

SUMMARY
Repulsive signaling by Semaphorins and Plexins is crucial for the development and homeostasis of the nervous, immune and cardiovascular systems. Sema7A acts as both an immune and a neural Semaphorin through PlexinC1, and A39R is a Sema7A mimic secreted by poxviruses. We report the structures of Sema7A and A39R complexed with the Semaphorin-binding module of PlexinC1. Both structures show two PlexinC1 molecules symmetrically bridged by Semaphorin dimers, in which the Semaphorin and PlexinC1 β-propellers interact in an edge-on, orthogonal orientation. Both binding interfaces are dominated by the insertion of the Semaphorin’s 4c-4d loop into a deep groove in blade 3 of the PlexinC1 propeller. A39R appears to achieve Sema7A mimicry by preserving key Plexin-binding determinants seen in the mammalian Sema7A complex, that have evolved to achieve higher affinity binding to the host-derived PlexinC1. The complex structures support a conserved Semaphorin-Plexin recognition mode, and suggest Plexins are activated by dimerization.
INTRODUCTION
Semaphorins and their receptors, Plexins, are two families of widely expressed proteins whose respective structures and functions are conserved across the animal kingdom. Originally identified as ligand-receptor pairs that control axon guidance by repulsion during CNS development (Kolodkin et al., 1993; Luo et al., 1993; Tamagnone et al., 1999; Tessier-Lavigne and Goodman, 1996), Semaphorins and Plexins have been shown to serve as path-finding controls for a diverse array of additional functions in physiology, including vascularization and angiogenesis (normal and pathological), organogenesis, and immune responses (Kruger et al., 2005; Suzuki et al., 2008). Semaphorins are also associated with tumor progression and other diseases (reviewed in (Kruger et al., 2005; Tamagnone and Comoglio, 2000)). Within the neural system, Semaphorin-Plexin signaling is implicated well beyond axon guidance, ranging from axon pruning to synaptic formation, specificity and plasticity (reviewed in (He et al., 2002; Waimey and Cheng, 2006)). While Plexins are the predominant receptors for Semaphorins, the alternative Semaphorin receptors, Neuropilin-1 and −2, differ from Plexins in structure and function, and appear to serve as obligate co-receptors for either Semaphorin-signaling or VEGF-signaling in a variety of specific cellular (neuronal, vascular, etc.) contexts (Pellet-Many et al., 2008; Uniewicz and Fernig, 2008).
Semaphorins have been grouped into eight classes on the basis of primary sequence and source, where classes 3-7 are vertebrate Semaphorins and class V are viral Semaphorins (Semaphorin_Nomenclature_Committee, 1999). The Semaphorins are characterized by an N-terminal ~500 amino acid (a.a.) Sema domain which is essential for signaling through Plexins (Koppel et al., 1997). The crystal structures of the Sema domains of Sema3A and Sema4D (also known as CD100) have been determined, showing that the Sema domain is a seven-bladed β-propeller, and appear to exist as a homodimer by virtue of conserved structural elements (Antipenko et al., 2003; Love et al., 2003). There are four subfamilies of vertebrate Plexins (A, B, C, and D), which are all type I transmembrane glycoproteins featuring an extracellular segment containing an N-terminal Sema domain, followed by variable numbers of PSI (‘found in Plexins, Semaphorins and Integrins’) domains, immunoglobulin-like (Ig) domains (Bork et al., 1999), and an intracellular GTPase-activating (GAP) domain that regulates Rho-family GTPases (Oinuma et al., 2004). The structures of the intracellular domains of two Plexins have been solved (He et al., 2009; Tong et al., 2009), but three-dimensional structural information does not exist for the Plexin extracellular segment, nor for the Semaphorin-Plexin interaction.
In the immune system, a subset of Semaphorins are active, and are designated as “immune Semaphorins”. These include Sema4D, Sema4A, and Sema7A, which appear to modulate a variety of immune responses ranging from thymic selection to B cell homing (Suzuki et al., 2008). Sema7A is a GPI-anchored cell-surface glycoprotein expressed on activated lymphocytes and thymocytes (Xu et al., 1998; Yamada et al., 1999), and its receptor is PlexinC1 (also named VESPR or CD232) (Tamagnone et al., 1999). The Sema7A/PlexinC1 interaction was originally shown to induce the activation of monocytes (Holmes et al., 2002), but recently this receptor-ligand pair has been shown to regulate melanocyte adhesion, and silencing of PlexinC1 is seen during the development and progression of melanoma (Scott et al., 2009). Sema7A also has been suggested to have neuronal functions, by promoting axon growth through integrins (Pasterkamp et al., 2003). Underscoring the importance of Semaphorin/Plexin interactions in the immune system, viruses have evolved proteins that engage the immune Semaphorin/Plexin system, presumably to enhance virus survival in the host. The most well characterized examples derive from Vaccinia virus and Alcelaphine herpesvirus, which encode secreted Semaphorin homologues A39R and AHVsema (Comeau et al., 1998; Kolodkin et al., 1993). These viral Semaphorins show sequence similarity to Sema7A and can also bind to, and activate PlexinC1 (Comeau et al., 1998). The viral Semaphorins probably play an immunomodulatory role by mimicking Sema7A; an antibody against PlexinC1 inhibits A39R-induced induction of inflammatory cytokines by monocytes (Comeau et al., 1998).
We present here the crystal structures of both the Sema7A/PlexinC1 complex and the A39R/PlexinC1 complex, revealing the basic architecture of a Semaphorin/Plexin recognition complex. Our data point to a conserved mode of recognition of Plexins by Semaphorins, and provide insights into Plexin activation and viral mimicry.
RESULTS
Biochemical analysis of PlexinC1 binding by the mammalian Semaphorin Sema7A and the viral Semaphorin A39R
The extracellular domains of Semaphorins and Plexins are large, and highly glycosylated, thus we expressed recombinant forms of PlexinC1, Sema7A and A39R using baculovirus-mediated mammalian cell gene transduction (BacMam) (Dukkipati et al., 2008), and purified the secreted proteins from HEK293H cells or N-acetylglucosaminyltransferase I-deficient HEK293 (GnTI- HEK293) cells (Reeves et al., 2002). Our construct designs were informed by prior structural analysis of Sema4D (Love et al., 2003), which showed that the first membrane-distal PSI domain is integrally associated with the Sema domain. Deletion of the PSI domains in either PlexinC1 or Sema7A destabilized the proteins. Ultimately, after testing a variety of constructs, for PlexinC1, we expressed the Sema domain plus the first PSI domain (SemaPSI), and the entire extracellular segment (ECD). For Sema7A, we expressed the entire extracellular segment excluding the GPI anchor; for A39R, we expressed the full-length protein, which is a “minimal” secreted Semaphorin that does not contain a PSI domain (Figure 1A).
Figure 1. Characterization of the binding of Sema7A and A39R to PlexinC1.
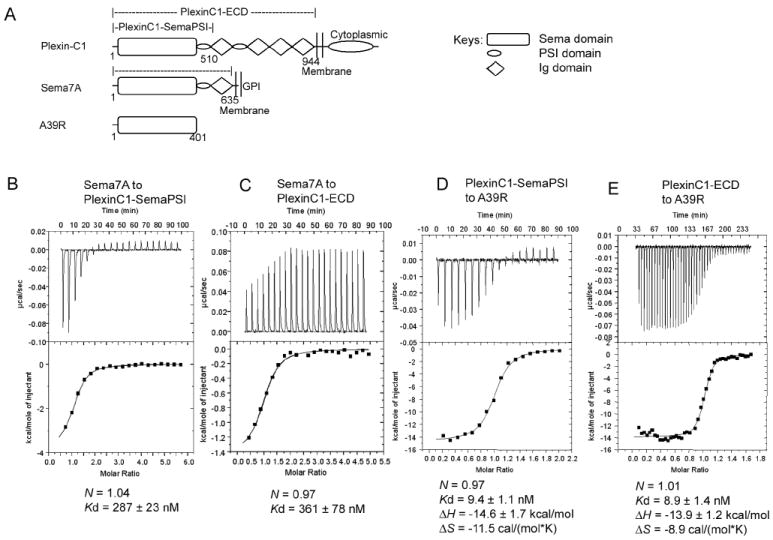
(A) Domain diagram of PlexinC1, Sema7A and A39R and the constructs used in characterization. The cartoon key for the diagrams is shown to the right.
