

Abstract
Background and purpose:
The transcription factor early growth response-1 (Egr-1) and the acidic fibroblast growth factor (FGF-1) are involved in many regulatory processes, including hippocampus-associated learning and memory. However, the intracellular signalling mechanisms regulating Egr-1 in hippocampal cells are not entirely understood.
Experimental approach:
We used primary mouse hippocampal neurons and the mouse hippocampal neuronal cell line HT22 to investigate how FGF-1 transiently induces Egr-1 protein. This was accomplished by a range of techniques including Western blotting, immunofluorescence, specific protein kinase inhibitors and transfectable constitutively active protein kinase constructs.
Key results:
Protein kinase B (PKB) and mitogen-activated protein kinase (MAPK) were both initially phosphorylated and activated by FGF-1 treatment, but when phosphorylated MAPK reached maximal activation, phosphorylated PKB was at its lowest levels, suggesting an interaction between MAPK kinase (MEK-1/2) and phosphatidyl inositol-3-kinase (PI3K) during Egr-1 induction. Interestingly, pharmacological inhibition of MEK-1/2 resulted in a robust increase in the phosphorylation of PKB, which was repressed in the presence of increasing doses of a PI3K inhibitor. FGF-1-mediated Egr-1 induction was impaired by inhibition of MEK-1/2, but not of PI3K. However, elevated levels of PKB, induced by transfection of constitutively active PKB (myrAkt) into hippocampal neuronal HT22 cells, led to reduced levels of Egr-1 after FGF-1 application.
Conclusions and implications:
Our data indicate a contribution of inactive (dephosphorylated) PKB to FGF-1-mediated induction of Egr-1, and strongly suggest a functionally and pharmacologically interesting cross-talk between MEK-1/2 and PI3K signalling in hippocampal neurons after FGF-1 stimulation that may play a role in hippocampal synaptic plasticity.
Keywords: MEK1/2, PI3K, PKB/Akt, HT22, hippocampus, FGF-1, Egr-1, U0126, LY294002
Introduction
Mitogen-activated protein kinase (MAPK) kinase (MEK-1/2) signalling is essential for hippocampal learning processes as demonstrated by the effects of MEK-1/2-specific inhibitors on long-term potentiation (LTP) (English and Sweatt, 1997). MAPK phosphorylation is also associated with chemically induced LTP in hippocampal slice cultures (Roberson et al., 1996). According to behavioural models for hippocampal learning, phosphorylated MAPK (pMAPK) appears essential for both consolidation and re-consolidation of long-term recognition memory in rats (Kelly et al., 2003). Recently, pMAPK has also been described to vary in the hippocampus in a circadian manner linking MAPK signalling to memory processes that depend on the time of day (Eckel-Mahan et al., 2008).
The phosphatidyl inositol 3-kinase (PI3K) pathway is also involved in hippocampal memory (Sanna et al. 2002; Opazo et al. 2003; Horwood et al. 2006). Mice lacking the p85α-regulatory subunit of PI3K show impaired learning compared to wild-type animals (Tohda et al., 2007). Phosphorylation of protein kinase B (PKB; also known as Akt) in the hippocampal regions CA1, CA3 and dentate gyrus was observed in mice during memory retrieval, and PI3K signalling was reported to be essential for the consolidation of contextual memory contents (Chen et al., 2005).
Both the MEK-1/2 and the PI3K pathways participate in the signalling processes initiated by acidic fibroblast growth factor (aFGF or FGF-1), which has been shown to enhance memory consolidation in the hippocampus (Oomura et al., 1992; Sasaki et al., 1994; 1995; Oomura, 2008). The MEK-1/2 and PI3K cascades are also involved in the regulation of the learning-associated transcription factor ‘early growth response’-1 [Egr-1, also known as zif268, Krox-24 or NGFI-A (Jones et al., 2001; Ko et al., 2005)]. FGF-1 application improves the performance of mice in the passive avoidance test, when administered into the CSF (Oomura et al., 1992), and is transiently released from ependymal cells into the CSF 2 h after food intake (Hanai et al., 1989; Oomura et al., 1992). In addition, FGF-1 enhances LTP in rat hippocampal slice cultures (Sasaki et al., 1994), and supports neurite outgrowth in rat hippocampal neurons (Flajolet et al., 2008; Hausott et al., 2008).
FGF-1 exerts many of its effects via the MEK-1/2- or PI3K signalling cascades (Reuss and von Bohlen und Halbach, 2003; Eswarakumar et al., 2005). It is, however, difficult to investigate such specific signal transduction processes in primary cultures of the hippocampus because these cultures consist of multiple different types of neurons and can contain contaminating cell types, such as astrocytes, fibroblasts or endothelial cells. Therefore (and because we are interested in the regulatory mechanisms on the single cell and mechanisms at an intracellular level), we mostly used the mouse hippocampal cell line HT22, which reportedly exhibits a cholinergic phenotype typical for a large subpopulation of hippocampal neurons (Liu et al., 2009) which is most relevant for the neurodegenerative processes described for Alzheimer's disease.
HT22 cells have served as a valuable model to study glutamate- and staurosporin-induced oxidative toxicity, and the protection from these neurodegenerative processes (Stanciu et al., 2000; Stanciu and DeFranco, 2002; Ho et al., 2008; Spahn et al., 2008; Steiger-Barraissoul and Rami, 2009). Intriguingly, HT22 cells under glutamate treatment exert a chronic activation of the MAPK pathway with prolonged nuclear location of pMAPK and deregulated MAPK phosphatases (MKPs) apparently being responsible for glutamate-induced HT22 cell death (Stanciu et al., 2000; Stanciu and DeFranco, 2002; Ho et al., 2008).
In the CNS, MEK-1/2 can be activated via FGF receptors and a variety of different protein kinases (Reuss and von Bohlen und Halbach, 2003; Eswarakumar et al., 2005). Finally, FGF-1 application leads to phosphorylation of MAPK, which phosphorylates various cytosolic targets, but also partly translocates into the nuclei of hippocampal neurons via passive diffusion (Wiegert et al., 2007). PI3K is also activated via FGF receptors and protein kinase cascades like phospho-inositide-dependent-kinase-1, which in turn phosphorylates PKB at serine-473 (pPKB–S473) and threonine-308 (pPKB–T308) (Eswarakumar et al., 2005). Activated PKB phosphorylates downstream targets such as glycogen synthase kinase-3β, which is involved in neuroprotective processes, bipolar disorder and circadian rhythms (Hashimoto et al., 2002; Kaladchibachi et al., 2007; Vougogiannopoulou et al., 2008). The PI3K/PKB pathway also regulates dendrite arborization in hippocampal neurons, a process thought to contribute to memory processing (Jaworski et al., 2005; Flajolet et al., 2008).
