipoprotein and other agents. J. Lipid Res. 21: 391–398. [PubMed] [Google Scholar]
- 3.Goodman D. S. 1965. Cholesterol ester metabolism. Physiol. Rev. 45: 747–839. [DOI] [PubMed] [Google Scholar]
- 4.Small C. A., Goodacre J. A., Yeaman S. J. 1989. Hormone-sensitive lipase is responsible for the neutral cholesterol ester hydrolase activity in macrophages. FEBS Lett. 247: 205–208. [DOI] [PubMed] [Google Scholar]
- 5.Escary J. L., Choy H. A., Reue K., Schotz M. C. 1998. Hormone-sensitive lipase overexpression increases cholesteryl ester hydrolysis in macrophage foam cells. Arterioscler. Thromb. Vasc. Biol. 18: 991–998. [DOI] [PubMed] [Google Scholar]
- 6.Okazaki H., Osuga J., Tsukamoto K., Isoo N., Kitamine T., Tamura Y., Tomita S., Sekiya M., Yahagi N., Iizuka Y., et al. 2002. Elimination of cholesterol ester from macrophage foam cells by adenovirus-mediated gene transfer of hormone-sensitive lipase. J. Biol. Chem. 277: 31893–31899. [DOI] [PubMed] [Google Scholar]
- 7.Escary J. L., Choy H. A., Reue K., Wang X. P., Castellani L. W., Glass C. K., Lusis A. J., Schotz M. C. 1999. Paradoxical effect on atherosclerosis of hormone-sensitive lipase overexpression in macrophages. J. Lipid Res. 40: 397–404. [PubMed] [Google Scholar]
- 8.Osuga J., Ishibashi S., Oka T., Yagyu H., Tozawa R., Fujimoto A., Shionoiri F., Yahagi N., Kraemer F. B., Tsutsumi O., et al. 2000. Targeted disruption of hormone-sensitive lipase results in male sterility and adipocyte hypertrophy, but not in obesity. Proc. Natl. Acad. Sci. USA. 97: 787–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Contreras J. A. 2002. Hormone-sensitive lipase is not required for cholesteryl ester hydrolysis in macrophages. Biochem. Biophys. Res. Commun. 292: 900–903. [DOI] [PubMed] [Google Scholar]
- 10.Sekiya M., Osuga J., Nagashima S., Ohshiro T., Igarashi M., Okazaki H., Takahashi M., Tazoe F., Wada T., Ohta K., et al. 2009. Ablation of neutral cholesterol ester hydrolase 1 accelerates atherosclerosis. Cell Metab. 10: 219–228. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh S. 2000. Cholesteryl ester hydrolase in human monocyte/macrophage: cloning, sequencing, and expression of full-length cDNA. Physiol. Genomics. 2: 1–8. [DOI] [PubMed] [Google Scholar]
- 12.Ghosh S., St. Clair R. W., Rudel L. L. 2003. Mobilization of cytoplasmic CE droplets by overexpression of human macrophage cholesteryl ester hydrolase. J. Lipid Res. 44: 1833–1840. [DOI] [PubMed] [Google Scholar]
- 13.Soni K. G., Lehner R., Metalnikov P., O'Donnell P., Semache M., Gao W., Ashman K., Pshezhetsky A. V., Mitchell G. A. 2004. Carboxylesterase 3 (EC 3.1.1.1) is a major adipocyte lipase. J. Biol. Chem. 279: 40683–40689. [DOI] [PubMed] [Google Scholar]
- 14.Shamir R., Johnson W. J., Morlock-Fitzpatrick K., Zolfaghari R., Li L., Mas E., Lombardo D., Morel D. W., Fisher E. A. 1996. Pancreatic carboxyl ester lipase: a circulating enzyme that modifies normal and oxidized lipoproteins in vitro. J. Clin. Invest. 97: 1696–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li F., Hui D. Y. 1997. Modified low density lipoprotein enhances the secretion of bile salt-stimulated cholesterol esterase by human monocyte-macrophages. species-specific difference in macrophage cholesteryl ester hydrolase. J. Biol. Chem. 272: 28666–28671. [DOI] [PubMed] [Google Scholar]
- 16.Hui D. Y., Howles P. N. 2002. Carboxyl ester lipase: structure-function relationship and physiological role in lipoprotein metabolism and atherosclerosis. J. Lipid Res. 43: 2017–2030. [DOI] [PubMed] [Google Scholar]