(B), (C), (D) and (E) Profiles of the binding of PlexinC1 to Sema7A and A39R as measured by isothermal titration calorimetry. The binding parameters are indicated below the traces.
Gel filtration profiles showing that Sema7A and A39R exist as dimers are in Figure S1.
To analyze the solution binding of PlexinC1 to its A39R and Sema7A ligands, we used isothermal titration calorimetry (ITC) (Figures 1B-E). In Hepes-buffered saline (HBS), Sema7A bound PlexinC1-SemaPSI with a KD of ~290nM (Figure 1B). The ITC measurements for Sema7A/PlexinC1 were complicated by the small enthalpy change (~-2 kcal/mol), which resulted in shallow titration curves with poor signal-to-noise ratio. Therefore we used high concentrations of the samples and injected large amounts of protein so as to maximize the heat per injection, and also to fully saturate the titration curve. The Sema7A/PlexinC1 KD measured here by calorimetry using soluble proteins, albeit in the nanomolar range, is weaker than the reported KD of 2.1nM from Scatchard analysis using intact receptors in the cell membrane (Tamagnone et al., 1999). Previously reported Semaphorin/Plexin affinity measurements are primarily cell-based, and generally in the low nanomolar KD range. We attribute the lower affinity values of the soluble recombinant proteins to the lack of membrane confinement in the ITC format, not to structural differences in the recombinant proteins. Similar differences between solution-based and cell-based affinities have been seen for ligand binding to other receptors, such as SCF/KIT (55nM vs 2nM)(Lemmon et al., 1997; Lev et al., 1992), CNP/NPRB (2.7nM vs 0.03nM) (He et al., 2006; Koller and Goeddel, 1992), and MHC/TCR (micromolar vs nanomolar) (Huppa et al.; Krogsgaard et al., 2003). We also determined that the affinity of Sema7A for the truncated PlexinC1-SemaPSI and full-length PlexinC1 ECD were nearly identical (Figures 1B, 1C), indicating the Ig- and PSI-domains C-terminal to the PlexinC1-SemaPSI module are not significantly involved in Semaphorin recognition. Thus for structural studies we proceeded with crystallization of the Sema7A/PlexinC1-SemaPSI complex.
The viral-derived A39R bound PlexinC1-SemaPSI with a KD of ~9.4nM (Figure 1D). The large enthalpy change (-14.6 kcal/mol) facilitated obtaining high quality binding curves. We also characterized A39R for the effect of various PlexinC1 ECD truncations on affinity. A39R affinity and thermodynamic parameters for full-length PlexinC1-ECD (KD ~8.9nM) were nearly identical to that of A39R binding to PlexinC1-SemaPSI (Figures 1D, 1E). Thus, similar to Sema7A/PlexinC1 binding, this indicated that the N-terminal Sema-PSI domains of PlexinC1 were sufficient for Semaphorin binding and represented an appropriate form for co-crystallization of both the Sema7A/PlexinC1 and A39R/PlexinC1 complexes.
Structures of the Sema7A/PlexinC1 complex, free A39R, and the A39R/PlexinC1 complex
To obtain diffraction quality crystals of the heavily-glycosylated Sema7A/PlexinC1-SemaPSI complex (4 N-linked glycosylated sites on Sema7A, 7 on PlexinC1-SemaPSI), we used Endo-H to trim the N-linked glycans attached to the GnTI- HEK293-expressed Sema7A and PlexinC1-SemaPSI proteins. This treatment leaves a single N-acetylglucosamine (GlcNAc) residue remaining at each occupied N-linked glycan site, preserving the core glycosylation of the proteins. The glycan-trimmed proteins exhibited the same solution behavior to wild-type proteins, e.g., the Semaphorins existed exclusively as dimers after glycan-trimming, as assessed by gel filtration chromatography (Figure S1). The Sema7A/PlexinC1-SemaPSI complex structure was determined to a resolution of 2.4Å using the method of single isomorphous replacement with anomalous scattering (SIRAS) (Table S1, Figure S2). The asymmetric unit of the crystal contains one dimeric complex in a 2:2 stoichiometry, comprised of a central Sema7A dimer and two monomeric PlexinC1-SemaPSI molecules (Figure 2A).
Figure 2. Structures of the Sema7A/PlexinC1-SemaPSI and A39R/PlexinC1-SemaPSI complexes.
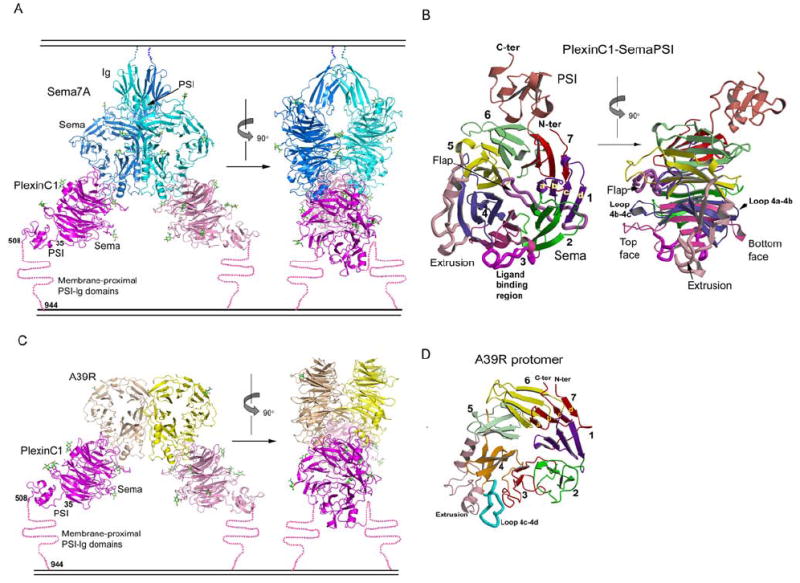
(A) Ribbon models of the Sema7A/PlexinC1-SemaPSI complex in front view (left) and side view (right), with the Sema7A protomers colored in cyan and blue, and the PlexinC1-SemaPSI protomers in pink and magenta. The N-linked glycans are depicted as sticks with carbon atoms colored in green. A cartoon of a membrane is drawn above and below the complex to indicate where the respective proteins would be attached to the cell surfaces.
(B) The structure of an individual PlexinC1-SemaPSI molecule from the complex in two orthogonal views, with each of the 7 β-propeller blades, the extrusion, the flap and the PSI domain individually colored.
(C) Ribbon model of the A39R/PlexinC1-Sema-PSI complex in front view (left) and side view (right), with the A39R protomers colored in yellow and wheat, and the other components colored similarly to panel A.
(D) Ribbon model of an A39R protomer from the free A39R dimer, with the structural modules colored in the same format as PlexinC1 shown in panel (B).
See Table S1 and Figure S2 for crystallographic statistics and structural comparisons.
Overall, the shape of the complex resembles a crab where Sema7A comprises the body, and the two PlexinC1 molecules resemble pincers emanating radially from the body at four and eight o’clock. The approximate dimensions of the complex are 80 Å × 140 Å × 160 Å (Figure 2A). The head-to-head docking architecture of the complex is in an ideal orientation for interaction of two cell-surface associated proteins across a cell-cell junction. In the complex, both Sema7A and PlexinC1 contain large, disk-shaped Sema domains composed of 7-bladed β-propellers, each of which is intimately associated with its respective PSI domain and, in the case of Sema7A, a single Ig domain that would lead to the membrane. In the complex, the Sema domains mediate the vast majority of the receptor-ligand contact. The center-to-center distances between the Sema propellers are 50 Å for Sema7A-Sema7A, 55 Å for Sema7A-PlexinC1 and 80 Å for PlexinC1-PlexinC1.
Interestingly, the Sema domains of Sema7A and PlexinC1 interact “edge-on” using their sides to contact one another, rather than the top or bottom faces, as was generally expected. The planes of the Sema7A and PlexinC1 β-propellers are orthogonally related. The orientation shown in Figure 2 places the C-termini of Sema7A close to the cell membrane, leading to the GPI-anchor. The C-terminus of the PSI domain of PlexinC1, which is ~160 Å from the Semaphorin C-terminus, leads to the opposing cell membrane, albeit through one additional PSI and four Ig-domains not present in the current structure. Because the remaining PSI and Ig-domains of PlexinC1 would appear to emanate away from the central dyad axis of the complex, they are unlikely to contact Sema7A.
