

Abstract
Animal cells must maintain a membrane cholesterol-to-phospholipid ratio within tight limits for normal function. Elaborate mechanisms control the cellular input of cholesterol from endogenous synthesis or uptake from plasma lipoproteins. Much less is known about the factors that regulate the output of cholesterol from cells. On pages 1566 and 1570 of this issue, Najafi-Shoushtari et al. (1) and Rayner et al. (2) show that cholesterol output is controlled by the same genes that regulate cholesterol input, but in a reciprocal manner and through an unexpected mechanism.
Animal cells must maintain a membrane cholesterol-to-phospholipid ratio within tight limits for normal function. Elaborate mechanisms control the cellular input of cholesterol from endogenous synthesis or uptake from plasma lipoproteins. Much less is known about the factors that regulate the output of cholesterol from cells. On pages 1566 and 1570 of this issue, Najafi -Shoushtari et al. (1) and Rayner et al. (2) show that cholesterol output is controlled by the same genes that regulate cholesterol input, but in a reciprocal manner and through an unexpected mechanism.
The regulatory genes in question direct the synthesis of sterol regulatory element-binding proteins (SREBPs), which are membrane-bound transcriptional activators (3). Vertebrates have two SREBP genes. SREBP-2 preferentially activates the synthesis and uptake of cholesterol, whereas SREBP-1 preferentially activates the synthesis of fatty acids (4). Najafi -Shoushtari et al. and Rayner et al. reveal that both genes also encode, within their introns, a microRNA (miR-33) that has the reciprocal effect. miR-33 blocks the egress of cholesterol from cells by reducing the mRNA and protein levels for ABCA1, a transporter in the plasma membrane that secretes cholesterol from cells (5). When cells are depleted of cholesterol, both the transcription of SREBPs and the intron-encoded miR-33 rise modestly.
SREBP-1 and SREBP-2 encode miR-33b and miR-33a, respectively, which differ in only 2 of 19 nucleotides that constitute the mature microRNA. Both miR-33 isoforms target for destruction several mRNAs—most prominently the mRNA encoding ABCA1— that contain a highly conserved target sequence in their 3′-untranslated regions. When cultured mammalian cells were transfected with miR-33, the amount of mRNA encoding ABCA1 decreased, whereas the opposite occurred when cells were transfected with RNA molecules that specifically reduced miR-33 levels.
ABCA1 functions most prominently in macrophages and hepatocytes (5). In macrophages, it excretes cholesterol that accumulates as a result of the uptake of oxidized cholesterol-carrying lipoproteins. In liver, ABCA1 is essential for the production of the precursor forms of high-density lipoprotein (HDL). Indeed, Najafi -Shoushtari et al. and Rayner et al. show that delivery of a miR-33 antagonist leads to a small but significant increase in plasma HDL. So far, the most remarkable feature of the miR-33a/b story is the pattern of evolutionary conservation. The precursor for mature miR-33a is found within the same intron of SREBP-2 from many animal species, including large and small mammals, chickens, and frogs. There is even a perfectly conserved mature form of miR-33a in the single SREBP-like gene of the fruit fly Drosophila melanogaster. The latter is most remarkable because insects do not synthesize sterols. Their single SREBP gene controls fatty acid production (6). Moreover, the fruit fly genome does not contain ABCA1, so the target of miR-33a in Drosophila is unknown.
In contrast to the uniform conservation of miR-33a in SREBP-2, there is a gap in the evolutionary conservation of miR-33b in SREBP-1 (according to the U.S. National Center for Biotechnology Information database). The SREBP-1 genes from large mammals encode miR-33b, but there is no trace of miR-33b in the SREBP-1 genes of small mammals (rats and mice) or chickens.
Although the amount of mature miR-33a rises and falls in concert with SREBP-2 mRNA, the amplitude of variation is quite small in the systems studied by Najafi-Shoushtari et al. and Rayner et al. This is likely because variations in cellular cholesterol levels cause relatively minor changes in the transcription of the SREBP genes. Cholesterol regulates SREBP activity most profoundly at the level of protein processing (3). SREBPs are synthesized as membrane proteins in the endoplasmic reticulum and transported to the Golgi complex, where they are proteolyzed to release active fragments that enter the nucleus. There, they enhance transcription of cholesterol-synthesizing genes, such as those encoding 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) synthase and HMG CoA reductase. When cells are depleted of cholesterol, the level of nuclear SREBP-2 increases by orders of magnitude owing to increased proteolytic processing, and mRNAs encoding HMG CoA synthase and reductase increase correspondingly (4). By contrast, the mRNA encoding SREBP-2 increases by less than a factor of 2, explaining why miR-33a also shows relatively small changes. Whether or not such small changes influence plasma HDL in humans is yet to be determined.