- 17.Okazaki H., Igarashi M., Nishi M., Sekiya M., Tajima M., Takase S., Takanashi M., Ohta K., Tamura Y., Okazaki S., et al. 2008. Identification of neutral cholesterol ester hydrolase, a key enzyme removing cholesterol from macrophages. J. Biol. Chem. 283: 33357–33364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jessani N., Liu Y., Humphrey M., Cravatt B. F. 2002. Enzyme activity profiles of the secreted and membrane proteome that depict cancer cell invasiveness. Proc. Natl. Acad. Sci. USA. 99: 10335–10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jessani N., Niessen S., Wei B. Q., Nicolau M., Humphrey M., Ji Y., Han W., Noh D. Y., Yates J. R., 3rd, Jeffrey S. S., et al. 2005. A streamlined platform for high-content functional proteomics of primary human specimens. Nat. Methods. 2: 691–697. [DOI] [PubMed] [Google Scholar]
- 20.Nomura D. K., Leung D., Chiang K. P., Quistad G. B., Cravatt B. F., Casida J. E. 2005. A brain detoxifying enzyme for organophosphorus nerve poisons. Proc. Natl. Acad. Sci. USA. 102: 6195–6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiang K. P., Niessen S., Saghatelian A., Cravatt B. F. 2006. An enzyme that regulates ether lipid signaling pathways in cancer annotated by multidimensional profiling. Chem. Biol. 13: 1041–1050. [DOI] [PubMed] [Google Scholar]
- 22.Nomura D. K., Fujioka K., Issa R. S., Ward A. M., Cravatt B. F., Casida J. E. 2008. Dual roles of brain serine hydrolase KIAA1363 in ether lipid metabolism and organophosphate detoxification. Toxicol. Appl. Pharmacol. 228: 42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haemmerle G., Zimmermann R., Hayn M., Theussl C., Waeg G., Wagner E., Sattler W., Magin T. M., Wagner E. F., Zechner R. 2002. Hormone-sensitive lipase deficiency in mice causes diglyceride accumulation in adipose tissue, muscle, and testis. J. Biol. Chem. 277: 4806–4815. [DOI] [PubMed] [Google Scholar]
- 24.Haemmerle G., Zimmermann R., Strauss J. G., Kratky D., Riederer M., Knipping G., Zechner R. 2002. Hormone-sensitive lipase deficiency in mice changes the plasma lipid profile by affecting the tissue-specific expression pattern of lipoprotein lipase in adipose tissue and muscle. J. Biol. Chem. 277: 12946–12952. [DOI] [PubMed] [Google Scholar]
- 25.Nomura D. K., Durkin K. A., Chiang K. P., Quistad G. B., Cravatt B. F., Casida J. E. 2006. Serine hydrolase KIAA1363: toxicological and structural features with emphasis on organophosphate interactions. Chem. Res. Toxicol. 19: 1142–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haemmerle G., Lass A., Zimmermann R., Gorkiewicz G., Meyer C., Rozman J., Heldmaier G., Maier R., Theussl C., Eder S., et al. 2006. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 312: 734–737. [DOI] [PubMed] [Google Scholar]
- 27.Basu S. K., Goldstein J. L., Anderson G. W., Brown M. S. 1976. Degradation of cationized low density lipoprotein and regulation of cholesterol metabolism in homozygous familial hypercholesterolemia fibroblasts. Proc. Natl. Acad. Sci. USA. 73: 3178–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Eck M., Herijgers N., Yates J., Pearce N. J., Hoogerbrugge P. M., Groot P. H., Van Berkel T. J. 1997. Bone marrow transplantation in apolipoprotein E-deficient mice. Effect of ApoE gene dosage on serum lipid concentrations, (beta)VLDL catabolism, and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 17: 3117–3126. [DOI] [PubMed] [Google Scholar]