We expressed A39R in baculovirus, and determined the unliganded A39R structure, to a resolution of 2.0Å, by SIRAS (Table S1). The structure of A39R shows a dimer of minimal Sema domains with no PSI or Ig domains (Figure 2D). The Sema domains of A39R and Sema7A are clearly built on the same scaffold, with an r.m.s deviation of 1.5 Å for matching Cα atoms (Figure S3). Major structural differences are located only at the N-terminal segment and several loops remote from the Plexin-binding site of Sema7A. Sema7A, like Sema3A and Sema4D, has a long N-terminal segment that provides an additional, fifth β-strand on the outer edge of blade 6, whereas A39R lacks this segment (Figure S3).
We followed the same approach as Sema7A/PlexinC1-SemaPSI to obtain the crystals of the A39R/PlexinC1-SemaPSI complex, which we solved by molecular replacement (Table S1). The architecture of the A39R/PlexinC1 complex is similar to that of Sema7A/PlexinC1, except that due to the lack of PSI and Ig domains in A39R, it is shorter in its longest dimension (Figure 2C). As in the Sema7A/PlexinC1 complex, the Sema domains mediate all the A39R-PlexinC1 contact. The orientations of the Sema propellers in the complex are nearly identical to those in Sema7A/PlexinC1, as is the “edge-on” interaction mode between the ligand and receptor β-propellers.
Dimerizations of Sema7A and A39R are mediated by conserved structural elements with varied chemistry
Dimerization appears to be a general, and likely important property of Semaphorins (Antipenko et al., 2003; Love et al., 2003) that we can now examine within the context of a receptor complex. The Sema7A molecules in the complex, consisting of Sema, PSI, and Ig domains, form a dimer roughly similar to the Sema4D dimer (Love et al., 2003) and the Sema3A dimers that only contain the Sema domains (Antipenko et al., 2003) (Figure S2C). This is consistent with our gel filtration analysis (Figure S1). The Sema7A dimer interface buries 2860 Å2 of solvent-accessible surface area. Two sets of three loops on the top face of the Sema domain, 4b-4c, 4d-5a and 5b-5c are intertwined at the dimer interface. In comparison, both Sema4D and Sema3A use an additional loop for dimerization (Figure S2C). The Sema7A Sema-Sema interface is primarily hydrophobic. The Ig domain of Sema7A is also involved in the dimer interface, but only contributes ~15% to the total buried surface area. The Ig domain of Sema7A, unlike the Sema4D Ig domain which is canonical, is an unusual variation of the Ig-fold, with a 5-on-2 topology where strand D switches from the BED β-sheet to join the AFGC β-sheet (Figure S2D).
The viral Semaphorin A39R also exists as a dimer both in solution and in the crystal (Figure S1). Dimerization of A39R is exclusively mediated by its Sema domain, since this protein does not have PSI or Ig domains, and the dimer interface is considerably smaller (~1990 Å2) than that seen on the canonical Semaphorins (Figure S2C). A39R uses the same set of loops as the Sema7A Sema for dimerization, but in contrast to the hydrophobic Sema7A dimer interface, the A39R dimer interface is composed almost exclusively of hydrophilic interactions involving six salt bridges.
The Sema7A and A39R structures, together with the previous Sema3A and Sema4D structures, indicate that although the dimer interfaces of Semaphorins can vary significantly in sequence and chemical nature, the general structural mode of domain dimerization is conserved, and this is likely important for the appropriate dimerization geometry of the bound Plexin receptors for signaling. The preservation of the dimerization geometry despite vastly different interface chemistries suggests that there is evolutionary pressure to achieve this dimerization mode for Semaphorin function, presumably to orient bound Plexins for signaling.
The structure of PlexinC1-SemaPSI has unique features
Given that the extracellular segments of Plexins have not been structurally characterized, the fold of the PlexinC1 Sema-PSI domains merits some description. The Sema domain of PlexinC1 in the complex is generally similar to other 7-bladed β-propeller domains in topology, but has unique features that are distinct from Semaphorins or MET (Stamos et al., 2004) (Figure 2B). Following the common nomenclature used by other propeller proteins, blade 7 of the PlexinC1 Sema domain is C-terminally adjacent to blade 1; each of the 7 blades is formed by four anti-parallel β-strands with strands a-d from the inside to the outside of the β-propeller (Figure 2B). The surface bears the loops linking strands b and c (e.g., loop 4b-4c) and linking strands d and a (e.g., loop 5d-6a) on the top face. Loop 1d-2a traverses the top of the propeller like a flap, sealing the central channel of the propeller, which is hollow in Semaphorins, MET and integrins (Figure S2E) (Antipenko et al., 2003; Love et al., 2003; Stamos et al., 2004; Xiao et al., 2004; Xiong et al., 2002). A long insertion, termed the ‘extrusion’ (Love et al., 2003), is located between strands 5c and 5d. A major structural difference is that the extrusion of PlexinC1 is shorter than that of Semaphorins. The structure of the complex shows that this extrusion in Semaphorins is critical for binding Plexins, but the extrusion in Plexins is not a central part of the interface. It appears, then, that Plexins have lost the prominence of this structural element as the relative functions of the Sema domain in Semaphorins versus Plexins specialized over the course of evolution (Figure S2E) (Antipenko et al., 2003; Love et al., 2003; Stamos et al., 2004).
The PSI domain of PlexinC1 is a small cysteine-rich domain similar to that of Semaphorins and integrins, but its orientation relative to the Sema domain is different from that in Semaphorins by a ~20° rotation (Figure S2E). The PSI domain is intimately packed against the Sema domain by a broad array of primarily hydrophobic interactions, which would facilitate the sensitive structural transmission of Sema binding from the membrane-distal to the membrane-proximal regions of PlexinC1.
The Sema7A-PlexinC1 interaction features a “loop-in-groove” recognition mode
The edge-on, orthogonal stacking of the respective β-propellers in the Sema7A-PlexinC1 interface (Figure 3A) buries a total of ~2100 Å2 solvent-accessible surface area. The extensive interface can be divided into three principal regions: 1- The 4c-4d loop of Sema7A inserting into the groove on PlexinC1 that is bounded by walls composed of the PlexinC1 loop 3b-3c and the bulged strand 3d. This PlexinC1 groove has an open and a closed (obstructed) end (Figure 3B). 2- The extrusion helix 2 of Sema7A contacting the loop 3b-3c of PlexinC1 at one side of the groove. 3- A small area of contact between the Sema7A blade 3, and the PlexinC1 strand 3d, that forms the opposing wall of the groove (Figures 3C, 3D and 3E).
Figure 3. The interface between Sema7A and PlexinC1-SemaPSI.

(A) The overall structure of an interacting pair of Sema7A and PlexinC1-SemaPSI protomers, highlighting the structural elements involved in their binding, colored cyan for Sema7A and pink for PlexinC1-SemaPSI.
(B) Closeup view of the interface with Sema7A depicted as ribbons and PlexinC1-SemaPSI in a surface representation, showing how the protruding loop 4c-4d of Sema7A inserts into a groove in PlexinC1 blade 3, and is flanked by the contacts between the Sema7A extrusion helix 2 and the PlexinC1 loop 3b-3c, and the contacts between the blade 3 of Sema7A and the bulged strand 3d of PlexinC1.
(C) The interactions of residues near the obstructed end of the PlexinC1 groove, with Sema7A in chocolate and PlexinC1 in pink.
(D) The interaction between residues of the 4c-4d loop of Sema7A (cyan) and the PlexinC1 groove (pink).
(E) The interaction between the residues of blade 3 of Sema7A (green) and strand 3d of PlexinC1 (pink). Note that residues 218-220 of PlexinC1 comprise a bulge from strand 3d.
The interactions are listed in Table S2. Mutagenesis/binding data on this interface are in Figure S4.