In the present study, we investigated the effects of FGF-1 on Egr-1 protein levels using a model of cholinergic hippocampal neurons, the cell line HT22 (Davis and Maher, 1994; Liu et al., 2009), and primary neuronal hippocampal cultures of C3H-mice. The involvement of MEK-1/2 and PI3K signalling in FGF-1-mediated Egr-1 protein induction was dissected in a dose- and time-dependent manner. Furthermore, kinase inhibitors, as well as a transfectable constitutively active form of PKB, were used to determine this interaction and clarify the relative contribution of these two pathways for regulation of Egr-1 protein levels.
Methods
Animals
Animal experiments were conducted in accordance with the Policy on the Use of Animals in Neuroscience Research, and the Policy on Ethics, as approved by the Society for Neuroscience and by the European Communities Council Directive (89/609/EEC).
Cell culture and reagents
HT22 cells were a kind gift from Dr David Schubert, Salk Institute, San Diego, CA, USA. The cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Karlsruhe, Germany), containing 10% fetal bovine serum (vol/vol) (PAA, Pasching, Austria), 1% (vol/vol) penicillin–streptomycin mix and 1% (vol/vol) Glutamax (both Invitrogen). For experiments, 50.000 cells were seeded in each well of a 24-well plate. The medium was replaced with serum-free DMEM 18–24 h prior to the start of the experiments. This 24 h of serum depletion before cell stimulation reportedly shifts the cells into a cholinergic phenotype (Liu et al., 2009).
Primary hippocampal neuronal cultures were prepared as described before (Brewer et al., 1993; Annaert et al., 1999). In brief, C3H mice were killed at postnatal day 3, and hippocampi were removed and transferred to DMEM at 4°C. Dissociation of cells was performed by trituration in papain-rich medium at 37°C. The cells were cultivated on glass coverslips coated with poly-l-lysine (Sigma-Aldrich, Munich, Germany) in 24-well plates in Neurobasal-A (Invitrogen) medium including 1% (vol/vol) penicillin–streptomycin mix, 1% (vol/vol) GlutaMax and B-27 supplement (Invitrogen) until the day of stimulation (a total of 14 days).
Transfection experiments
The constitutively active myristoylated PKB pcDNA3.1 construct (myrAkt) was a kind gift from Professor Brian Hemmings, Friedrich Miescher Institute, Basel, Switzerland. Plasmid purification was performed using the Qiagen Plasmid Midi Kit according to the manufacturer's instructions (Qiagen, Hilden, Germany).
FuGENE HD transfection reagent (Roche Applied Science, Mannheim, Germany) was used to introduce the plasmid DNA into HT22 cells according to the manufacturer's protocol. In brief, 50.000 HT22 cells were seeded in serum-enriched DMEM in a 24-well plate. The medium was replaced with serum-free DMEM after 24 h. Four hours after serum deprivation, the transfection complex consisting of FuGENE HD transfection reagent, plasmid DNA and Opti-MEM I (Invitrogen) was added to the medium. Another 18 h later, the experiments were performed as described above.
Western blot analysis
At the end of each experiment, the medium was removed and cells were immediately lysed in lithium dodecylsulphate sample buffer (Invitrogen). Cell extracts were sonified five times for 3 s using a 24 kHz ultrasound sonifier (Dr Hielscher, Teltow, Germany), heated for 10 min at 70°C, chilled on ice and centrifuged for 5 min at 8000× g in an Eppendorf cap centrifuge (Eppendorf, Hamburg, Germany). Samples were either used immediately for gel electrophoretic separation on Bis/Tris gradient gels (Invitrogen) or stored at –20°C. Gels were blotted onto polyvinylidenfluoride membrane (Millipore, Billerica, MA, USA), washed with Tris-buffered saline (pH 7.6) containing 0.1% (vol/vol) Tween-20 (TBS-T; Sigma-Aldrich), blocked for 1 h at room temperature using Rotiblock (Roth, Karlsruhe, Germany) and incubated with primary antibodies diluted in Rotiblock overnight at 4°C [anti-Egr-1 (1:30.000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-β-actin (1:40.000; Sigma-Aldrich), anti-phospho-p42/44 MAPK (1:10.000), anti-total-p42/44 MAPK (1:5.000), anti-pPKB–S473 (1:5.000) and anti-pPKB–T308 (1:3.000; all from Cell Signaling Technology, Bad Nauheim, Germany). After incubation with the first antibody, membranes were washed three times for 3 min with TBS-T, and incubated with the appropriate secondary HRP-coupled antibodies against rabbit (1:50.000; Santa Cruz Biotechnology) or mouse (1:25.000; DAKO, Hamburg, Germany) in Rotiblock for 1 h at room temperature. Membranes were washed with TBS-T five times for 3 min. Signal detection was performed using chemiluminescent substrate Immobilon (Millipore) and a CCD camera-equipped luminescence analysis system (Quantity One, ChemiDoc XRS, Bio-Rad, Hercules, CA, USA) as described (Wicht et al., 1999; Maronde et al., 2007).
Immunocytochemistry and confocal laser scanning microscopy
HT22 cells and primary hippocampal neurons were seeded on glass coverslips and treated as described above. For fixation, the medium was removed at the end of each experiment and replaced with phosphate buffer (0.1 M) containing 4% (wt/vol) paraformaldehyde for 15 min at room temperature. Samples were washed with phosphate-buffered saline (PBS) for 15 min, and blocked for 45 min at room temperature using 5% (vol/vol) normal goat serum (Sigma-Aldrich) in PBS. Cells were incubated overnight with a mixture of the primary antibodies in PBS, containing 1% (wt/vol) BSA (Sigma-Aldrich) at 4°C. The primary antibodies used were anti-Egr-1 (1:10.000; Santa Cruz Biotechnology), anti-p42/44 MAPK (1:5.000) and anti-pPKB–S473 (1:800; both Cell Signaling Technology). For double-labelling experiments, primary antibodies were combined with antiserum against microtubule-associated protein-2 (MAP2; 1:300; Sigma-Aldrich) as a neuronal marker or nuclear pore complex protein (NPCP; 1:1.000; Abcam, Cambridge, UK). Samples were washed three times for 10 min in PBS and incubated in fluorochrome-coupled secondary antibodies against rabbit (Alexa-568; 1:500, Invitrogen) and mouse (Chromeo-488; 1:1.000; Active Motif, Rixensart, Belgium) in PBS containing 1% (wt/vol) BSA for 1 h. Coverslips were washed three times for 10 min in PBS, mounted on slides using fluorescent mounting medium (DakoCytomation, DAKO) and stored at 4°C.
Intracellular distribution of proteins was determined by confocal laser scanning microscopy (LSM510, Zeiss, Göttingen, Germany). Proteins of interest were visualized together with NPCP or MAP2, using monochromatic light at 488/543 nm with a dichroic beam splitter (FT 488/543) and an emission bandpass filter of 505–530/585–605 nm. Images were acquired at 1024 × 1024 pixel resolution in multitracking mode using electronic zoom of LSM system (Dehghani et al., 2000).