One circumstance in which transcription of an SREBP gene is profoundly regulated in vivo and where changes in miR-33 are likely to be important clinically is in the liver (see the figure). Hepatocytes produce two alternatively spliced transcripts of SREBP-1, called SREBP-1a and SREBP-1c. The promoter and first exon of SREBP-1c differ from those of SREBP-1a, the isoform that predominates in most nonhepatic tissues (7). Transcription of SREBP-1c in liver is enhanced by insulin, working in concert with nuclear liver X receptors (8, 9). When insulin levels are high, SREBP-1c is transcribed at extremely high levels, and the resultant nuclear SREBP-1c activates genes necessary to produce fatty acids, which are incorporated into triglycerides (4). As a result, in states of hyperinsulinemia, the liver becomes engorged with fat, and plasma triglyceride levels rise. The usual cause of hyperinsulinemia is peripheral insulin resistance, which leads to hyperglycemia and enhanced insulin secretion. Inasmuch as miR-33b is encoded in human (but not in rodent) SREBP-1c, the hepatic level of miR-33b would be predicted to be markedly elevated in insulin-resistant states in humans, but not in mice and rats.
In humans, insulin resistance is a hallmark of metabolic syndrome, which is provoked by obesity (10). In addition to hyperinsulinemia, hyperglycemia, and fatty liver, cardinal features of metabolic syndrome include an increase in plasma triglyceride levels, owing to elevated very-low-density lipoproteins (VLDL), and a decrease in plasma HDL. Low HDL is believed to contribute to the increase in coronary heart disease in these subjects. Evidence suggests that the hypertriglyceridemia is caused by the insulin-induced increase in SREBP-1c mRNA and protein. Is it possible that the reduction in HDL is caused by a decrease in ABCA1, owing to the increased production of miR-33b from the insulin-stimulated SREBP-1c gene?
Unfortunately, this question cannot be answered by study of hepatic miR-33b in the usual models of insulin resistance in obese rats or mice, because the SREBP-1c genes of these model animals lack miR-33b. This is consistent with the observation that obese insulin-resistant mice manifest all of the cardinal features of metabolic syndrome except a reduction in HDL (11). Here is a situation where geneticists may come to the rescue, by searching for mutations in miR-33b in patients with metabolic syndrome who manifest inappropriately elevated HDL. Alternatively, the hypothesis can be tested by treating metabolic syndrome subjects with agents that antagonize miR-33b and testing for increased HDL.
Potential dual role of SREBP in metabolic syndrome.
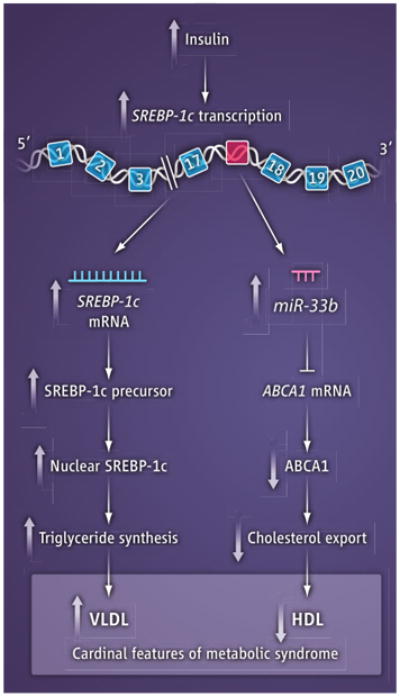
In obese subjects with metabolic syndrome, insulin resistance leads to elevated insulin, which activates transcription of SREBP-1c in the liver. This leads to increased VLDL triglycerides and decreased HDL cholesterol in the blood. miR-33b is encoded in intron 17 (red); exons are in blue.
Acknowledgments
Supported by NIH grant HL20948.
References and Notes
- 1.Najafi -Shoushtari SH, et al. Science. 2010;328:1566. doi: 10.1126/science.1189123. published online 13 May 2010 (10.1126/science. 1189123) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rayner KJ, et al. Science. 2010;328:1570. doi: 10.1126/science.1189862. published online 13 May 2010 (10.1126/science.1189862) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown MS, Goldstein JL. Cell. 1997;89:331. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 4.Horton JD, Goldstein JL, Brown MS. J Clin Invest. 2002;109:1125. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Attie AD. Trends Biochem Sci. 2007;32:172. doi: 10.1016/j.tibs.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 6.Kunte AS, Matthews KA, Rawson RB. Cell Metab. 2006;3:439. doi: 10.1016/j.cmet.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 7.Shimomura I, Shimano H, Horton JD, Goldstein JL, Brown MS. J Clin Invest. 1997;99:838. doi: 10.1172/JCI119247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Repa JJ, et al. Genes Dev. 2000;14:2819. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen G, Liang G, Ou J, Goldstein JL, Brown MS. Proc Natl Acad Sci USA. 2004;101:11245. doi: 10.1073/pnas.0404297101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reaven GM. Cell Metab. 2005;1:9. doi: 10.1016/j.cmet.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 11.Kennedy AJ, Ellacott KLJ, King VL, Hasty AH. Dis Model Mech. 2010;3:156. doi: 10.1242/dmm.003467. [DOI] [PMC free article] [PubMed] [Google Scholar]