The central “loop-in-groove” interaction (Figure 3D) features 10 hydrogen bonds (all hydrogen bonds discussed are predicted from geometry) between the protruding 4c-4d loop of Sema7A and the PlexinC1 groove (Table S2). The polar interactions are supplemented by two hydrophobic residues (Leu276 and Val278) in the Sema7A 4c-4d loop, which contact several hydrophobic residues in the PlexinC1 groove through van der Waals interactions. Overall, the PlexinC1/Sema7A loop-in-groove interaction includes a mixture of hydrophobic residues lining the groove wall, interspersed with hydrophilic residues, to presumably provide ligand specificity through polar interactions. Importantly, at the edge of this groove in PlexinC1, Sema7A Lys280 forms a salt bridge with PlexinC1 Asp200, and this interaction appears to be a key component of A39R mimicry (discussed below).
At the obstructed (closed) end of the groove (Figure 3B), the Sema7A extrusion helix 2 interacts in a roughly parallel manner with the PlexinC1 3b-3c loop, also showing several ancillary long-range interactions with the tips of the PlexinC1 2b-2c and 2d-3a loops (Figure 3C). The interaction is intimate only at the PlexinC1 Ala197-Ala198-Ser199 bulge. The obstructed end of the PlexinC1 groove is largely due to the steric bulk sidechain of Arg131, which forms a salt bridge with Sema7A Glu376 (Figure 3C).
In the third region of the interface, at the open end of the PlexinC1 groove (Figure 3B), the outer edge of Sema7A blade 3 forms extended interactions with the bulged strand 3d of PlexinC1 (Figure 3E). There are two spatially separate clusters of salt bridges in this region of the interface. The first cluster involves Sema7A residues Arg204 and Arg202 both forming salt bridges with PlexinC1 residue Glu219. Sema7A Tyr213 occupies the space between this salt bridge and a salt-bridge involving the important Sema7A residue Lys280 and PlexinC1 Asp200 (Figure S2A). The second cluster of charged interactions involve Sema7A Asp216, which forms bifurcated salt bridges with Arg222 and Lys224 of PlexinC1. Mutagenesis data supports that the interactions at this region are important for Sema7A-PlexinC1 binding (Figure S4). In the complex structure, the location of the ridges flanking the groove in the Semaphorin binding site is consistent with a previous mutagenesis study implicating the region between residues 166-235 of the Sema domain of Sema3A in its Plexin-binding specificity (Koppel et al., 1997). In summary, the Sema7A/PlexinC1 interface is extensive and varied in chemical and structural character, and dominated by the insertion of a long loop in Sema7A into a deep groove in PlexinC1.
The A39R-PlexinC1 interaction globally resembles the Sema7A-PlexinC1 interaction
With strikingly similar orientations of the respective β-propellers in the two complexes, the A39R-PlexinC1 interface involves the same set of structural elements as the Sema7A-PlexinC1 interface (Figure 4). The A39R-PlexinC1 interface buries a total of 1890 Å2 solvent-accessible surface area, slightly smaller than the Sema7A-PlexinC1 interface. The protruding loop 4c-4d of A39R is in an almost identical conformation as that of Sema7A, inserting into the groove of the blade 3 surface of PlexinC1 (Figures 4A, 4B, and 5). The neighboring segments of this loop, including the extrusion helix 2 of A39R that contacts the loop 3b-3c of PlexinC1, and the A39R blade 3 that contacts the PlexinC1 strand 3d, have undergone some relatively minor structural accommodations at the periphery of the interface (Figure 4A). These small movements result in remodeled pairwise interactions by these surrounding structural elements, relative to the Sema7A/PlexinC1 interface, but still preserving several key contacts that presumably are important for the cross-reactivity (Discussed below).
Figure 4. The interface between A39R and PlexinC1-SemaPSI.

(A) Structural superposition of the A39R/PlexinC1-SemaPSI and Sema7A/PlexinC1-SemaPSI complexes indicating the closely related docking modes of the viral and mammalian Semaphorins to the PlexinC1 receptor. The complexes have been aligned on the PlexinC1 component in order to visualize the respective overlap of the two different Semaphorins. The A39R is in yellow, Sema7A in cyan.
(B) Closeup view of the interface with A39R depicted as ribbons and PlexinC1-SemaPSI in a surface representation.
(C) The interactions of residues near the obstructed end of the PlexinC1 groove, with A39R in chocolate and PlexinC1-SemaPSI in pink.
(D) The interaction between residues of the 4c-4d loop of A39R (blue) and the PlexinC1 groove (pink).
(E) The interaction between the residues of blade 3 of A39R (green) and strand 3d of PlexinC1-SemaPSI (pink).
The interactions are listed in Table S3.
Figure 5. The conformation of the 4c-4d loop of Semaphorins is central for Plexin recognition.
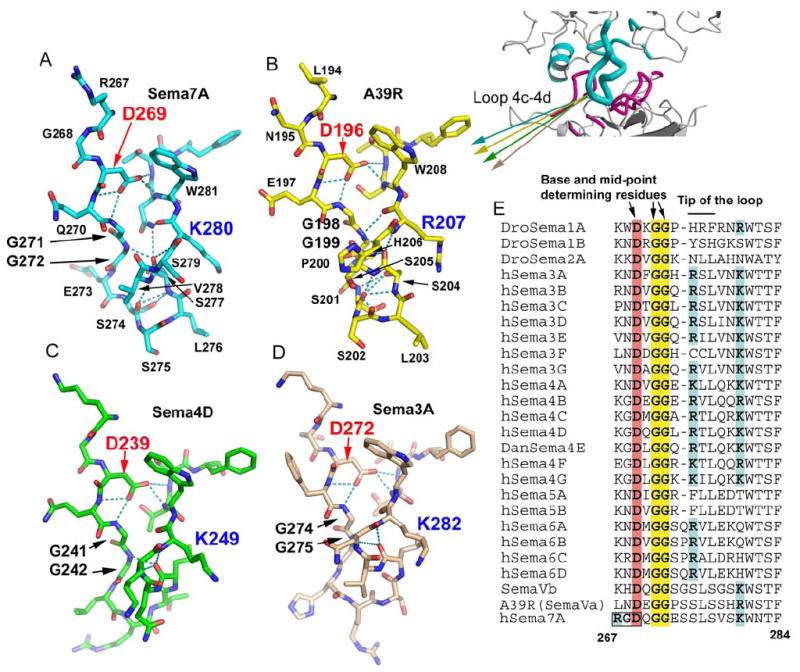
(A), (B), (C) and (D) Stick models of the isolated 4c-4d loops of Sema7A (cyan), A39R (yellow), Sema4D (green), and Sema3A (pink), showing its conserved conformation at the bases (top) and the mid-points of the loops, and variable conformation at the tips of the loops (bottom).
(E) Sequence comparison of the 4c-4d loop of Semaphorins, with the key conserved structural determinants highlighted.
See also Figure S5 for the RGD motif at the base of the Sema7A 4c-4d loop.
The “loop-in-groove” interaction in A39R/PlexinC1 (Figure 4D) has 6 hydrogen bonds between the A39R 4c-4d loop and the PlexinC1 groove, 4 less than in Sema7A/PlexinC1 (Table S3). Only one of these hydrogen bonds is conserved between Sema7A/PlexinC1 and A39R/PlexinC1. The diversity of amino acid contacts with PlexinC1 formed by the Sema7A versus A39R 4c-d loop indicates that the PlexinC1 pocket has the capacity for highly degenerate interactions, which facilitates cross-reactivity. At the edge of this groove in PlexinC1, A39R presents an arginine (Arg207), as apposed to a lysine (Lys280) in Sema7A, to form a salt bridge with PlexinC1 Asp200 (discussed below).
Peripheral to the 4c-4d loop, the structural chemistry of the A39R/PlexinC1 interactions at the obstructed (closed) end of the PlexinC1 groove (Figures 4B, 4C) are quite different than what is seen for Sema7A/PlexinC1 due to a slight rigid-body repositioning (Figure 4A). Compared to Sema7A, the A39R extrusion helix 2 is shifted outwards relative to the center of the interface, and tilted away from PlexinC1 (Figures 4A, 4C). Consequently, the A39R/PlexinC1 interaction is not as intimate as the Sema7A/PlexinC1 interaction at this region. However, the shifting of the helix also allows A39R Asp300, corresponding to Sema7A Gln379 (Figures 3C, 4C), to move into the position to form a salt bridge with PlexinC1 Arg131, which is not present in Sema7A/PlexinC1.