Statistical analysis
Images of protein bands were digitized using a Bio-Rad Universal Hood equipped with a CCD camera, Quantity One and ChemiDoc XRS software. Signal intensities of the digitized images were analysed using a combination of densitometry and volumetry as implemented in the QuantiScan software (BioSoft, Cambridge, UK), and described in detail before (Wicht et al., 1999). Each area/density value for a specific protein band was normalized against the corresponding β-actin signal of each extract. Groups were analysed using one-way anova, followed by the Bonferroni post hoc test. The criterion of significance was P < 0.05, with analysis performed using GraphPad Prism 3.0 (GraphPad, San Diego, CA, USA).
Materials
FGF-1 was purchased from Sigma-Aldrich and dissolved in water containing 50% (vol/vol) glycerol. U0126 and LY294002 (both from Cell Signaling Technology) were dissolved in dimethylsulphoxide. Reagents or appropriate vehicle were applied to the media for the indicated periods and concentrations. The nomenclature of molecules, drugs, protein kinases and other proteins follows Alexander et al. (2009).
Results
FGF-1 application induces Egr-1 in C3H mouse primary hippocampal cultures
The transcription factor Egr-1 is elevated in C3H mouse primary hippocampal neurons after 90 min of treatment with FGF-1 (10 ng·mL−1), whereas it was not different from basal levels after 10 and 240 min (Figure 1A). Accordingly, an increased number of Egr-1-positive nuclei were seen in immunofluorescence images (Figure 1B). Note that Egr-1 was detectable under control conditions. The levels of the transcription factor Egr-1 and pMAPK were both elevated approximately threefold after 90 min of FGF-1-treatment (10 ng·mL−1) (both P < 0.001 vs. control; Figure 1). These FGF-1-mediated effects were both fully inhibited by co-application of the MEK-1/2 inhibitor, U0126 (both P < 0.001). It is also evident that the PI3K inhibitor, LY294002, was as effective as FGF-1 or FGF-1 plus LY294002 application in the induction of Egr-1 (P < 0.01). PKB was phosphorylated at serine-473 under all conditions, except those where LY294002 was present.
Figure 1.
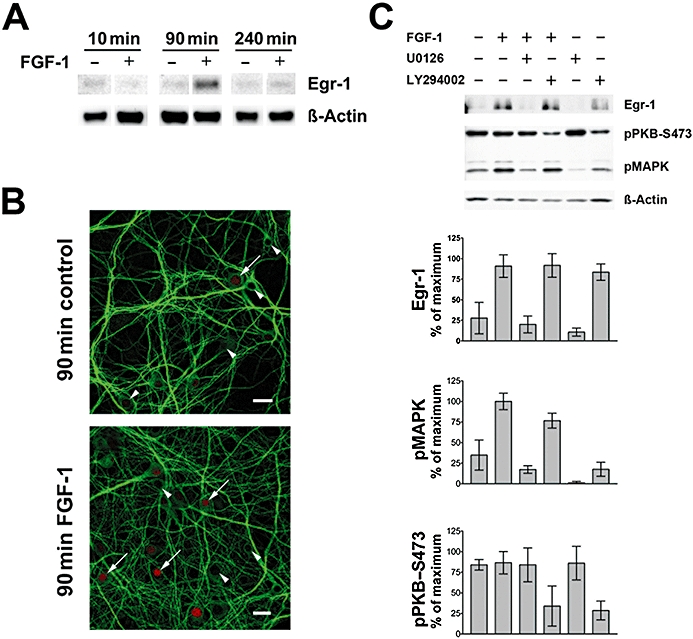
FGF-1 induces Egr-1 protein in primary hippocampal cell cultures. (A) The transcription factor Egr-1 was elevated in C3H mouse primary hippocampal neurons after 90 min of FGF-1 (10 ng·mL−1) treatment as judged by Egr-1 immunoreaction in the Western blot. Corresponding signals for β-actin are shown as loading control. (B) The number of Egr-1-positive nuclei (red) is elevated after application of FGF-1 for 90 min. Arrowheads indicate Egr-1-negative and arrows Egr-1-positive nuclei. An antibody against microtubule-associated protein 2 (MAP2, green) is used as a counter-stain (bars = 20 µm). (C) Upper panel: level of the transcription factor Egr-1, PKB and MAPK phosphorylation after 90 min of FGF-1-treatment (10 ng·mL–1) with or without the protein kinase inhibitors U0126 and LY294002 (both 10 µM) as shown in a representative Western blot. Lower panel: quantification of Western blot data for pMAPK, pPKB and Egr-1 normalized for β-actin (mean ± SD, n = 3).
FGF-1 regulates Egr-1 in HT22 cells in a dose-dependent manner
In mouse hippocampal HT22 cells, induction of Egr-1 protein by FGF-1 application was concentration dependent, reaching a plateau around 10 ng·mL−1 after 90 min (Figure 2A,B). We used this concentration (10 ng·mL−1) of FGF-1 throughout all the following experiments, unless otherwise indicated. Egr-1 protein induction by FGF-1 application was abolished by U0126, but not by LY294002 (Figure 2C,E). FGF-1-induced Egr-1 immunoreactivity in the nucleus was strongly decreased by U0126, but not by LY294002 (Figure 2D).
Figure 2.
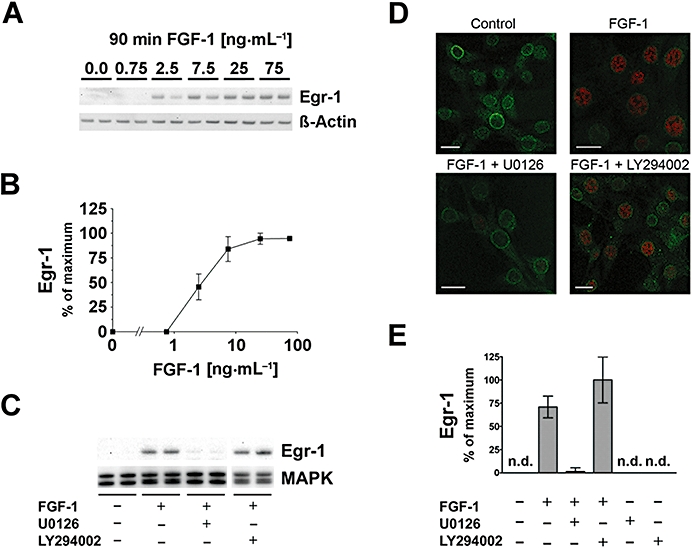
FGF-1 induces Egr-1 in hippocampal HT22 cells. (A) The transcription factor Egr-1 was concentration-dependently elevated after 90 min of FGF-1 treatment, as shown in a representative Western blot. Corresponding signals for β-actin are shown as loading control. (B) Quantification of Western blot dose–response actin-normalized data ± SD, n = 4–10. (C) FGF-1-mediated Egr-1 was strongly inhibited by the MEK-1/2 inhibitor U0126, but not by the PI3K inhibitor LY294002 (both 10 µM). Total MAPK is shown as a loading control. (D) Immunofluorescence images reveal that after 90 min of FGF-1-induced Egr-1, immunoreactivity (red) in the nucleus was strongly inhibited by the MEK-1/2 inhibitor U0126, but not by the PI3K inhibitor LY294002. An antibody against NPCP (green) is used as a counter-stain (bars = 20 µm). (E) Quantitative analysis of Western blot data showing that FGF-1 induced Egr-1 protein levels which were not significantly affected by PI3K inhibition, but were abolished by MEK-1/2 inhibition. Neither U0126 nor LY294002 influenced Egr-1 when applied alone. All bar graphs show the mean ± SD of three independent experiments (n = 6–9).