- 29.Kratzer A., Buchebner M., Pfeifer T., Becker T. M., Uray G., Miyazaki M., Miyazaki-Anzai S., Ebner B., Chandak P. G., Kadam R. S., et al. 2009. Synthetic LXR agonist attenuates plaque formation in apoE−/− mice without inducing liver steatosis and hypertriglyceridemia. J. Lipid Res. 50: 312–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kienesberger P. C., Lass A., Preiss-Landl K., Wolinski H., Kohlwein S. D., Zimmermann R., Zechner R. 2008. Identification of an insulin-regulated lysophospholipase with homology to neuropathy target esterase. J. Biol. Chem. 283: 5908–5917. [DOI] [PubMed] [Google Scholar]
- 31.Holm C., Osterlund T. 1999. Hormone-sensitive lipase and neutral cholesteryl ester lipase. Lipase and Phospholipase Protocols. Doolittle M. H., Reue K., Vol. 109 Methods in molecular biology (series) Humana Press, Totowa, NJ: 109–121. [DOI] [PubMed] [Google Scholar]
- 32.Schmidinger H., Birner-Gruenberger R., Riesenhuber G., Saf R., Susani-Etzerodt H., Hermetter A. 2005. Novel fluorescent phosphonic acid esters for discrimination of lipases and esterases. Chembiochem. 6: 1776–1781. [DOI] [PubMed] [Google Scholar]
- 33.Birner-Gruenberger R., Susani-Etzerodt H., Waldhuber M., Riesenhuber G., Schmidinger H., Rechberger G., Kollroser M., Strauss J. G., Lass A., Zimmermann R., et al. 2005. The lipolytic proteome of mouse adipose tissue. Mol. Cell. Proteomics. 4: 1710–1717. [DOI] [PubMed] [Google Scholar]
- 34.Kraemer F. B., Shen W. J. 2002. Hormone-sensitive lipase: control of intracellular tri-(di-)acylglycerol and cholesteryl ester hydrolysis. J. Lipid Res. 43: 1585–1594. [DOI] [PubMed] [Google Scholar]
- 35.Fredrikson G., Stralfors P., Nilsson N. O., Belfrage P. 1981. Hormone-sensitive lipase of rat adipose tissue. Purification and some properties. J. Biol. Chem. 256: 6311–6320. [PubMed] [Google Scholar]
- 36.Hajjar D. P., Minick C. R., Fowler S. 1983. Arterial neutral cholesteryl esterase. A hormone-sensitive enzyme distinct from lysosomal cholesteryl esterase. J. Biol. Chem. 258: 192–198. [PubMed] [Google Scholar]
- 37.Sekiya M., Osuga J., Yahagi N., Okazaki H., Tamura Y., Igarashi M., Takase S., Harada K., Okazaki S., Iizuka Y., et al. 2008. Hormone-sensitive lipase is involved in hepatic cholesteryl ester hydrolysis. J. Lipid Res. 49: 1829–1838. [DOI] [PubMed] [Google Scholar]
- 38.Vallet-Erdtmann V., Tavernier G., Contreras J. A., Mairal A., Rieu C., Touzalin A. M., Holm C., Jegou B., Langin D. 2004. The testicular form of hormone-sensitive lipase HSLtes confers rescue of male infertility in HSL-deficient mice. J. Biol. Chem. 279: 42875–42880. [DOI] [PubMed] [Google Scholar]
- 39.Igarashi M., Osuga J-i., Isshiki M., Sekiya M., Okazaki H., Takase S., Takanashi M., Ohta K., Kumagai M., Nishi M., et al. 2010. Targeting of neutral cholesterol ester hydrolase to the endoplasmic reticulum via its N-terminal sequence. J. Lipid Res. 51: 274–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khoo J. C., Mahoney E. M., Steinberg D. 1981. Neutral cholesterol esterase activity in macrophages and its enhancement by cAMP-dependent protein kinase. J. Biol. Chem. 256: 12659–12661. [PubMed] [Google Scholar]
- 41.Stralfors P., Belfrage P. 1983. Phosphorylation of hormone-sensitive lipase by cyclic AMP-dependent protein kinase. J. Biol. Chem. 258: 15146–15152. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.