The A39R/PlexinC1 interactions at the open end of the PlexinC1 groove (Figures 4B, 4E) involve the same set of PlexinC1 residues as in Sema7A/PlexinC1, but the pattern of interaction is altered. While there are four salt bridges in two clusters in Sema7A/PlexinC1, there are only two (A39R Arg132/Arg134 to PlexinC1 Glu219) in A39R/PlexinC1 at this region (Figure 4E). In A39R/Sema7A, the loss of the other cluster of salt bridges seen in Sema7A/PlexinC1 (PlexinC1 Arg222/Lys224 to Sema7A Asp216) is due to the raised A39R blade 3. While PlexinC1 Lys224 is not directly bonded to A39R in the A39R/PlexinC1 complex, PlexinC1 Arg222 manages to form hydrogen bonds with the hydroxyl of A39R Tyr145. The loss of these two salt bridges from the mammalian complex may be further compensated by the strengthening of the A39R Arg132 – PlexinC1 Glu219 salt bridge, which is more deeply buried than in Sema7A/PlexinC1, due to the neighboring hydrophobic interaction between A39R Ile125 and PlexinC1 Leu220 (Figure 4E). Collectively, The A39R/PlexinC1 interface shows variations from the Sema7A/PlexinC1 interface due to some intermolecular repositioning around the central 4c-4d loop. Nevertheless, the general scheme of inserting the long 4c-4d loop into the blade 3 groove in PlexinC1 is identical for Sema7A and A39R.
The similarities and variations of the central recognition loops of Semaphorins
The structural features of the 4c-4d interaction loop of Sema7A and A39R are similar in all Semaphorin structures determined to date (Figure 5). The conformation of this loop is stabilized by an extensive intra-loop hydrogen-bonding network, which involves residues highly conserved in most Semaphorins (Figures 5A-E). At the base of the loop, Asp269 (Sema7A numbering), a buried aspartate conserved in all Semaphorins (highlighted in Figures 5A-E), forms a network of four hydrogen bonds with main chain amides, which play a central role in organizing the loop conformation. At the mid-point, a serine (274 in Sema7A and 201 in A39R) hydrogen bonds with 2 main chain amides on the opposing strand to narrow the loop. The kinking of the loop is also facilitated by the presence of two consecutive glycines (Gly271-Gly272) conserved in all Semaphorins (Figure 5E). At the tip of the loop, the Sema7A Ser274 main chain carbonyl forms hydrogen bonds with the main chain amides of Ser277 and Val278, a pattern reminiscent of reverse-turns, which may help to maintain a rigid and protruding conformation. Similar conformation is observed for the tip of this A39R loop (Figure 5A, Figure 5B).
In Sema3A and Sema4D, however, the tip of this loop has a variation from Sema7A/A39R (Figures 5C and 5D). Class 3-5 Semaphorins including Sema3A and Sema4D have a one-residue deletion at the Ser274 position. Sema4D and Sema3A also lack the main-chain hydrogen bonds at the tip seen in Sema7A and A39R. Consequently, the tips of the 4c-4d loops of these Semaphorins can adopt different main chain conformations, probably reflecting the structural requirements of their specific Semaphorin-Plexin interactions, yet all appear to contain a polar residue at the loop tips for hydrogen bonding. Because the residues which reside at the apex of the loop are the most deeply embedded in the base of the Plexin groove, as exemplified by Sema7A/PlexinC1, it is likely that these are major determinants for Semaphorin-Plexin binding specificity between classes. Indeed, there is extensive sequence divergence between different classes of Semaphorins at these positions, but few within the same class of Semaphorins (Figure 5E).
The mimicry of the mammalian Semaphorin Sema7A by the viral Semaphorin A39R
The mammalian and viral Semaphorins have arrived at nearly identical binding modes despite substantial variations in sequence (31% identity between Sema7A and A39R). Visualization of the PlexinC1 binding surfaces of Sema7A and A39R (Figures 6A and 6B) indicates that there are several key corresponding regions of structural mimicry that likely serve as the foundation for their cross-reactivity with a common receptor. There is obvious structural conservation of the centrally located 4c-4d loop and the presence of identical residues at the tips of the loop (Ser-Leu) that engage the deepest portion of the PlexinC1 groove. The preservation of the di-Glycine pair in both Sema7A and A39R 4c-4d loops (Figure 5) ensures similar loop conformations presented to PlexinC1. There is mimicry of an array of interactions peripheral to the tip of the 4c-4d loop, as is clear from comparison of the respective binding surfaces. For example, the Lys280 in Sema7A that salt-bridges to Asp200 in PlexinC1 is mimicked by Arg207 in A39R that also salt bridges to Asp200 on PlexinC1. Also found in both Semaphorins is a cluster of salt bridges on blade 3 that interact with residues on strand d of PlexinC1. Here, Arg202 of Sema7A salt bridges with Glu219 in PlexinC1, this interaction is mimicked by Arg134 of A39R salt bridging to PlexinC1 Glu219. Arg202 and Asp216 in Sema7A are spatially mimicked by Arg132 and Asp148 in A39R, but the hydrogen bonding network with the same PlexinC1 residues is remodeled. Key to this cluster is a Tyrosine residue (Tyr213 in Sema7A, Tyr145 in A39R) (Figure 6F) that is in a nearly identical position in both complexes, in the center of the cluster of salt bridges.
Figure 6. Viral mimicry of Sema7A by A39R.
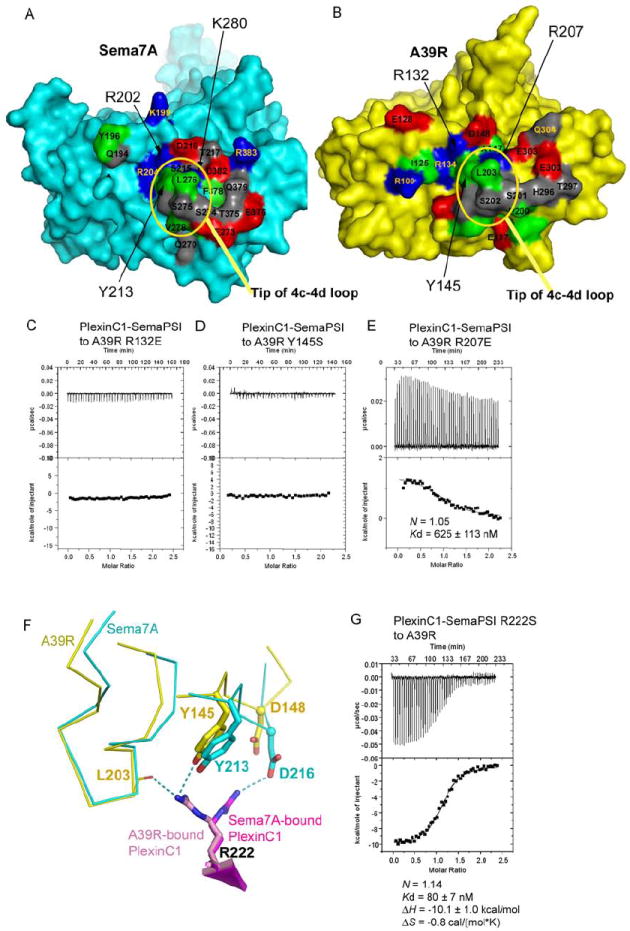
(A) and (B) The respective PlexinC1-binding surfaces of Sema7A (left, cyan) and A39R (right, yellow). The residues mapped at the interface are colored red for acidic residues, blue for basic residues, gray for polar, non-charged residues, and green for apolar residues.
(C), (D) and (E) Mutations of the A39R residues mapped to the Sema7A-PlexinC1 interface abolished or dramatically reduced A39R-PlexinC1 binding.
(F) The different conformation of PlexinC1 Arg222 in binding A39R and Sema7A.
(G) Calorimetric measurement of the binding between A39R and the Arg222Ser mutant of PlexinC1-SemaPSI.
A structural and sequence comparison of Sema7A and A39R is shown in Figure S3.