FGF-1 regulates pMAPK and PKB in HT22 cells
Phospho-MAPK is induced by FGF-1 application (Figure 3A–C). These FGF-1-elevated pMAPK signals were fully inhibited by application of U0126, but not changed by LY294002 (Figure 3A–C). Accordingly, immunofluorescence revealed that FGF-1-induced pMAPK immunoreactivity, which was mostly cytosolic in location, was diminished by U0126, but not by LY294002 (Figure 3B). Quantitative analysis of the immunoblotting data showed that FGF-1 (10 ng·mL−1) elevated pMAPK levels 5.7-fold, and that these levels were not significantly affected by PI3K inhibition, but were abolished by MEK-1/2 inhibition (Figure 3C).
Figure 3.
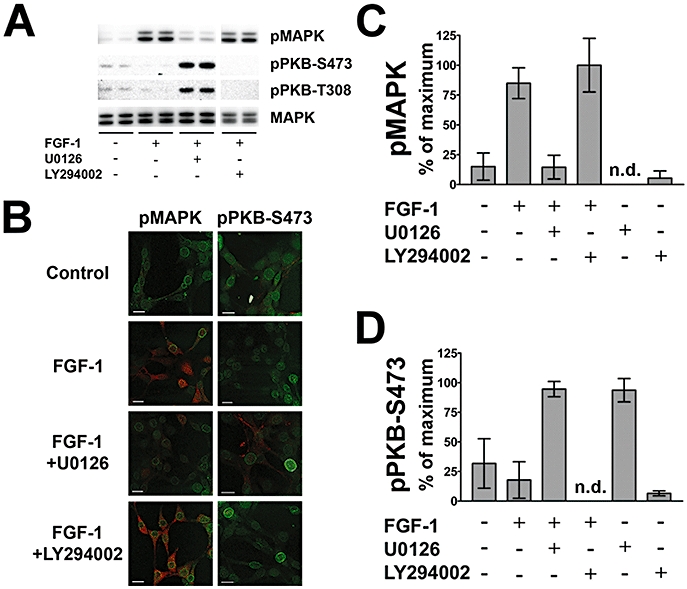
FGF-1 effects on p42/44 MAPK and pPKB phosphorylation. (A) Representative Western blot data reveal that pMAPK induction by FGF-1 application is strongly inhibited by the MEK-1/2 inhibitor U0126, but not changed by the PI3K inhibitor LY294002. Basal levels of pPKB–S473 and –T308 appear reduced by FGF-1 application. The MEK-1/2 inhibitor U0126 elevates both pPKB–S473 and –T308. Total MAPK is shown as a loading control. (B) Representative immunofluorescence images reveal that pMAPK induction by FGF-1 application is inhibited by the MEK-1/2 inhibitor U0126, but not changed by the PI3K inhibitor LY294002. Levels of pPKB–S473 are higher under U0126 treatment. An antibody against NPCP (green) is used as a counter-stain for the immunofluorescence images (bars = 20 µm). (C) Quantitative analysis of Western blot data reveals that pMAPK induction by FGF-1 application is inhibited to basal levels by U0126, but not changed by LY294002. The inhibitors applied alone have no significant effect. (D) Quantitative analysis of Western blot data shows significant induction of pPKB–S473 by U0126 given alone or in combination with FGF-1. All bar graphs show the mean ± SD of three independent experiments (n = 6–9).
The relatively high basal levels of both pPKB–S473 and –T308 were repressed by FGF-1 application (Figure 3A,B). Surprisingly, the application of U0126 alone (Figure 3D) or combined with FGF-1 (Figure 3A,B,D) induced the levels of both pPKB–S473 and –T308 about threefold. The PI3K inhibitor LY294002 alone did not reduce basal pPKB levels, but abolished pPKB immunoreactivity in combination with FGF-1 (Figure 3B,D). Immunofluorescence images confirm that U0126 induces pPKB–S473 (Figure 3B).
Concentration-dependent responses to protein kinase inhibitors
The effects of U0126 and LY294002 in HT22 cells were used to characterize the interaction of the two investigated signalling pathways in the absence of FGF-1. Different doses of the MEK-1/2 inhibitor U0126 were applied for 90 min alone or in combination with 10 µM of the PI3K inhibitor LY294002 (Figure 4A). Application of 1–30 µM of U0126 revealed that pPKB–S473 is induced in a concentration-dependent manner, reaching a maximum at 10 µM. At 30 µM U0126, no further elevation of pPKB–S473 levels occurred (P > 0.05, compared with 10 µM U0126). In the presence of 10 µM of LY294002, basal pPKB–S473 levels were decreased compared to controls (Figure 4B, P < 0.01). By adding increasing concentrations of U0126, pPKB–S473 levels were again elevated with a maximal effect at 10 and 30 µM of U0126. Thus, the addition of a fixed concentration of LY294002 to increasing concentrations of U0126 revealed a similar pattern of pPKB induction, which was generally lower when compared with application of U0126 alone (Figure 4A,B). These data indicate an intrinsic inhibitory influence of MEK-1/2 signalling on the PI3K/PKB pathway.
Figure 4.