In order to ask whether the residues A39R uses to ‘mimic’ Sema7A were energetically important for the viral Semaphorin binding, we mutated several of them and tested binding by ITC (Figure 6). Mutations of A39R Arg132 or Tyr145 (corresponding to Sema7A Arg202 and Tyr213) to glutamate or serine abolished PlexinC1-binding (Figures 6C and 6D), whereas mutation of A39R Arg207 (corresponding to Sema7A Lys280) to glutamate reduced PlexinC1-binding by >60 fold (Figure 6E). The A39R Arg207 has a stronger charge and a larger head group than a Lys residue, and its aliphatic stem is kinked, contacting the neighbouring hydrophobic moieties more intimately. Arg207 is a more complementary fit in the A39R/PlexinC1 interface compared to the Lys280 in the Sema7A-PlexinC1 interface, where there is more solvent-occupied space adjacent to the side chain (Figure S2A, S2B). Our data (Figure 6E) suggest that the difference at this position could be an important contributing factor to the higher affinity binding to PlexinC1 achieved by A39R, although additional interactions no doubt also contribute. We also mutated PlexinC1 Arg222, which is hydrogen bonded to A39R Tyr145, and serves as shared contact with both Semaphorins. We found a 9-fold affinity loss in A39R-PlexinC1 binding (Figure 6G). While Sema7A and A39R have exactly the same residues at the local region contacting PlexinC1, PlexinC1 Arg222 adopts very different rotamers in the two complexes (Figure 6F). The 9-fold affinity-loss upon PlexinC1 Arg222Ser mutation suggests this is an important determinant in A39R/PlexinC1 binding (Figure 6G). Therefore the residues we mutated, which are strikingly coincident in the viral and mammalian Sema complexes, while certainly not giving us a comprehensive energetic map of the A39R/PlexinC1 interface landscape, are indeed energetically important in ligand-receptor binding.
Collectively, our structures suggest that A39R, built on a smaller scaffold, has used a limited set of key, highly coincident amino acid contacts to evolve a more efficient binding interaction with PlexinC1 through both the enhancement of particularly important polar interactions, such as the substitution of A39R Arg207 for the Sema7A Lys280, and slight adjustments of side rotamer and main chain positions throughout the binding surface. These adjustments sum to a substantial optimization of binding energetics, and a concomitant gain in binding affinity.
DISCUSSION
We present here the structures of two Semaphorin-Plexin complexes, establishing the general recognition paradigm between two protein families with important and wide-ranging physiological functions involving deliverance of repulsive guidance cues. Unexpectedly, the two β-propellers of the Semaphorin and Plexin Sema domains contact each other edge-on. In comparison, MET uses the bottom face of the propeller for HGF-β chain interaction (Stamos et al., 2004), whereas integrins bind their ligands with the top face of the propeller (Xiao et al., 2004; Xiong et al., 2002). Thus, as for other recognition modules in biology, such as leucine-rich repeats, immunoglobulin folds, and even MHC-folds, the β-propeller reveals itself to be capable of a diverse array of ligand interaction modes. The moderate-to-low sequence identity between different classes of Semaphorins and Plexins, especially at the binding segments, suggests that there is variability in the specific inter-atomic contacts in Semaphorin/Plexin interfaces between families that is buttressed by a framework of conserved interactions. This variability may be necessary for coding the complex pattern of specificity in the growth- or motility- guiding activities. Nevertheless, the orientation of each component in the complex, the Sema-Sema docking geometry, and the global usage of structural elements should be generally conserved for Semaphorins and Plexins, as has been found in other ligand-receptor families with similar extents of sequence identity.
The poxvirus-derived A39R has evolved a more efficient PlexinC1 binding surface, yet it is smaller than that of Sema7A, with fewer overall contacts. Our structural analysis showed that the central binding elements of Sema7A are rigorously maintained in the viral complex, therefore how does A39R achieve higher affinity binding? This is very difficult to clearly answer, as the sources of free energy differences between interacting proteins are not obviously identified by inspection of structures, and are contributed by many subtle effects such as intramolecular side chain cooperativity, solvent restructuring at the binding surface, and enthalpy-entropy compensation. Thus, we suspect that it is due to a collective reduction of energetic penalties across the entire Semaphorin/Plexin interface, perhaps in combination with an optimization of protein dynamics, such as lowering overall temperature factors (Table S1), to gain favor in entropic change. The high similarity of Sema7A and A39R suggests the likelihood that the poxviruses have acquired the A39R gene by hijacking and mutating the Sema7A gene, rather than through convergent evolution where the virus would have evolved a structurally distinct scaffold for PlexinC1 binding (Gewurz et al., 2001). Thus, this does not seem to be a case of molecular “mimicry” as much as simply modifying an existing binding scaffold with subtle improvements. Given the lack of involvement of the PSI and Ig domains in Plexin interactions, these domains may have been lost in A39R over the course of evolution.
The dimerized Sema7A/PlexinC1 and A39R/PlexinC1 complexes support a model of PlexinC1 activation via dimerization akin to many other cell-surface receptors. This, in itself, is an important advance since the downstream mechanisms of Semaphorin/Plexin repulsive signaling are not well understood, and the knowledge that Plexins are active as dimers should help refine our thinking about possible downstream effectors. The quiescent PlexinC1 possibly exists as pre-formed oligomers, given the evidence that some other Plexins, e.g., plexin B, exist as stable homodimers in the absence of ligands (Tong et al., 2007). We have also found that unliganded PlexinC1 ECD exists as a mixture of oligomeric states, as assessed by gel filtration chromatography and cross-linking, although we have not quantitated the exact stochiometries of these states. We propose that constitutively dimeric ligands such as Sema7A and A39R orient the inactive PlexinC1 into a dimerized state, in which the distance, geometry and conformation enables the activation of the intracellular GAP activity. This PlexinC1 activation scheme is simpler than a previously proposed model for Sema3A-induced PlexinA1 activation. That model proposed that the Plexin Sema domain associates intramolecularly with its additional extracellular domains in an autoinhibited conformation, analogous to the LDL receptor (Rudenko et al., 2002), and that Sema3A disrupts this intra-molecular association (Antipenko et al., 2003; Takahashi and Strittmatter, 2001). Although we have no unequivocal data to support or refute such a model, our ITC experiments of A39R binding to full-length versus truncated PlexinC1 ECD show identical binding energetics and mono-phasic association, indirectly implying that PlexinC1 is in the same conformation in the full-length ECD and truncated forms (Figures 1D and 1E). An important caveat to our experiments is that thermodynamic measurements would not detect a potential biphasic binding kinetics indicative of a two-step binding event involving a Plexin conformational change. Nevertheless, whether Plexin undergoes a conformational change prior to Semaphorin binding, clearly a Semaphorin-induced arrangement of Plexin into a specifically orientated dimer is the means of receptor activation. Future structural and mechanistic studies can now address whether the pre-activated Plexin is involved in an intramolecular association that contributes to the maintenance of an autoinhibited state.
The Sema7A/PlexinC1 and A39R/PlexinC1 complexes evoke several important questions. First, where is the Neuropilin-docking site in a class 3 Semaphorin-Plexin complex? Although predictions could be made based on our structures and the previous mutagenesis mapping data (Antipenko et al., 2003), accurate insights may require a complex structure encompassing Neuropilin. Second, does Sema7A bind integrins directly? It has been suggested Sema7A promotes axon growth and initiates T-cell-mediated inflammation responses through integrins, but not PlexinC1, using an RGD-dependent mechanism (Pasterkamp et al., 2003; Suzuki et al., 2007). The RGD motif of Sema7A (Arg267-Gly268-Asp269) is buried (Figure S5), and is unlikely to be recognized by integrins. Finally, the complex structures can now be used as a basis to interrogate Sema/Plexin interactions in vivo with high precision. While mutating the key determinants of both Sema7A-PlexinC1 and A39R-PlexinC1 interfaces clearly compromise binding (Figure 6 and Figure S4), further work will be necessary to demonstrate the consequences of such perturbation in a cellular or organismal context. Such functional validation should determine whether modulating Semaphorin-Plexin recognition holds clinical promise for applications involving cell guidance, e.g., directional nerve regeneration.
EXPERIMENTAL PROCEDURES
Cloning, cell culture and baculovirus generation
Human Sema7A, PlexinC1-SemaPSI and PlexinC1-ECD, were expressed in the BacMam system as described previously (Dukkipati et al., 2008), with a 7-His tag directly attached to the C-terminus of each expression construct. Full length Vaccinia virus A39R was expressed in both the BacMam system and the baculovirus-insect cells system. Sf9 and hi5 cells were maintained in the HyQ-SFX (HyClone) and InsectXpress (Lonza) media. HEK-293H cells (Invitrogen) and GnTI- HEK293 cells (Reeves et al., 2002) were maintained in CDM4HEK293 media (HyClone). To generate recombinant viruses, the expression constructs and the BacVector-3000 baculovirus DNA (EMD Chemicals) were used to co-transfect sf9 cells in 6-well plates. After 5 days, the resulted low titer virus stock was harvested, and was used to infect Sf9 cells at 2 × 106 cell/ml for amplification.
Protein preparation
The amplified baculoviruses were used to infect 1-6 liters of HEK293 cells or hi5 cells at a density of 1.0-1.5 × 106 cells/ml. After 72 hours, the cells were pelleted, and the supernatants were concentrated. The recombinant proteins in the supernatant were captured by Ni-NTA metal affinity resin (Qiagen) and eluted with 300mM imidazole pH7.5. The eluted proteins were further purified with gel filtration.
Glycan-trimming, crystallization, data collection and processing
For the Sema7A-PlexinC1 complex, GnTI- HEK293-expressed proteins were trimmed with carboxypeptidase-A and Endo-H. The digested products were further purified with size exclusion columns, and concentrated to 10mg/ml. Crystallization was performed using the sitting-drop vapor-diffusion method. The Sema7A and PlexinC1-SemaPSI proteins, at 1:1 molar ratio, were mixed with an equal volume of reservoir solution, and equilibrated against the reservoir solution containing 12% PEG1000, 0.1 M Tris pH 7.5 and 0.2 M Li2SO4.
The A39R used for crystallization was expressed from hi5 cells using the pAcGP67A vector. A39R was His-tagged at the N-terminus and expressed in the presence of 10μM kifunensine in the expression media (InsectXpress). The His-tag was removed from A39R with TEV protease, the glycans were trimmed with Endo-H, and the protein was further purified by gel filtration. A39R crystallized in the following condition: 50mM sodium citrate pH 5.0, 0.2 M Li2SO4, 15% PEG3350 and 20% glycerol.
For the A39R-PlexinC1-SemaPSI complex, the A39R protein was expressed from the BacMam system following the same procedure for PlexinC1-SemaPSI. The complex was crystallized in the following condition: 6% PEG8000, 0.1 M CaCl2, 0.1 M Hepes pH 7.0 and 0.2 M NaCl.
The Sema7A/PlexinC1 and A39R/PlexinC1 data were measured at the Life Sciences Collaborative Access Team (LS-CAT) beamlines 21-ID-D and 21-ID-G at the Advanced Photon Source (APS), and were processed with HKL2000 (Otwinowski and Minor, 1997) (Table S1). The A39R data were measured at both the Stanford Synchrotron Research Lab (SSRL) and the Advanced Light Source (ALS), and were processed with Mosflm and scaled with Scala of the CCP4 program suite (Collaborative Computational Project, 1994). The statistics on the data are presented in Table S1.
Structure determination and model refinement
For structure determinations of Sema7A/PlexinC1-SemaPSI and free A39R, experimental phases were calculated using heavy atom derivatives by the SIRAS method, as implemented in the program SOLVE (Terwilliger and Berendzen, 1999) and SHARP (Bricogne et al., 2003). For structure determination of the A39R/PlexinC1-SemaPSI complex, the components from the above two structures were used as search models in molecular replacement. The models were refined with CNS (Brunger et al., 1998). The final models include 7 N-linked glycans per PlexinC1-SemaPSI molecule (Asn86, Asn141, Asn149, Asn241, Asn252, Asn386 and Asn407), 4 per Sema7A (Asn105, Asn157, Asn258, and Asn330), and one per A39R (Asn51). Each glycan was modeled with a single GlcNAc residue, as expected, due to the Endo-H treatment of the proteins. A summary of the refinement statistics is given in Table S1.
Isothermal titration calorimetry
The proteins used for calorimetry were all expressed from HEK-293H cells. ITC was carried out on a VP-ITC calorimeter (MicroCal, Northhampton, MA) at 30°C. All samples were thoroughly degassed before titration. The titration data were processed with MicroCal Origin software, Version 5.0.
Supplementary Material
Acknowledgments
We thank the staff at the LS-CAT beamlines at APS and the staffs of ALS and SSRL for the support in X-ray data collection. We also thank L. Colf, and N. Goriatcheva for help with the expression of A39R. X.H. is supported by the NIH grant 1R01GM07805, and K.C.G. is an Investigator of the Howard Hughes Medical Institute. The Structural Biology Facility is supported by the R.H. Lurie Comprehensive Cancer Center of Northwestern University.
Footnotes
ACCESSION NUMBER The structure factors and coordinates for the Sema7A/PlexinC1-SemaPSI complex, free A39R and the A39R/PlexinC1-SemaPSI complex have been deposited in the Protein Data Bank (accession codes 3NVQ, 3NVX, and 3NVN).
SUPPLEMENTAL INFORMATION Supplemental Information includes Extended Experimental Procedures, five figures and three tables.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antipenko A, Himanen JP, van Leyen K, Nardi-Dei V, Lesniak J, Barton WA, Rajashankar KR, Lu M, Hoemme C, Puschel AW, et al. Structure of the semaphorin-3A receptor binding module. Neuron. 2003;39:589–598. doi: 10.1016/s0896-6273(03)00502-6. [DOI] [PubMed] [Google Scholar]
- Bork P, Doerks T, Springer TA, Snel B. Domains in plexins: links to integrins and transcription factors. Trends Biochem Sci. 1999;24:261–263. doi: 10.1016/s0968-0004(99)01416-4. [DOI] [PubMed] [Google Scholar]
- Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W. Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr D Biol Crystallogr. 2003;59:2023–2030. doi: 10.1107/s0907444903017694. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Comeau MR, Johnson R, DuBose RF, Petersen M, Gearing P, VandenBos T, Park L, Farrah T, Buller RM, Cohen JI, et al. A poxvirus-encoded semaphorin induces cytokine production from monocytes and binds to a novel cellular semaphorin receptor, VESPR. Immunity. 1998;8:473–482. doi: 10.1016/s1074-7613(00)80552-x. [DOI] [PubMed] [Google Scholar]
- Dukkipati A, Park HH, Waghray D, Fischer S, Garcia KC. BacMam system for high-level expression of recombinant soluble and membrane glycoproteins for structural studies. Protein Expr Purif. 2008;62:160–170. doi: 10.1016/j.pep.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewurz BE, Gaudet R, Tortorella D, Wang EW, Ploegh HL. Virus subversion of immunity: a structural perspective. Curr Opin Immunol. 2001;13:442–450. doi: 10.1016/s0952-7915(00)00239-9. [DOI] [PubMed] [Google Scholar]
- He H, Yang T, Terman JR, Zhang X. Crystal structure of the plexin A3 intracellular region reveals an autoinhibited conformation through active site sequestration. Proc Natl Acad Sci U S A. 2009;106:15610–15615. doi: 10.1073/pnas.0906923106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XL, Dukkipati A, Garcia KC. Structural determinants of natriuretic peptide receptor specificity and degeneracy. J Mol Biol. 2006;361:698–714. doi: 10.1016/j.jmb.2006.06.060. [DOI] [PubMed] [Google Scholar]
- He Z, Wang KC, Koprivica V, Ming G, Song HJ. Knowing how to navigate: mechanisms of semaphorin signaling in the nervous system. Sci STKE. 2002;2002:re1. doi: 10.1126/stke.2002.119.re1. [DOI] [PubMed] [Google Scholar]