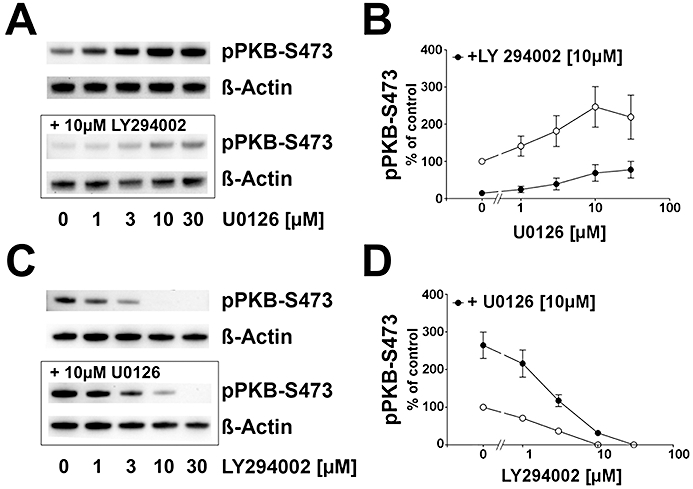
Effect of MEK-1/2 and PI3K inhibition on pPKB–S473 in HT22 cells. (A) Typical Western blot demonstrating that after 90 min, control samples displayed a basal level of pPKB–S473 which was elevated with increasing concentrations of U0126 (upper panel). Pre-incubation of 10 µM LY294002 (lower panel) led to a down-regulation of pPKB–S473. β-Actin immunoreaction served as loading control. (B) Analysis of β-actin-normalized pPKB signals showed that the maximum pPKB–S473 level was reached at 10 µM of the MEK-1/2 inhibitor. Pre-incubation of 10 µM LY294002 led to a down-regulation of pPKB–S473 in the absence of U0126 (P < 0.01). U0126 application led to an induction of pPKB–S473 with a maximum at 10 µM U0126 (P < 0.01) with no further induction at 30 µM U0126. Compared with the levels in the absence of LY294002, U0126-mediated induction of pPKB was generally suppressed in the presence of the PI3K inhibitor. Shown are the mean ± SD of two separate experiments with three repeats. (C) Typical Western blot showing that basal pPKB–S473 was reduced by increasing concentrations of LY294002 (upper panel). Pre-incubating 10 µM of the MEK-1/2 inhibitor U0126 induced pPKB–S473 levels which were reduced when rising concentrations of LY294002 were added. A full suppression of pPKB–S473 below the detection limit was seen at 30 µM PI3K inhibitor (10 µM U0126 + 30 µM LY294002). β-Actin immunoreaction served as loading control. (D) Statistical analysis of pPKB–S473 signals normalized to β-actin showing that basal pPKB–S473 was linearly reduced with increasing concentrations of LY294002. A reduction below detection limit was observed at 10 and 30 µM LY294002. U0126-mediated increase in pPKB–S473 was concentration-dependently reduced by the PI3K inhibitor. A full suppression was seen at 30 µM of LY294002. Shown are the mean ± SD of two separate experiments with three repeats.
However, pPKB–S473 was still inducible by U0126 in the presence of 10 µM LY294002 (Figure 4A,B). To address the question of whether the interaction occurred upstream or downstream of PI3K, we performed experiments with increasing doses of LY294002 with 10 µM of U0126 as inducer of pPKB (Figure 4C,D). Basal pPKB–S473 levels in HT22 cells were reduced by increasing doses of the PI3K inhibitor LY294002 alone (Figure 4C); 10 µM of LY294002 was sufficient to abolish basal pPKB–S473 immunoreactivity. With 10 µM U0126 and no LY294002, pPKB–S473 was elevated compared with control (no LY294002 and no U0126, P < 0.001). Increasing doses of LY294002 were able to reduce these elevated levels of pPKB–S473. A complete suppression of U0126-induced pPKB–S473 occurred at 30 µM LY294002, indicating that the inhibitory influence of MEK-1/2 on the PI3K/PKB cascade occurs at a level or upstream of PI3K (Figure 4C,D).
Influence of constitutively active PKB (myrAkt) on the FGF-1-induced Egr-1 elevation
HT22 cells were transiently transfected with increasing amounts of a plasmid encoding the constitutively active form of PKB (myrAkt) 18 h prior to 90 min of FGF-1 treatment (Figure 5). In untreated control samples, low levels of pMAPK were detected and did not change when 3–30 ng of myrAkt was introduced to the cells (P > 0.05 each, compared with mock-transfected cells). FGF-1 application led to an induction of pMAPK above control values as seen before (Figure 3). However, elevated pMAPK values were not affected by increasing amounts of myrAkt (Figure 5B lower part; P > 0.05 each, compared to mock-transfected cells).
Figure 5 In.
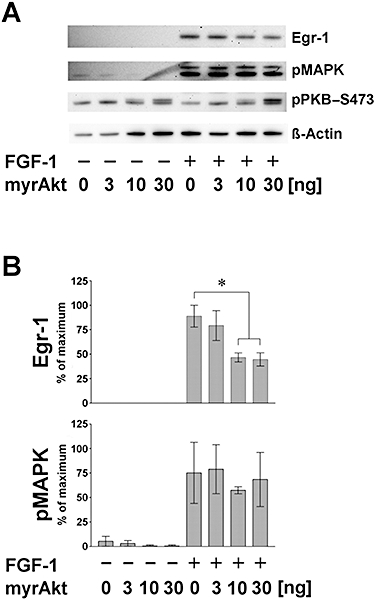
Influence of transfected constitutively active PKB (myrAkt) on FGF-1 effects in HT22 cells. Cells were transiently transfected with the indicated amounts of myrAkt plasmid DNA and stimulated 18 h later for 90 min. (A) Representative Western blot showing that FGF-1-mediated Egr-1 induction is reduced under increasing amounts of myrAkt. Phospho-MAPK signals were not affected by the presence of myrAkt, neither in the presence (+) nor in the absence (−) of FGF-1. After 90 min, similar levels of pPKB–S473 were obtained comparing FGF-1-stimulated samples and controls. Using anti-pPKB–S473, at 10 and 30 ng of myrAkt-DNA, a second, larger signal appears in controls and FGF-1-treated samples. (B) Statistical analysis of β-actin-normalized Western blot data obtained from three independent experiments displayed as per cent of maximum (mean ± SD; n = 4). In untreated samples, Egr-1 protein was not detectable. Egr-1 was induced by FGF-1 addition. At 10 and 30 ng of myrAkt-DNA, a significant reduction of Egr-1 was observed (*P < 0.05). pMAPK levels were low in controls, but were strongly elevated by application of FGF-1. Introducing myrAkt prior to FGF-1 treatment did not change pMAPK levels (P > 0.05).
Elevated Egr-1 levels were only detected when FGF-1 was administered. Moreover, within the group of FGF-1-treated samples, transfection of 10 and 30 ng DNA led to a suppression of FGF-1-mediated Egr-1 (Figure 5A,B, upper parts; P < 0.05, compared with mock-transfected cells respectively). These data indicate that the FGF-1-mediated transient inactivation of PKB contributes to Egr-1 protein induction. If the transient inactivation of PKB, induced by FGF-1 application, is compensated by constitutively active myrAkt transfection, Egr-1 protein does not reach maximal levels. Thus, this inhibitory effect of pPKB on Egr-1 activity is not accompanied with a suppression of pMAPK levels (Figure 5B, lower part; see also Figure 7).
Figure 7.
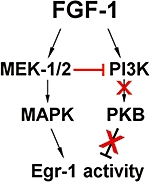
Scheme of signalling events following FGF application to hippocampal cells. In HT22 cells, application of FGF-1 leads to an initial activation of pMAPK and pPKB. Activation of the MEK-1/2 cascade down-regulates PI3K, which in turn disinhibits Egr-1-activity via dephosphorylation of PKB. Thus, activation of MAPK plus inhibition of PKB contribute to the transient expression of Egr-1 protein in hippocampal neurons.
Time dependency of the effect of FGF-1 application on Egr-1, pMAPK and pPKB–S473
FGF-1-induced Egr-1, pMAPK and pPKB–S473 levels varied in a time-dependent manner (Figure 6). Egr-1 levels were below limit of detection up to 30 min of FGF-1 application, were strongly elevated after 60 and 90 min, barely detectable after 240 min and below limit of detection after 360 min (Figure 6A,B). Compared with controls, pMAPK levels were induced by FGF-1 after 3, 15, 30 and 90 min with a maximum between 15 and 60 min. Phospho-PKB–S473 was elevated after 3 and 15 min of FGF-1, but then declined significantly below untreated time-matched controls after 60 min. It is significant that maximal levels of Egr-1 first appeared when pMAPK was maximally elevated and pPKB–S473 was maximally repressed. These data were confirmed in experiments where after 60 min of incubation, PKB dephosphorylation fell below control levels in the presence of increasing concentrations of FGF-1 (data not shown).
Figure 6.
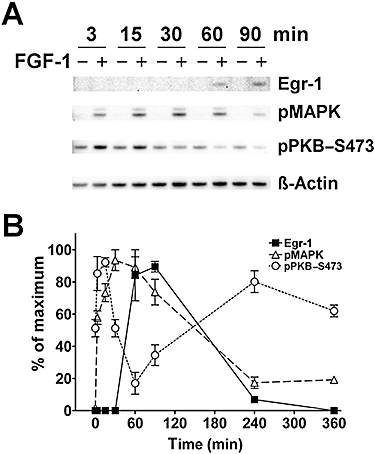
Time dependency of the effect of FGF-1 application on Egr-1, pMAPK and pPKB–S473. (A) Representative Western blot of the FGF-1 effect on Egr-1, pMAPK and pPKB–S473 levels up to 90 min. β-Actin is shown as a loading control. (B) Statistical analysis of Western blot data showing the time relation of signalling events from 0 to 360 min of FGF-1 application. Note that maximal levels of Egr-1 first appear where pMAPK is maximally elevated and pPKB–S473 is maximally repressed. For greater clarity, control values are not shown (mean ± SEM, n = 6–9).
Discussion
In this study, we have demonstrated an intrinsic cross-talk between the MEK-1/2 and the PI3K pathways that contributes to the level of expression of Egr-1 protein in the presence of FGF-1 in the mouse hippocampal neuronal cell line HT22 and in primary hippocampal neuronal cultures from C3H mice.
Pharmacologically, we show that inhibition of MEK-1/2 by U0126 may not only inhibit the intended target, but might also, as seen here for HT22 cells, strongly elevate PKB phosphorylation. These observations may have significant consequences for the use of U0126 and the interpretation of data utilizing this compound.
The effect of UO126 occurs as soon as 60–90 min of incubation, which may well be sufficient time for some of the immediate early genes to appear and interact with the phosphorylation state of the proteins we observed. However, the total protein levels of both MAPK and PKB were not changed in our experiments (data not shown), which should be the case if a direct transcriptional regulation was involved. It is therefore probable that a post-translational mechanism, rather than a transcriptional mechanism, was responsible for the observed influence of MEK-1/2 inhibition on pPKB levels.
Our data show that application of FGF-1 initially elevated both pMAPK and pPKB-levels. Similar events have been described before in other non-neuronal cellular systems (Hashimoto et al., 2002; Eswarakumar et al., 2005). Interestingly, all observed effects in the continuous presence of FGF-1 were of transient nature: activation of pMAPK and Egr-1 was accompanied by a time-matched suppression of PKB phosphorylation. These observations suggested a potentially negative influence of the MEK-1/2 pathway on PI3K/PKB signalling. This negative influence was investigated in more detail to address the question as to whether only the increase in pMAPK levels, or the decline in pPKB, or both contribute to the temporally gated elevation and subsequent suppression of Egr-1. These findings may be of wider interest because not only FGF-1, but also the PKC activator, phorbol 12-myristate 13-acetate, shows a similar pattern of suppression of pPKB accompanied by sustained MAPK phosphorylation (data not shown).
The present study revealed that under full suppression of PI3K activity, pPKB was no longer inducible by MEK-1/2 inhibition, leading to the suggestion that the negative impact of MEK-1/2 signalling on the PI3K/PKB pathway occurs on the same level, or even upstream of PI3K, but probably not on downstream mediators. An interesting implication of this finding is the observation in hippocampal neuronal cells [as shown here and similar to findings in NIH3T3 cells (Hayashi et al., 2008)] that MEK-1/2 inhibitors may not exclusively exert their effect by inhibition of the MEK-1/2 pathway, but also via an induction of PKB phosphorylation. It also implies that basal levels of MEK-1/2 activity already suppress PI3K, or upstream processes, which are dis-inhibited upon MEK-1/2 inhibition (see also the model in Figure 7).
How widespread the interaction between MEK-1/2 and PI3K described here for HT22 cells is represented is not clear. Similar attempts to influence these pathways using inhibitors for MEK-1/2 did not result in an increase of pPKB in rat cortical neurons (Hashimoto et al., 2002), and only to a slight elevation of pPKB levels in Neuro2A cells (Graham et al., 2006). Moreover, in the human hepatoma cell line HepG2, MEK-1/2 inhibition did not lead to an increase of pPKB–S473 levels (Malmlöf et al., 2007). However, in Neuro2A cells, Egr-1 induction by the cannabinoid CB1 receptor agonist, Hu-210, is under negative control of PI3K, leading to a further increase of Egr-1 during PI3K inhibition (Graham et al., 2006).
In primary neuronal hippocampal cultures, Egr-1 protein levels were elevated by application of FGF-1, as in HT22 cells (Figures 1C and 2). However, PI3K inhibition by LY294002 elevated Egr-1 protein levels only in primary cultures, but not in HT22 cells. Whereas pMAPK regulation in primary cultures is very similar to HT22 cells, pPKB–S473 is constitutively high in primary cells and not further elevated by U0126 application. In this respect, our findings are similar to those of others (Hashimoto et al., 2002; Graham et al., 2006) and may be a result of the mixed neuronal phenotype of the primary cultures used, where cellular coupling and other factors beyond control in these primary culture systems differ from the conditions in a cell line like the HT22 used here which mostly represents cholinergic neurons of hippocampal origin (Davis and Maher, 1994; Liu et al., 2009).
In HT22 cells, FGF-1-induced Egr-1 levels were not significantly affected by PI3K inhibition, but abolished by MEK-1/2 inhibition. The fact that the combination of FGF-1 and LY294002 did not result in an increase of Egr-1 may be due to silencing of the PI3K/PKB pathway, mediated by FGF-1 itself (or the activated or basally active MEK-1/2 cascade) so that an additional pharmacological inhibition would be without further effect on downstream targets.