- Holmes S, Downs AM, Fosberry A, Hayes PD, Michalovich D, Murdoch P, Moores K, Fox J, Deen K, Pettman G, et al. Sema7A is a potent monocyte stimulator. Scand J Immunol. 2002;56:270–275. doi: 10.1046/j.1365-3083.2002.01129.x. [DOI] [PubMed] [Google Scholar]
- Huppa JB, Axmann M, Mortelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, Klein LO, Schutz GJ, Davis MM. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 463:963–967. doi: 10.1038/nature08746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koller KJ, Goeddel DV. Molecular biology of the natriuretic peptides and their receptors. Circulation. 1992;86:1081–1088. doi: 10.1161/01.cir.86.4.1081. [DOI] [PubMed] [Google Scholar]
- Kolodkin AL, Matthes DJ, Goodman CS. The semaphorin genes encode a family of transmembrane and secreted growth cone guidance molecules. Cell. 1993;75:1389–1399. doi: 10.1016/0092-8674(93)90625-z. [DOI] [PubMed] [Google Scholar]
- Koppel AM, Feiner L, Kobayashi H, Raper JA. A 70 amino acid region within the semaphorin domain activates specific cellular response of semaphorin family members. Neuron. 1997;19:531–537. doi: 10.1016/s0896-6273(00)80369-4. [DOI] [PubMed] [Google Scholar]
- Krogsgaard M, Prado N, Adams EJ, He XL, Chow DC, Wilson DB, Garcia KC, Davis MM. Evidence that structural rearrangements and/or flexibility during TCR binding can contribute to T cell activation. Mol Cell. 2003;12:1367–1378. doi: 10.1016/s1097-2765(03)00474-x. [DOI] [PubMed] [Google Scholar]
- Kruger RP, Aurandt J, Guan KL. Semaphorins command cells to move. Nat Rev Mol Cell Biol. 2005;6:789–800. doi: 10.1038/nrm1740. [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Pinchasi D, Zhou M, Lax I, Schlessinger J. Kit receptor dimerization is driven by bivalent binding of stem cell factor. J Biol Chem. 1997;272:6311–6317. doi: 10.1074/jbc.272.10.6311. [DOI] [PubMed] [Google Scholar]
- Lev S, Yarden Y, Givol D. Dimerization and activation of the kit receptor by monovalent and bivalent binding of the stem cell factor. J Biol Chem. 1992;267:15970–15977. [PubMed] [Google Scholar]
- Love CA, Harlos K, Mavaddat N, Davis SJ, Stuart DI, Jones EY, Esnouf RM. The ligand-binding face of the semaphorins revealed by the high-resolution crystal structure of SEMA4D. Nat Struct Biol. 2003;10:843–848. doi: 10.1038/nsb977. [DOI] [PubMed] [Google Scholar]
- Luo Y, Raible D, Raper JA. Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell. 1993;75:217–227. doi: 10.1016/0092-8674(93)80064-l. [DOI] [PubMed] [Google Scholar]
- Oinuma I, Ishikawa Y, Katoh H, Negishi M. The Semaphorin 4D receptor Plexin-B1 is a GTPase activating protein for R-Ras. Science. 2004;305:862–865. doi: 10.1126/science.1097545. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pasterkamp RJ, Peschon JJ, Spriggs MK, Kolodkin AL. Semaphorin 7A promotes axon outgrowth through integrins and MAPKs. Nature. 2003;424:398–405. doi: 10.1038/nature01790. [DOI] [PubMed] [Google Scholar]
- Pellet-Many C, Frankel P, Jia H, Zachary I. Neuropilins: structure, function and role in disease. Biochem J. 2008;411:211–226. doi: 10.1042/BJ20071639. [DOI] [PubMed] [Google Scholar]
- Reeves PJ, Callewaert N, Contreras R, Khorana HG. Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc Natl Acad Sci U S A. 2002;99:13419–13424. doi: 10.1073/pnas.212519299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudenko G, Henry L, Henderson K, Ichtchenko K, Brown MS, Goldstein JL, Deisenhofer J. Structure of the LDL receptor extracellular domain at endosomal pH. Science. 2002;298:2353–2358. doi: 10.1126/science.1078124. [DOI] [PubMed] [Google Scholar]
- Scott GA, McClelland LA, Fricke AF, Fender A. Plexin C1, a receptor for semaphorin 7a, inactivates cofilin and is a potential tumor suppressor for melanoma progression. J Invest Dermatol. 2009;129:954–963. doi: 10.1038/jid.2008.329. [DOI] [PubMed] [Google Scholar]
- Semaphorin_Nomenclature_Committee. Unified nomenclature for the semaphorins/collapsins. Cell. 1999;97:551–552. [Google Scholar]
- Stamos J, Lazarus RA, Yao X, Kirchhofer D, Wiesmann C. Crystal structure of the HGF beta-chain in complex with the Sema domain of the Met receptor. EMBO J. 2004;23:2325–2335. doi: 10.1038/sj.emboj.7600243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Kumanogoh A, Kikutani H. Semaphorins and their receptors in immune cell interactions. Nat Immunol. 2008;9:17–23. doi: 10.1038/ni1553. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Okuno T, Yamamoto M, Pasterkamp RJ, Takegahara N, Takamatsu H, Kitao T, Takagi J, Rennert PD, Kolodkin AL, et al. Semaphorin 7A initiates T-cell-mediated inflammatory responses through alpha1beta1 integrin. Nature. 2007;446:680–684. doi: 10.1038/nature05652. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Strittmatter SM. Plexina1 autoinhibition by the plexin sema domain. Neuron. 2001;29:429–439. doi: 10.1016/s0896-6273(01)00216-1. [DOI] [PubMed] [Google Scholar]
- Tamagnone L, Artigiani S, Chen H, He Z, Ming GI, Song H, Chedotal A, Winberg ML, Goodman CS, Poo M, et al. Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell. 1999;99:71–80. doi: 10.1016/s0092-8674(00)80063-x. [DOI] [PubMed] [Google Scholar]
- Tamagnone L, Comoglio PM. Signalling by semaphorin receptors: cell guidance and beyond. Trends Cell Biol. 2000;10:377–383. doi: 10.1016/s0962-8924(00)01816-x. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J. Automated structure solution for MIR and MAD. Acta Crystallogr D. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier-Lavigne M, Goodman CS. The molecular biology of axon guidance. Science. 1996;274:1123–1133. doi: 10.1126/science.274.5290.1123. [DOI] [PubMed] [Google Scholar]
- Tong Y, Chugha P, Hota PK, Alviani RS, Li M, Tempel W, Shen L, Park HW, Buck M. Binding of Rac1, Rnd1, and RhoD to a novel Rho GTPase interaction motif destabilizes dimerization of the plexin-B1 effector domain. J Biol Chem. 2007;282:37215–37224. doi: 10.1074/jbc.M703800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Hota PK, Penachioni JY, Hamaneh MB, Kim S, Alviani RS, Shen L, He H, Tempel W, Tamagnone L, et al. Structure and function of the intracellular region of the plexin-b1 transmembrane receptor. J Biol Chem. 2009;284:35962–35972. doi: 10.1074/jbc.M109.056275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uniewicz KA, Fernig DG. Neuropilins: a versatile partner of extracellular molecules that regulate development and disease. Front Biosci. 2008;13:4339–4360. doi: 10.2741/3008. [DOI] [PubMed] [Google Scholar]
- Waimey KE, Cheng HJ. Axon pruning and synaptic development: how are they per-plexin? Neuroscientist. 2006;12:398–409. doi: 10.1177/1073858406292631. [DOI] [PubMed] [Google Scholar]
- Xiao T, Takagi J, Coller BS, Wang JH, Springer TA. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature. 2004;432:59–67. doi: 10.1038/nature02976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong JP, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL, Arnaout MA. Crystal structure of the extracellular segment of integrin alpha Vbeta3 in complex with an Arg-Gly-Asp ligand. Science. 2002;296:151–155. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- Xu X, Ng S, Wu ZL, Nguyen D, Homburger S, Seidel-Dugan C, Ebens A, Luo Y. Human semaphorin K1 is glycosylphosphatidylinositol-linked and defines a new subfamily of viral-related semaphorins. J Biol Chem. 1998;273:22428–22434. doi: 10.1074/jbc.273.35.22428. [DOI] [PubMed] [Google Scholar]
- Yamada A, Kubo K, Takeshita T, Harashima N, Kawano K, Mine T, Sagawa K, Sugamura K, Itoh K. Molecular cloning of a glycosylphosphatidylinositol-anchored molecule CDw108. J Immunol. 1999;162:4094–4100. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.