The findings presented here may also provide a mechanistic basis for an interesting effect of FGF-1 on rat hippocampal slices. Using high frequency-induced LTP in rat hippocampal slice cultures, the maximal LTP-enhancing effect of co-applied FGF-1 occurred 20 min prior to LTP induction (Sasaki et al., 1994). Hence, it appears that for the effectiveness of FGF-1, the relative timing is most important, as the LTP-promoting effect was not observed when FGF-1 was added simultaneously, or after electric LTP induction, suggesting that FGF-1-dependent early signalling processes may be required for the gating of enhanced LTP. Furthermore, it provides a molecular basis for the effect of FGF-1 on LTP formation, because it appears approximately in the time window where parallel activation of both MEK-1/2 and PI3K changes into a further induction of the former and a suppression of the latter.
Taken together, our observations have shed new light on the role and interaction of the learning-associated factors FGF-1 and Egr-1, and their signal transduction cascades, and how a cross-talk such as the one described here may influence synaptic plasticity and contribute to the regulation of transcriptional targets of Egr-1 and ultimately of LTP and memory processes.
Acknowledgments
We thank Jörg H. Stehle for critically reviewing the manuscript, helpful discussions and constant support; Antje Jilg for expert support with Zotero; and Jhoy O. Pagulayan for editing the manuscript.
Glossary
Abbreviations:
- CA-1
hippocampal sub-region CA-1
- CA-3
hippocampal sub-region CA-3
- CCD
charge-coupled device
- DMEM
Dulbecco's modified Eagle's medium
- Egr-1
early growth response-1
- FGF-1
acidic fibroblast growth factor
- HRP
horseradish peroxidase
- LTP
long-term potentiation
- LY294002
2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one
- MAP2
microtubule-associated protein-2
- MAPK
mitogen-activated protein kinase
- MEK-1/2
mitogen-activated kinase kinase
- MKP
mitogen-activated kinase phosphatase
- NPCP
nuclear pore complex protein
- PBS
phosphate-buffered saline
- PFA
paraformaldehyde
- PI3K
phosphatidyl inositol-3-kinase
- PKB
protein kinase B
- pMAPK
double-phosphorylated mitogen-activated protein kinase
- pPKB
phosphorylated PKB
- TBS
Tris-buffered saline
- U0126
1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene
Conflict of interest
The authors declare no conflict of interest.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annaert WG, Levesque L, Craessaerts K, Dierinck I, Snellings G, Westaway D, et al. Presenilin 1 controls gamma-secretase processing of amyloid precursor protein in pre-Golgi compartments of hippocampal neurons. J Cell Biol. 1999;147:277–294. doi: 10.1083/jcb.147.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Chen X, Garelick MG, Wang H, Lil V, Athos J, Storm DR. PI3 kinase signaling is required for retrieval and extinction of contextual memory. Nat Neurosci. 2005;8:925–931. doi: 10.1038/nn1482. [DOI] [PubMed] [Google Scholar]
- Davis JB, Maher P. Protein kinase C activation inhibits glutamate-induced cytotoxicity in a neuronal cell line. Brain Res. 1994;652:169–173. doi: 10.1016/0006-8993(94)90334-4. [DOI] [PubMed] [Google Scholar]
- Dehghani F, Maronde E, Schachenmayr W, Korf H. Neurofilament H immunoreaction in oligodendrogliomas as demonstrated by a new polyclonal antibody. Acta Neuropathol (Berl) 2000;100:122–130. doi: 10.1007/s004010050003. [DOI] [PubMed] [Google Scholar]
- Eckel-Mahan KL, Phan T, Han S, Wang H, Chan GCK, Scheiner ZS, et al. Circadian oscillation of hippocampal MAPK activity and cAmp: implications for memory persistence. Nat Neurosci. 2008;11:1074–1082. doi: 10.1038/nn.2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Flajolet M, Wang Z, Futter M, Shen W, Nuangchamnong N, Bendor J, et al. FGF acts as a co-transmitter through adenosine A(2A) receptor to regulate synaptic plasticity. Nat Neurosci. 2008;11:1402–1409. doi: 10.1038/nn.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham ES, Ball N, Scotter EL, Narayan P, Dragunow M, Glass M. Induction of Krox-24 by endogenous cannabinoid type 1 receptors in neuro2A cells is mediated by the MEK–ERK MAPK pathway and is suppressed by the phosphatidylinositol 3-kinase pathway. J Biol Chem. 2006;281:29085–29095. doi: 10.1074/jbc.M602516200. [DOI] [PubMed] [Google Scholar]
- Hanai K, Oomura Y, Kai Y, Nishikawa K, Shimizu N, Morita H, et al. Central action of acidic fibroblast growth factor in feeding regulation. Am J Physiol. 1989;256:R217–R223. doi: 10.1152/ajpregu.1989.256.1.R217. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Sagara Y, Langford D, Everall IP, Mallory M, Everson A, et al. Fibroblast growth factor 1 regulates signalling via the glycogen synthase kinase-3beta pathway. Implications for neuroprotection. J Biol Chem. 2002;277:32985–32991. doi: 10.1074/jbc.M202803200. [DOI] [PubMed] [Google Scholar]
- Hausott B, Schlick B, Vallant N, Dorn R, Klimaschewski L. Promotion of neurite outgrowth by fibroblast growth factor receptor 1 overexpression and lysosomal inhibition of receptor degradation in pheochromocytoma cells and adult sensory neurons. Neuroscience. 2008;153:461–473. doi: 10.1016/j.neuroscience.2008.01.083. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Tsuchiya Y, Nakayama K, Satoh T, Nishida E. Down-regulation of the PI3-kinase/Akt pathway by ERK MAP kinase in growth factor signaling. Genes Cells. 2008;13:941–947. doi: 10.1111/j.1365-2443.2008.01218.x. [DOI] [PubMed] [Google Scholar]
- Ho Y, Samarasinghe R, Knoch ME, Lewis M, Aizenman E, DeFranco DB. Selective inhibition of mitogen-activated protein kinase phosphatases by zinc accounts for extracellular signal-regulated kinase 1/2-dependent oxidative neuronal cell death. Mol Pharmacol. 2008;74:1141–1151. doi: 10.1124/mol.108.049064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwood JM, Dufour F, Laroche S, Davis S. Signalling mechanisms mediated by the phosphoinositide 3-kinase/Akt cascade in synaptic plasticity and memory in the rat. Eur J Neurosci. 2006;23:3375–3384. doi: 10.1111/j.1460-9568.2006.04859.x. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci. 2005;25:11300–11312. doi: 10.1523/JNEUROSCI.2270-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MW, Errington ML, French PJ, Fine A, Bliss TV, Garel S, et al. A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat Neurosci. 2001;4:289–296. doi: 10.1038/85138. [DOI] [PubMed] [Google Scholar]
- Kaladchibachi SA, Doble B, Anthopoulos N, Woodgett JR, Manoukian AS. Glycogen synthase kinase 3, circadian rhythms, and bipolar disorder: a molecular link in the therapeutic action of lithium. J Circadian Rhythms. 2007;5:3. doi: 10.1186/1740-3391-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly A, Laroche S, Davis S. Activation of mitogen-activated protein kinase/extracellular signal-regulated kinase in hippocampal circuitry is required for consolidation and reconsolidation of recognition memory. J Neurosci. 2003;23:5354–5360. doi: 10.1523/JNEUROSCI.23-12-05354.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko S, Ao H, Mendel A, Qiu C, Wei F, Milbrandt J, et al. Transcription factor Egr-1 is required for long-term fear memory and anxiety. Sheng Li Xue Bao: [Acta Physiol Sinica] 2005;57:421–432. [PubMed] [Google Scholar]
- Liu J, Li L, Suo WZ. HT22 hippocampal neuronal cell line possesses functional cholinergic properties. Life Sci. 2009;84:267–271. doi: 10.1016/j.lfs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Malmlöf M, Roudier E, Högberg J, Stenius U. MEK–ERK-mediated phosphorylation of Mdm2 at Ser-166 in hepatocytes. Mdm2 is activated in response to inhibited Akt signalling. J Biol Chem. 2007;282:2288–2296. doi: 10.1074/jbc.M604953200. [DOI] [PubMed] [Google Scholar]
- Maronde E, Pfeffer M, Glass Y, Stehle JHJ. Transcription factor dynamics in pineal gland and liver of the Syrian hamster (Mesocricetus auratus) adapts to prevailing photoperiod. J Pineal Res. 2007;43:16–24. doi: 10.1111/j.1600-079X.2007.00438.x. [DOI] [PubMed] [Google Scholar]
- Oomura Y. Acidic fibroblast growth factor, a satiety substance, with diverse physiological significance. In. In: Phelps, Korneva, editors. NeuroImmune Biology: Cytokines and the Brain. Amsterdam: Elsevier; 2008. pp. 199–211. Vol. 6. Available at: http://www.sciencedirect.com/science/article/B8GWR-4SYS5CH-M/2/52713e908363dd792163aa259083a1cd (accessed 17 November 2008) [Google Scholar]
- Oomura Y, Sasaki K, Suzuki K, Muto T, Li AJ, Ogita Z, et al. A new brain glucosensor and its physiological significance. Am J Clin Nutr. 1992;55:278S–282S. doi: 10.1093/ajcn/55.1.278s. [DOI] [PubMed] [Google Scholar]
- Opazo P, Watabe AM, Grant SGN, O'Dell TJ. Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms. J Neurosci. 2003;23:3679–3688. doi: 10.1523/JNEUROSCI.23-09-03679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuss B, von Bohlen und Halbach O. Fibroblast growth factors and their receptors in the central nervous system. Cell Tissue Res. 2003;313:139–157. doi: 10.1007/s00441-003-0756-7. [DOI] [PubMed] [Google Scholar]
- Roberson ED, English JD, Sweatt JD. A biochemist's view of long-term potentiation. Learn Mem. 1996;3:1–24. doi: 10.1101/lm.3.1.1. [DOI] [PubMed] [Google Scholar]
- Sanna PP, Cammalleri M, Berton F, Simpson C, Lutjens R, Bloom FE, et al. Phosphatidylinositol 3-kinase is required for the expression but not for the induction or maintenance of long-term potentiation in the hippocampal CA1 region. J Neurosci. 2002;22:3359–3365. doi: 10.1523/JNEUROSCI.22-09-03359.2002. Erratum in: J Neurosci22:10507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K, Oomura Y, Figurov A, Yagi H. Acidic fibroblast growth factor facilitates generation of long-term potentiation in rat hippocampal slices. Brain Res Bull. 1994;33:505–511. doi: 10.1016/0361-9230(94)90075-2. [DOI] [PubMed] [Google Scholar]
- Sasaki K, Oomura Y, Li AJ, Hanai K, Tooyama I, Kimura H, et al. Actions of acidic fibroblast growth factor fragments on food intake in rats. Obes Res. 1995;3(Suppl 1):697S–706S. doi: 10.1002/j.1550-8528.1995.tb00488.x. [DOI] [PubMed] [Google Scholar]
- Spahn A, Blondeau N, Heurteaux C, Dehghani F, Rami A. Concomitant transitory up-regulation of X-linked inhibitor of apoptosis protein (XIAP) and the heterogeneous nuclear ribonucleoprotein C1–C2 in surviving cells during neuronal apoptosis. Neurochem Res. 2008;33:1859–1868. doi: 10.1007/s11064-008-9658-0. [DOI] [PubMed] [Google Scholar]
- Stanciu M, DeFranco DB. Prolonged nuclear retention of activated extracellular signal-regulated protein kinase promotes cell death generated by oxidative toxicity or proteasome inhibition in a neuronal cell line. J Biol Chem. 2002;277:4010–4017. doi: 10.1074/jbc.M104479200. [DOI] [PubMed] [Google Scholar]
- Stanciu M, Wang Y, Kentor R, Burke N, Watkins S, Kress G, et al. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J Biol Chem. 2000;275:12200–12206. doi: 10.1074/jbc.275.16.12200. [DOI] [PubMed] [Google Scholar]
- Steiger-Barraissoul S, Rami A. Serum deprivation induced autophagy and predominantly an AIF-dependent apoptosis in hippocampal HT22 neurons. Apoptosis. 2009;14:1274–1288. doi: 10.1007/s10495-009-0396-9. [DOI] [PubMed] [Google Scholar]
- Tohda C, Nakanishi R, Kadowaki M. Learning deficits and agenesis of synapses and myelinated axons in phosphoinositide-3 kinase-deficient mice. Neurosignals. 2007;15:293–306. doi: 10.1159/000108936. [DOI] [PubMed] [Google Scholar]
- Vougogiannopoulou K, Ferandin Y, Bettayeb K, Myrianthopoulos V, Lozach O, Fan Y, et al. Soluble 3′,6-substituted indirubins with enhanced selectivity toward glycogen synthase kinase-3 alter circadian period. J Med Chem. 2008;51:6421–6431. doi: 10.1021/jm800648y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicht H, Maronde E, Olcese J, Korf H. A semiquantitative image-analytical method for the recording of dose–response curves in immunocytochemical preparations. J Histochem Cytochem. 1999;47:411–419. doi: 10.1177/002215549904700315. [DOI] [PubMed] [Google Scholar]
- Wiegert JS, Bengtson CP, Bading H. Diffusion and not active transport underlies and limits ERK1/2 synapse-to-nucleus signalling in hippocampal neurons. J Biol Chem. 2007;282:29621–29633. doi: 10.1074/jbc.M701448200. [DOI] [PubMed] [Google Scholar]